Endothelial Aldehyde Dehydrogenase 2 as a Target to Maintain Vascular Wellness and Function in Ageing


Abstract
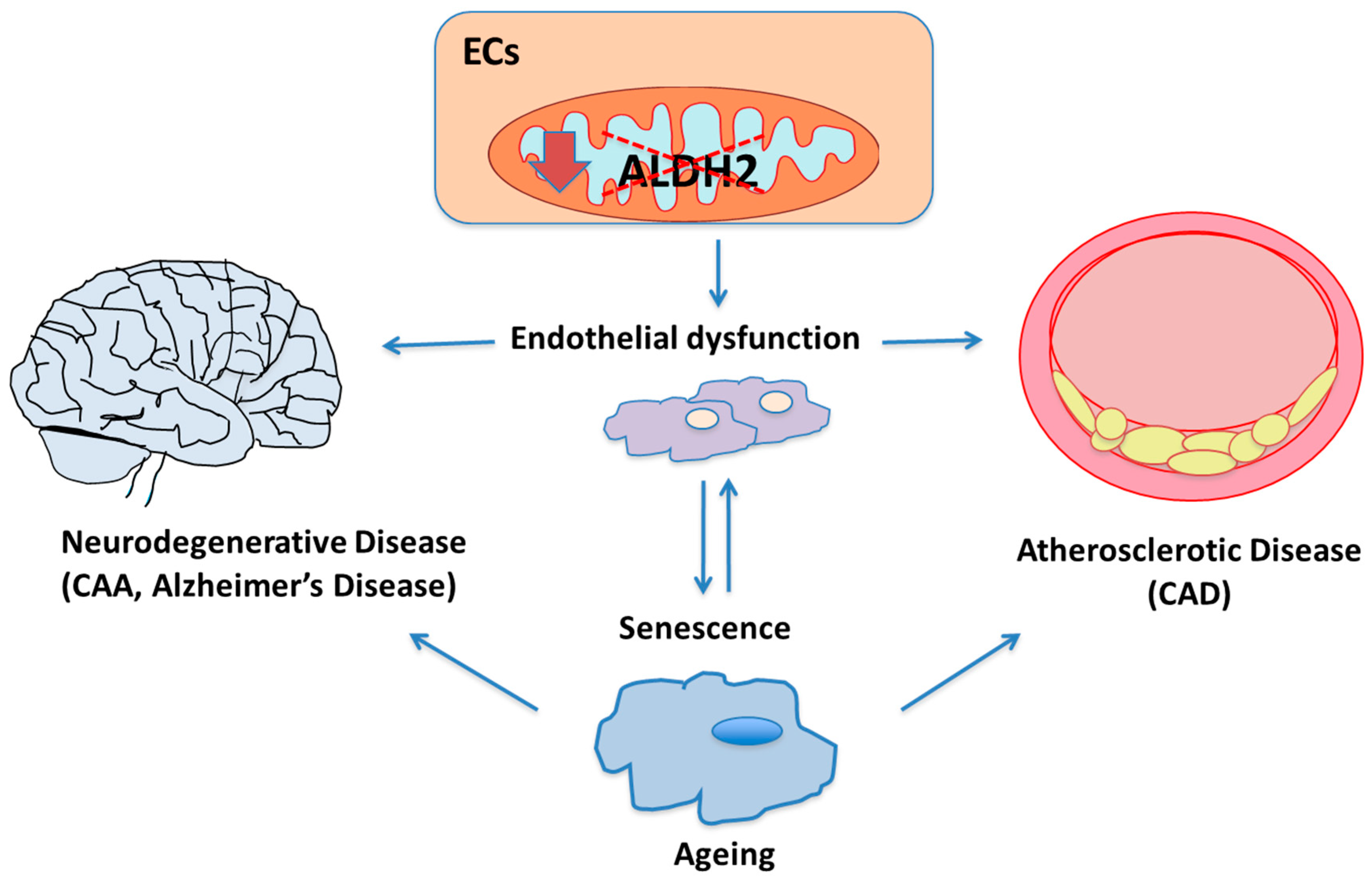
:1. Introduction
1.1. Endothelial Function and Dysfunction
1.2. Endothelial Senescence
1.3. Aldehyde Dehydrogenase-2 (ALDH2)
2. ALDH2 and Endothelial-Related Diseases
2.1. ALDH2, Oxidative Stress and Ageing
2.2. ALDH2 and Atherosclerosis
2.3. ALDH2 and Neurodegenerative Diseases
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wagner, D.D.; Frenette, P.S. The vessel wall and its interactions. Blood 2008, 111, 5271–5281. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daiber, A.; Oelze, M.; Wenzel, P.; Wickramanayake, J.M.; Schuhmacher, S.; Jansen, T.; Lackner, K.J.; Torzewski, M.; Münzel, T. Nitrate tolerance as a model of vascular dysfunction: Roles for mitochondrial aldehyde dehydrogenase and mitochondrial oxidative stress. Pharmacol. Rep. 2009, 61, 33–48. [Google Scholar] [CrossRef]
- Monti, M.; Donnini, S.; Giachetti, A.; Mochly-Rosen, D.; Ziche, M. deltaPKC inhibition or varepsilonPKC activation repairs endothelial vascular dysfunction by regulating eNOS post-translational modification. J. Mol. Cell. Cardiol. 2010, 48, 746–756. [Google Scholar] [CrossRef] [Green Version]
- Haybar, H.; Shahrabi, S.; Rezaeeyan, H.; Shirzad, R.; Saki, N. Endothelial Cells: From Dysfunction Mechanism to Pharmacological Effect in Cardiovascular Disease. Cardiovasc. Toxicol. 2019, 19, 13–22. [Google Scholar] [CrossRef]
- Donnini, S.; Cantara, S.; Morbidelli, L.; Giachetti, A.; Ziche, M. FGF-2 overexpression opposes the beta amyloid toxic injuries to the vascular endothelium. Cell Death Differ. 2006, 13, 1088–1096. [Google Scholar] [CrossRef] [Green Version]
- Morbidelli, L.; Donnini, S.; Ziche, M. Targeting endothelial cell metabolism for cardio-protection from the toxicity of antitumor agents. Cardio-Oncology 2016, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Tarantini, S.; Kiss, T.; Wren, J.; Giles, C.B.; Griffin, C.T.; Murfee, W.L.; Pacher, P.; Csiszar, A. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat. Rev. Cardiol. 2018, 15, 555–565. [Google Scholar] [CrossRef]
- Caruso, P.; Signori, R.; Moretti, R. Small vessel disease to subcortical dementia: A dynamic model, which interfaces ageing, cholinergic dysregulation and the neurovascular unit. Vasc. Health Risk Manag. 2019, 15, 259–281. [Google Scholar] [CrossRef]
- Trott, D.W.; Fadel, P.J. Inflammation as a mediator of arterial ageing. Exp. Physiol. 2019, 104, 1455–1471. [Google Scholar] [CrossRef]
- Hao, Y.M.; Yuan, H.Q.; Ren, Z.; Qu, S.L.; Liu, L.S.; Wei, D.-H.; Yin, K.; Fu, M.; Jiang, Z.S. Endothelial to mesenchymal transition in atherosclerotic vascular remodeling. Clin. Chim. Acta 2019, 490, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Machin, D.R.; Lesniewski, L.A. Mechanisms of Dysfunction in the Ageing Vasculature and Role in Age-Related Disease. Circ. Res. 2018, 123, 825–848. [Google Scholar] [CrossRef]
- Dantas, A.P.; Jiménez-Altayó, F.; Vila, E. Vascular ageing: Facts and factors. Front. Physiol. 2012, 3, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghebre, Y.T.; Yakubov, E.; Wong, W.T.; Krishnamurthy, P.; Sayed, N.; Sikora, A.G.; Bonnen, M.D. Vascular Ageing: Implications for Cardiovascular Disease and Therapy. Transl. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Sinclair, D.A. The intersection between ageing and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Masi, S.; Colucci, R.; Duranti, E.; Nannipieri, M.; Anselmino, M.; Ippolito, C.; Tirotta, E.; Georgiopoulos, G.; Garelli, F.; Nericcio, A.; et al. Ageing Modulates the Influence of Arginase on Endothelial Dysfunction in Obesity. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2474–2483. [Google Scholar] [CrossRef] [Green Version]
- Foote, K.; Reinhold, J.; Yu, E.P.K.; Figg, N.L.; Finigan, A.; Murphy, M.P.; Bennett, M.R. Restoring mitochondrial DNA copy number preserves mitochondrial function and delays vascular ageing in mice. Ageing Cell 2018, 17, e12773. [Google Scholar] [CrossRef]
- De Silva, T.M.; Li, Y.; Kinzenbaw, D.A.; Sigmund, C.D.; Faraci, F.M. Endothelial PPARγ (Peroxisome Proliferator-Activated Receptor-γ) Is Essential for Preventing Endothelial Dysfunction with Ageing. Hypertension 2018, 72, 227–234. [Google Scholar] [CrossRef]
- Cannizzo, E.S.; Clement, C.C.; Sahu, R.; Follo, C.; Santambrogio, L. Oxidative stress, inflamm-ageing and immunosenescence. J. Proteom. 2011, 74, 2313–2323. [Google Scholar] [CrossRef]
- Campisi, J.; Andersen, J.K.; Kapahi, P.; Melov, S. Cellular senescence: A link between cancer and age-related degenerative disease? Semin. Cancer Biol. 2011, 21, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lui, K.O.; Zhou, B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat. Rev. Cardiol. 2018, 15, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Thompson, D.C.; Smith, C.; Fujita, M.; Chen, Y. Aldehyde dehydrogenases: From eye crystallins to metabolic disease and cancer stem cells. Chem. Biol. Interact. 2013, 202, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Zavala, J.S.; Calleja, L.F.; Moreno-Sánchez, R.; Yoval-Sánchez, B. Role of Aldehyde Dehydrogenases in Physiopathological Processes. Chem. Res. Toxicol. 2019, 32, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Ferreira, J.C.; Gross, E.R.; Mochly-Rosen, D. Targeting aldehyde dehydrogenase 2: New therapeutic opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Rhinn, M.; Dollé, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [Green Version]
- Ciccone, V.; Terzuoli, E.; Donnini, S.; Giachetti, A.; Morbidelli, L.; Ziche, M. Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1α/VEGF signalling in MCF-7 breast cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 311, Correction in 2019, 38, 45. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, P.; Hink, U.; Oelze, M.; Seeling, A.; Isse, T.; Bruns, K.; Steinhoff, L.; Brandt, M.; Kleschyov, A.L.; Schulz, E.; et al. Number of nitrate groups determines reactivity and potency of organic nitrates: A proof of concept study in ALDH-2−/− mice. Br. J. Pharmacol. 2007, 150, 526–533. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Batista Ferreira, J.C.; Mochly-Rosen, D. ALDH2 activator inhibits increased myocardial infarction injury by nitroglycerin tolerance. Sci. Transl. Med. 2011, 3, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Opelt, M.; Eroglu, E.; Waldeck-Weiermair, M.; Russwurm, M.; Koesling, D.; Malli, R.; Graier, W.F.; Fassett, J.T.; Schrammel, A.; Mayer, B. Formation of Nitric Oxide by Aldehyde Dehydrogenase-2 Is Necessary and Sufficient for Vascular Bioactivation of Nitroglycerin. J. Biol. Chem. 2016, 291, 24076–24084. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhao, Q.; Ye, F.; Huang, C.Y.; Chen, W.M.; Huang, W.Q. Alda-1 an ALDH2 activator, protects against hepatic ischemia/reperfusion injury in rats via inhibition of oxidative stress. Free Radic. Res. 2018, 13, 1–10. [Google Scholar] [CrossRef]
- Wei, S.J.; Xing, J.H.; Wang, B.L.; Xue, L.; Wang, J.L.; Li, R.; Qin, W.D.; Wang, J.; Wang, X.P.; Zhang, M.X.; et al. Poly(ADPribose) polymerase inhibition prevents reactive oxygen species induced inhibition of aldehyde dehydrogenase2 activity. Biochem. Biophys. Acta 2013, 1833, 479–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.L.; Li, Y. Endothelial cell senescence and age-related vascular diseases. J. Genet. Genom. 2014, 41, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Sergiev, P.V.; Dontsova, O.A.; Berezkin, G.V. Theories of ageing: An ever-evolving field. Acta Nat. 2015, 7, 9–18. [Google Scholar] [CrossRef]
- Gladyshev, V.N. The free radical theory of ageing is dead. Long live the damage theory! Antioxid. Redox. Signal. 2014, 20, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I. Reactive oxygen species and the free radical theory of ageing. Free Radic. Biol. Med. 2013, 60, 1–4. [Google Scholar] [CrossRef]
- Gomes, K.M.; Campos, J.C.; Bechara, L.R.; Queliconi, B.; Lima, V.M.; Disatnik, M.H.; Magno, P.; Chen, C.H.; Brum, P.C.; Kowaltowski, A.J.; et al. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovasc. Res. 2014, 103, 498–508. [Google Scholar] [CrossRef]
- Gomes, K.M.; Bechara, L.R.; Lima, V.M.; Ribeiro, M.A.; Campos, J.C.; Dourado, P.M.; Kowaltowski, A.J.; Mochly-Rosen, D.; Ferreira, J.C. Aldehydic load and aldehyde dehydrogenase 2 profile during the progression of post-myocardial infarction cardiomyopathy: Benefits of Alda-1. Int. J. Cardiol. 2015, 179, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Panisello-Roselló, A.; Lopez, A.; Folch-Puy, E.; Carbonell, T.; Rolo, A.; Palmeira, C.; Adam, R.; Net, M.; Roselló-Catafau, J. Role of aldehyde dehydrogenase 2 in ischemia reperfusion injury: An update. World J. Gastroenterol. 2018, 24, 2984–2994. [Google Scholar] [CrossRef]
- Kimura, M.; Yokoyama, A.; Higuchi, S. Aldehyde dehydrogenase-2 as a therapeutic target. Expert Opin. Ther. Targets 2019, 23, 955–966. [Google Scholar] [CrossRef]
- Zhang, Y.; Mi, S.L.; Hu, N.; Doser, T.A.; Sun, A.; Ge, J.; Ren, J. Mitochondrial aldehyde dehydrogenase 2 accentuates ageing-induced cardiac remodeling and contractile dysfunction: Role of AMPK, Sirt1, and mitochondrial function. Free Radic. Biol. Med. 2014, 71, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Yu, L.; Wang, Y.; Wang, H.; Li, C.; Yin, Y.; Yang, J.; Wang, Z.; Zheng, Q.; Ma, H. Aldehyde dehydrogenase 2 activation in aged heart improves the autophagy by reducing the carbonyl modification on SIRT1. Oncotarget 2016, 7, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Nannelli, G.; Terzuoli, E.; Giorgio, V.; Donnini, S.; Lupetti, P.; Giachetti, A.; Bernardi, P.; Ziche, M. ALDH2 Activity Reduces Mitochondrial Oxygen Reserve Capacity in Endothelial Cells and Induces Senescence Properties. Oxid. Med. Cell Longev. 2018, 2018, 9765027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchidaand, K.; Stadtman, E.R. Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc. Natl. Acad. Sci. USA 1992, 89, 4544–4548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, F.-L.; Nath, R.G.; Ocando, J.; Nishikawa, A.; Zhang, L. Deoxyguanosine Adducts of t-4-Hydroxy-2-nonenal Are Endogenous DNA Lesions in Rodents and Humans: Detection and Potential Source. Cancer Res. 2000, 60, 1507–1511. [Google Scholar] [PubMed]
- Chen, C.H.; Joshi, A.U.; Mochly-Rosen, D. The Role of Mitochondrial Aldehyde Dehydrogenase 2 (ALDH2) in Neuropathology and Neurodegeneration. Acta Neurol. Taiwan 2016, 25, 111–123. [Google Scholar]
- Yoval-Sánchez, B.; Rodríguez-Zavala, J.S. Differences in susceptibility to inactivation of human aldehyde dehydrogenases by lipid peroxidation byproducts. Chem. Res. Toxicol. 2012, 25, 722–729. [Google Scholar] [CrossRef]
- Li, S.Y.; Gomelsky, M.; Duan, J.; Zhang, Z.; Gomelsky, L.; Zhang, X.; Epstein, P.N.; Ren, J. Overexpression of aldehyde dehydrogenase-2 (ALDH2) transgene prevents acetaldehyde-induced cell injury in human umbilical vein endothelial cells: Role of ERK and p38 mitogen-activated protein kinase. J. Biol. Chem. 2004, 279, 11244–11252. [Google Scholar] [CrossRef] [Green Version]
- Solito, R.; Corti, F.; Chen, C.H.; Mochly-Rosen, D.; Giachetti, A.; Ziche, M.; Donnini, S. Mitochondrial aldehyde dehydrogenase-2 activation prevents β-amyloid-induced endothelial cell dysfunction and restores angiogenesis. J. Cell Sci. 2013, 126 Pt 9, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Pang, J.; Wang, J.; Zhang, Y.; Xu, F.; Chen, Y. Targeting acetaldehyde dehydrogenase 2 (ALDH2) in heart failure-Recent insights and perspectives. Biochem. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1933–1941. [Google Scholar] [CrossRef]
- Zhang, L.L.; Wang, Y.Q.; Fu, B.; Zhao, S.L.; Kui, Y. Aldehyde dehydrogenase 2 (ALDH2) polymorphism gene and coronary artery disease risk: A meta-analysis. Genet. Mol. Res. 2015, 14, 18503–18514. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, S.; Wang, L.; Lei, M.; Wang, Y.; Miao, C.; Jin, Y. ALDH2 rs671 Polymorphism and coronary heart disease risk among Asian populations: A meta-analysis and meta-regression. DNA Cell Biol. 2013, 32, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Yasue, H.; Mizuno, Y.; Harada, E. Association of East Asian Variant Aldehyde Dehydrogenase 2 Genotype (ALDH2*2*) with Coronary Spasm and Acute Myocardial Infarction. Adv. Exp. Med. Biol. 2019, 1193, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, ageing, and neurodegeneration. Front. Ageing Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, L.; Xu, F.; Meng, L.; Wei, S.; Wang, J.; Hao, P.; Bian, Y.; Zhang, Y.; Chen, Y. Acetylation-dependent regulation of mitochondrial ALDH2 activation by SIRT3 mediates acute ethanol-induced eNOS activation. FEBS Lett. 2012, 586, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Xing, J.H.; Zhang, C.; Zhang, Y.M.; Zhang, L.T.; Wei, S.J.; Zhang, M.X.; Wang, X.P.; Yuan, Q.H.; Xue, L.; et al. Aldehyde dehydrogenase 2 inhibits inflammatory response and regulates atherosclerotic plaque. Oncotarget 2016, 7, 35562–35576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzuoli, E.; Nannelli, G.; Giachetti, A.; Morbidelli, L.; Ziche, M.; Donnini, S. Targeting endothelial-to-mesenchymal transition: The protective role of hydroxytyrosol sulfate metabolite. Eur. J. Nutr. 2019. [Google Scholar] [CrossRef]
- Tressera-Rimbau, A.; Arranz, S.; Eder, M.; Vallverdú-Queralt, A. Dietary Polyphenols in the Prevention of Stroke. Oxid. Med. Cell. Longev. 2017, 2017, 7467962. [Google Scholar] [CrossRef] [Green Version]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 2003, 64, 7–10. [Google Scholar]
- Piaceri, I.; Nacmias, B.; Sorbi, S. Genetics of familial and sporadic Alzheimer’s disease. Front. Biosci. 2015, 5, 167–177. [Google Scholar]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [Green Version]
- Lyros, E.; Bakogiannis, C.; Liu, Y.; Fassbender, K. Molecular links between endothelial dysfunction and neurodegeneration in Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Revesz, T.; Ghiso, J.; Lashley, T.; Plant, G.; Rostagno, A.; Frangione, B.; Holton, J.L. Cerebral amyloid angiopathies: A pathologic, biochemical, and genetic view. J. Neuropathol. Exp. Neurol. 2003, 62, 885–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.S.; Mathura, V.S.; Bachmeier, C.; Beaulieu-Abdelahad, D.; Laporte, V.; Weeks, O.; Mullan, M.; Paris, D. Alzheimer’s beta-amyloid peptide blocks vascular endothelial growth factor mediated signaling via direct interaction with VEGFR-2. J. Neurochem. 2010, 112, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Solito, R.; Corti, F.; Fossati, S.; Mezhericher, E.; Donnini, S.; Ghiso, J.; Giachetti, A.; Rostagno, A.; Ziche, M. Dutch and Arctic mutant peptides of beta amyloid(1-40) differentially affect the FGF-2 pathway in brain endothelium. Exp. Cell Res. 2009, 315, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Siegel, S.J.; Bieschke, J.; Powers, E.T.; Kelly, J.W. The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry 2007, 46, 1503–1510. [Google Scholar] [CrossRef] [Green Version]
- Donnini, S.; Solito, R.; Cetti, E.; Corti, F.; Giachetti, A.; Carra, S.; Beltrame, M.; Cotelli, F.; Ziche, M. Abeta peptides accelerate the senescence of endothelial cells in vitro and in vivo, impairing angiogenesis. FASEB J. 2010, 24, 2385–2395. [Google Scholar] [CrossRef]
- D’souza, Y.; Elharram, A.; Soon-Shiong, R.; Andrew, R.D.; Bennett, B.M. Characterization of Aldh2−/− mice as an age-related model of cognitive impairment and Alzheimer’s disease. Mol. Brain 2015, 8, 27. [Google Scholar] [CrossRef]


{kind=link}
| ALDH2 Status | Tissue/Cells | Molecular Mechanism | Function | Ref. |
|---|---|---|---|---|
| ALDH2 and vascular ageing | ||||
| ALDH2 activation | Heart (rat) | (−) aldehydic adducts (carbonylation) | (−) cardiac dysfunction | [28] |
| ALDH2 activation | Liver (rat) | (−) ROS (−) pro-inflammatory cytokines (−) 4-HNE and MDA | (−) tissue apoptosis (+) mitochondrial membrane potential | [30] |
| ALDH2 transgenic mice | Heart | (+) ROS (−) AMPK phosphorylation (−) SIRT1 | (+) ageing-induced cardiac hypertrophy (+) apoptosis (+) mitochondrial injury | [40] |
| ALDH2 activation | Heart (mouse) | (−) 4-HNE-protein adducts (−) protein carbonyls (+) autophagy flux (+) SIRT1 | (+) cardiac function | [41] |
| ALDH2 gene silencing | Endothelial cells | (+) ROS (+) 4-HNE | (−) respiration (+) senescence | [42] |
| ALDH2 gene transfection | Endothelial cells | (−) ROS | (−) apoptosis (−) ERK/p38-MAPK | [47] |
| ALDH2 and atherosclerosis | ||||
| ALDH2*2 loss-of-function | --- | (+) ox-LDLs | (+) coronary artery disease | [50,51,52] |
| ALDH2 gene silencing | ApoE−/− mice | (+) ROS | (+) vessel wall inflammation (+) plaque instability | [55] |
| ALDH2 activation | Endothelial cells | (−) ROS; (+) eNOS (−) SIRT3 activation | --- | [54] |
| ALDH2 inhibition | Endothelial cells | (+) NF-κB (+) AP-1 | (+) permeability (+) plaque formation | [55] |
| ALDH2 and neurodegenerative diseases | ||||
| ALDH2*2 loss-of-function | Brain | --- | (+) incidence of Alzheimer’s disease | [45] |
| ALDH2−/− mouse model | Brain | (+) 4-HNE adducts (+) Aβ | (+) cognitive deficits (+) endothelial dysfunction (+) Aβ in microvessels | [47] |
| ALDH2 inactivation | Endothelial cells | (+) 4-HNE | (−) angiogenesis (+) senescence | [48] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nannelli, G.; Ziche, M.; Donnini, S.; Morbidelli, L. Endothelial Aldehyde Dehydrogenase 2 as a Target to Maintain Vascular Wellness and Function in Ageing. Biomedicines 2020, 8, 4. https://doi.org/10.3390/biomedicines8010004
Nannelli G, Ziche M, Donnini S, Morbidelli L. Endothelial Aldehyde Dehydrogenase 2 as a Target to Maintain Vascular Wellness and Function in Ageing. Biomedicines. 2020; 8(1):4. https://doi.org/10.3390/biomedicines8010004
Chicago/Turabian StyleNannelli, Ginevra, Marina Ziche, Sandra Donnini, and Lucia Morbidelli. 2020. "Endothelial Aldehyde Dehydrogenase 2 as a Target to Maintain Vascular Wellness and Function in Ageing" Biomedicines 8, no. 1: 4. https://doi.org/10.3390/biomedicines8010004