
USP7 Deregulation Impairs S Phase Specific DNA Repair after Irradiation in Breast Cancer Cells

 , and
, and 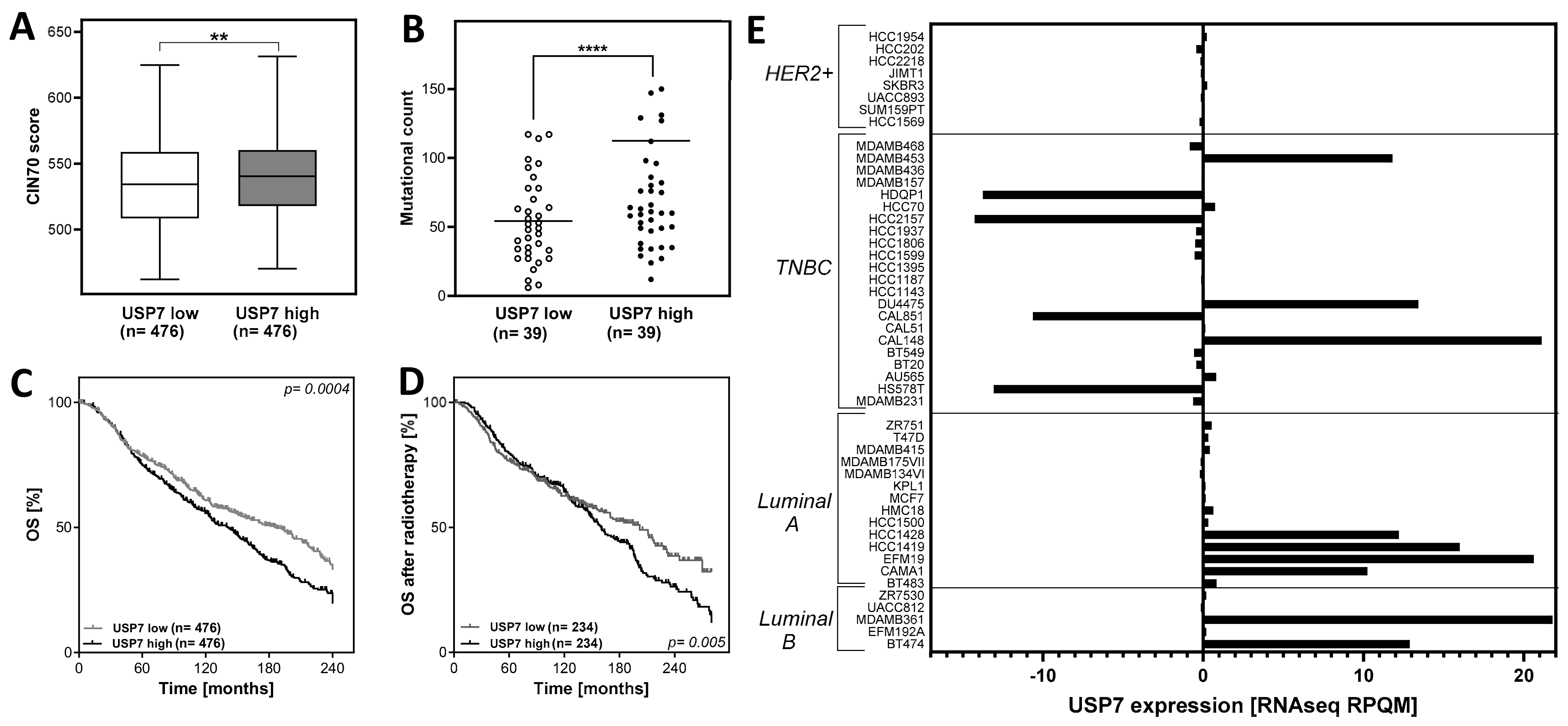{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Role of Ubiquitin Specific Protease 7 (USP7) in Cancer
1.2. The Impact of USP7 for Radiation Response
2. Materials and Methods
2.1. Clinical In Silico Analysis
2.2. Cell Culture and Treatments
2.3. Stable Transfection
2.4. Western Blot
2.5. Colony Formation Assay
2.6. Immunofluorescence
2.7. Cell Cycle Analysis
2.8. Statistical Analysis
3. Results
3.1. Abnormal USP7 Expression Affects CIN70 Dependent Chromosomal Instability and Overall Survival in Breast Cancer
3.2. Differences in USP7 Expression among Breast Cancer Subtypes and Cell Lines
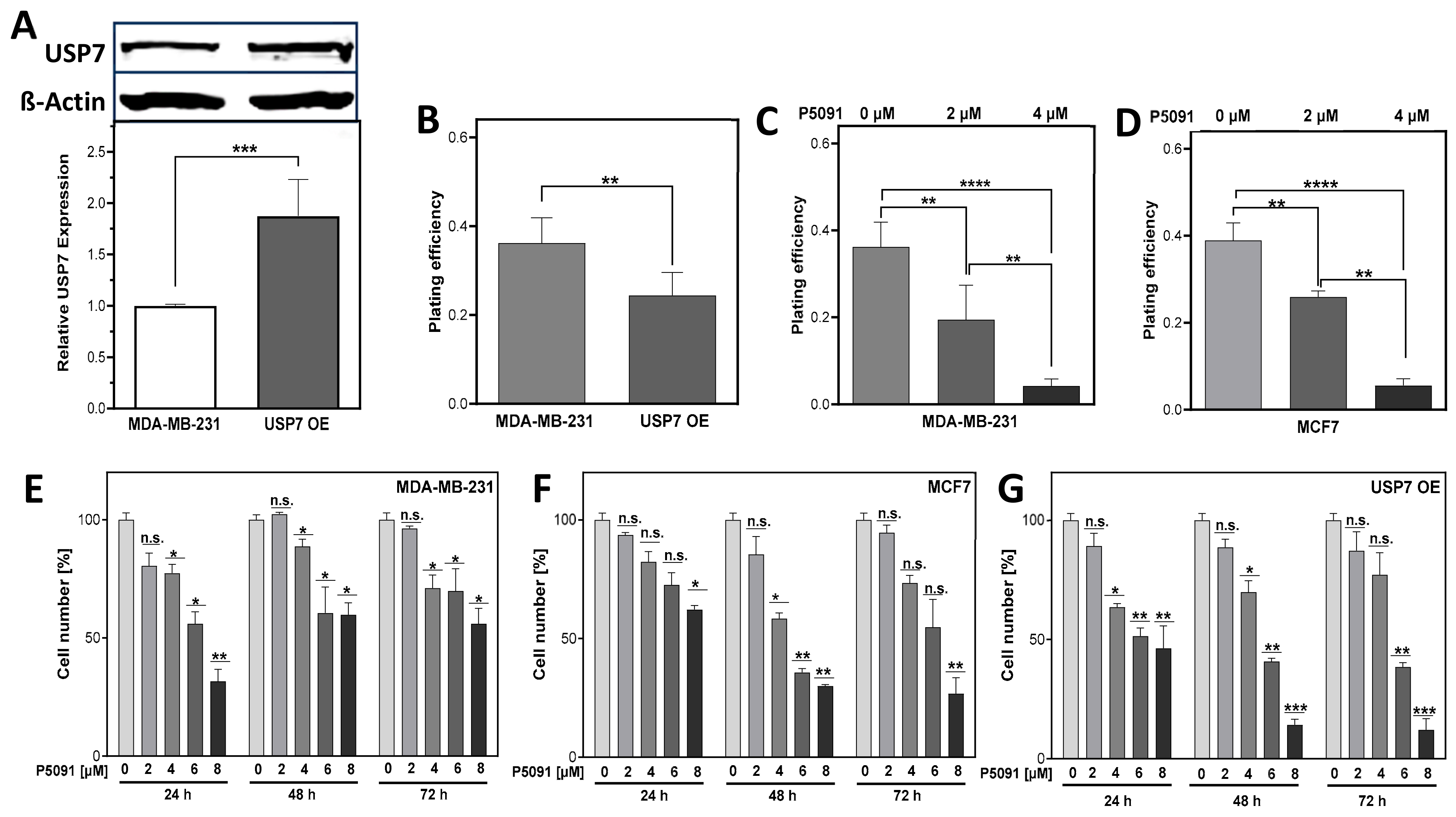
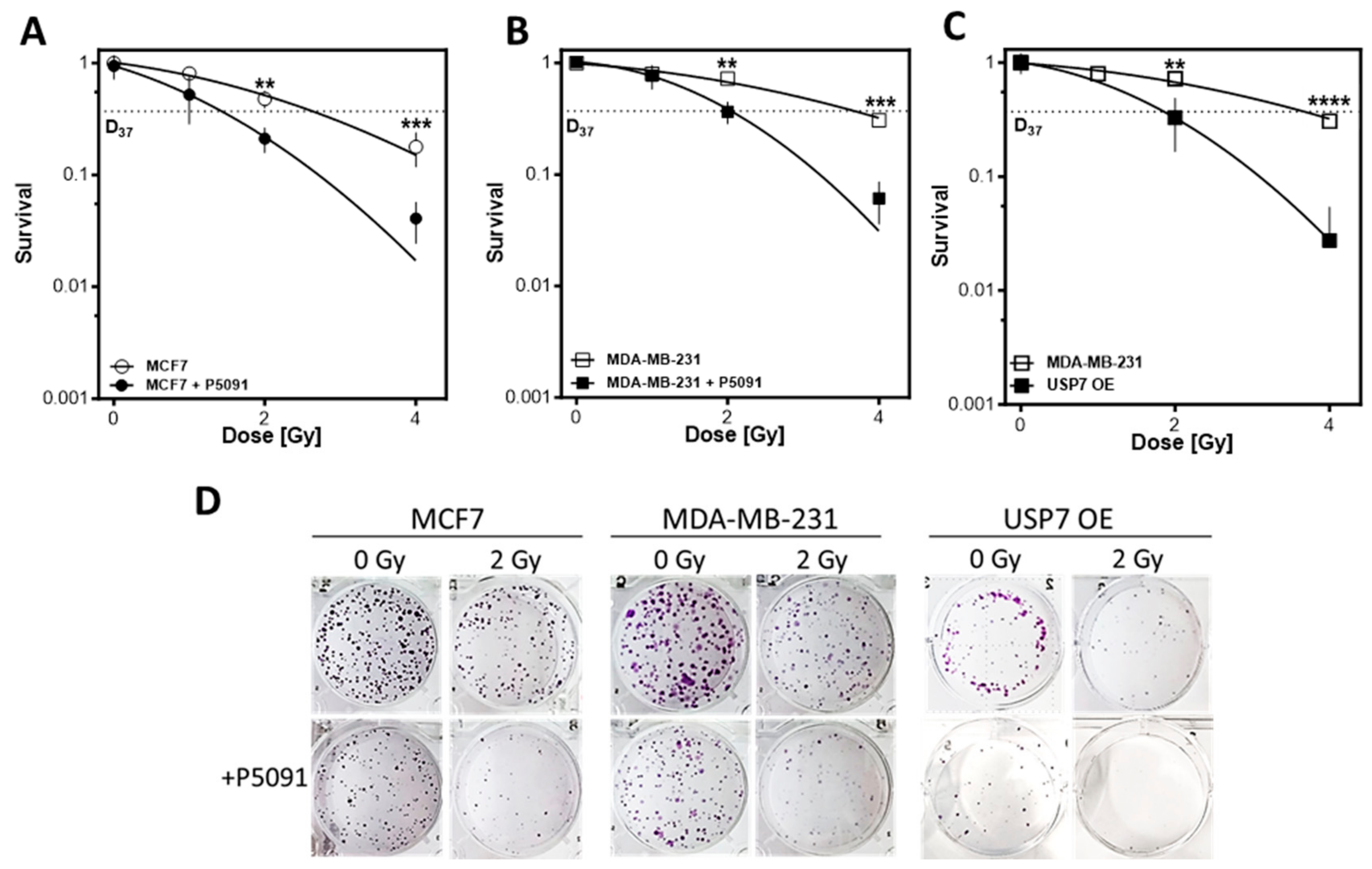
3.3. USP7 Inhibition and Overexpression Lead to Lower Cellular Survival
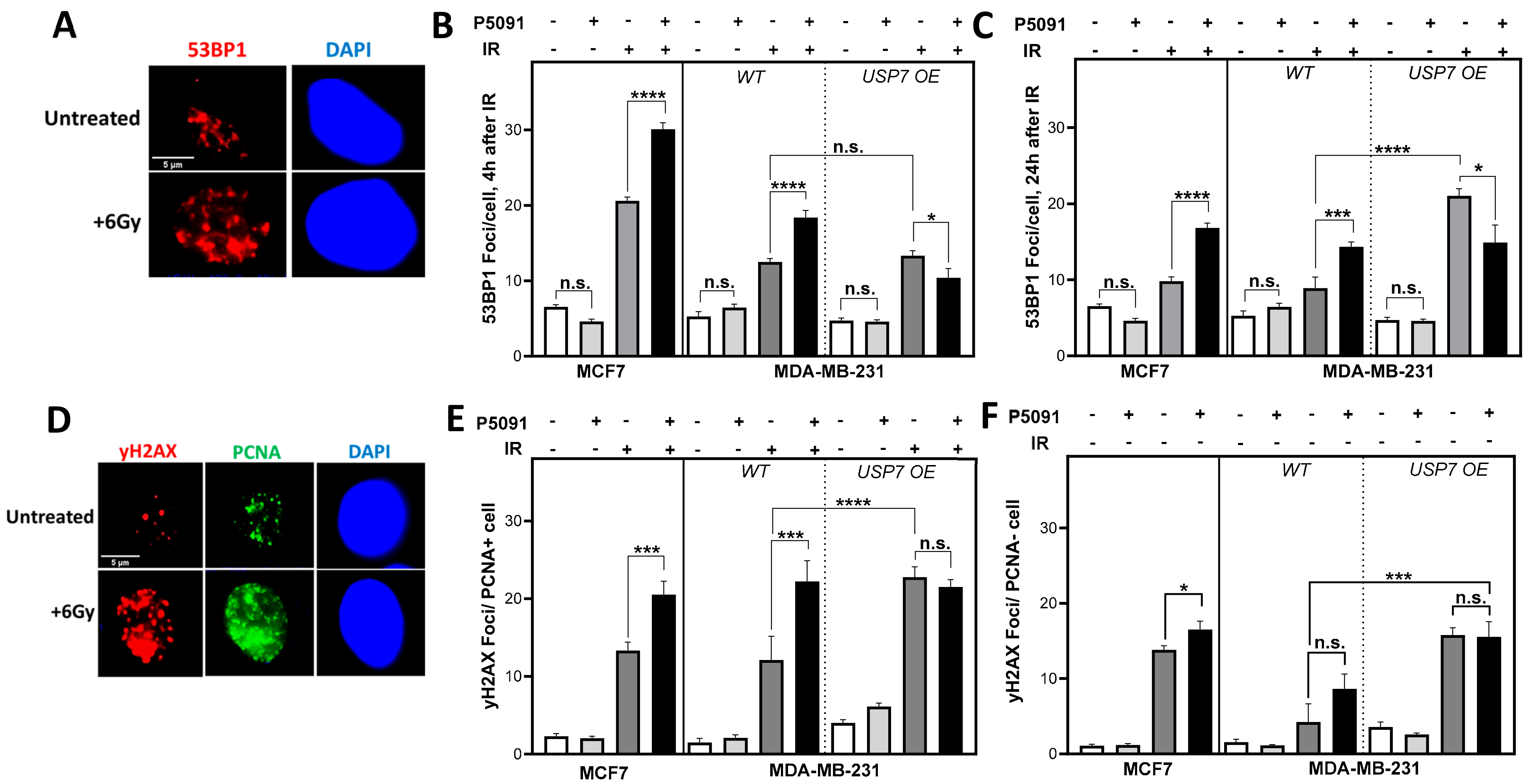
3.4. USP7 Inhibition and Overexpression Lead to Increased DNA Damage after Irradiation in S Phase
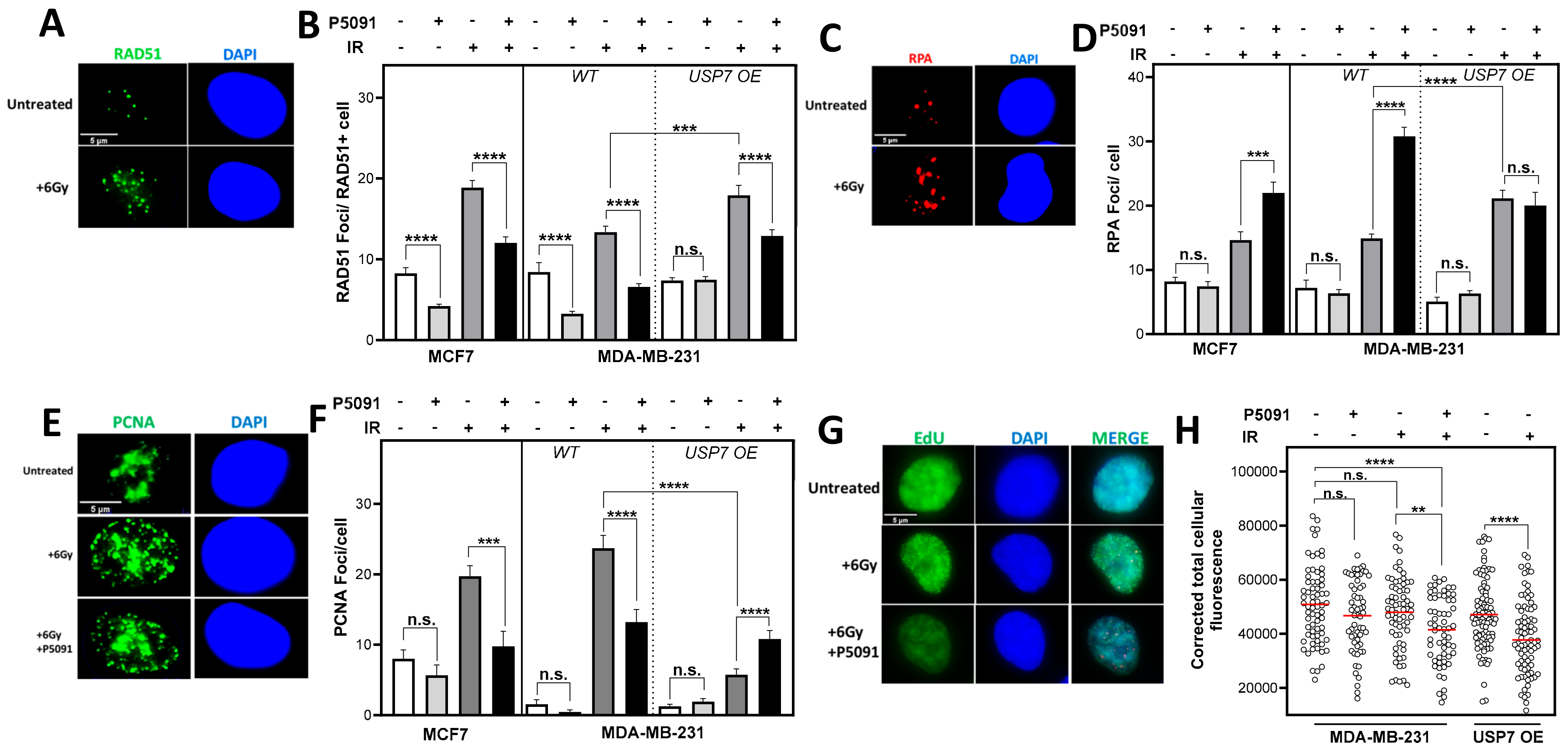
3.5. Altered USP7 Expression Affects RAD51 Foci Formation and Compromises DNA Replication
3.6. USP7 Deregulation Leads to Distinct Radiosensitization
4. Discussion
4.1. Effect of USP7 Overexpression on Survival and CIN70 of Breast Cancer Patients
4.2. USP7 Overexpression Enhances Radiosensitivity Due to Increased Replication Stress
4.3. USP7 Inhibition Affects Cell Survival and DNA Replication after Irradiation in Breast Cancer Cells
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. 2), S26–S35. [Google Scholar] [CrossRef]
- Ades, F.; Zardavas, D.; Bozovic-Spasojevic, I.; Pugliano, L.; Fumagalli, D.; de Azambuja, E.; Viale, G.; Sotiriou, C.; Piccart, M. Luminal B breast cancer: Molecular characterization, clinical management, and future perspectives. J. Clin. Oncol. 2014, 32, 2794–2803. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.M.; Cole, S.R.; Tse, C.-K.; Perou, C.M.; Carey, L.A.; Foulkes, W.D.; Dressler, L.G.; Geradts, J.; Millikan, R.C. Intrinsic breast tumor subtypes, race, and long-term survival in the Carolina Breast Cancer Study. Clin. Cancer Res. 2010, 16, 6100–6110. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Nofech-Mozes, S.; Trudeau, M.; Kahn, H.K.; Dent, R.; Rawlinson, E.; Sun, P.; Narod, S.A.; Hanna, W.M. Patterns of recurrence in the basal and non-basal subtypes of triple-negative breast cancers. Breast Cancer Res. Treat. 2009, 118, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Afifi, A.M.; Saad, A.M.; Al-Husseini, M.J.; Elmehrath, A.O.; Northfelt, D.W.; Sonbol, M.B. Causes of death after breast cancer diagnosis: A US population-based analysis. Cancer 2020, 126, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic Instability in Cancer: Teetering on the Limit of Tolerance. Cancer Res. 2017, 77, 2179–2185. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254. [Google Scholar] [CrossRef]
- Meyer, F.; Becker, S.; Classen, S.; Parplys, A.C.; Mansour, W.Y.; Riepen, B.; Timm, S.; Ruebe, C.; Jasin, M.; Wikman, H.; et al. Prevention of DNA Replication Stress by CHK1 Leads to Chemoresistance Despite a DNA Repair Defect in Homologous Recombination in Breast Cancer. Cells 2020, 9, 238. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining ‘chromosomal instability’. Trends. Genet 2008, 24, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; Hewish, M.; Lord, C.J.; Ashworth, A. Genomic instability and the selection of treatments for cancer. J. Pathol. 2010, 220, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A. The ubiquitin system for protein degradation and some of its roles in the control of the cell-division cycle (Nobel lecture). Angew. Chem. Int. Ed. Engl. 2005, 44, 5932–5943. [Google Scholar] [CrossRef] [PubMed]
- Nininahazwe, L.; Liu, B.; He, C.; Zhang, H.; Chen, Z.-S. The emerging nature of Ubiquitin-specific protease 7 (USP7): A new target in cancer therapy. Drug Discov. Today 2021, 26, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhao, H.; Yu, C.; Kang, Y.; Yang, X. Targeting Ubiquitin-Specific Protease 7 (USP7) in Cancer: A New Insight to Overcome Drug Resistance. Front. Pharmacol. 2021, 12, 648491. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Q.; Wang, Y.; Zhuang, H.; Chen, B. Clinical Significance of Ubiquitin Specific Protease 7 (USP7) in Predicting Prognosis of Hepatocellular Carcinoma and its Functional Mechanisms. Med. Sci. Monit. 2018, 24, 1742–1750. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; He, J.; Zhang, J.; Liu, B.; Liu, X.; Xie, L.; Wei, M.; Dong, R.; Li, K.; Ma, D.; et al. USP7 promotes hepatoblastoma progression through activation of PI3K/AKT signaling pathway. Cancer Biomark. 2021, 31, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Kisaï, K.; Koji, S. Prognostic role of USP7 expression in cancer patients: A systematic review and meta-analysis. Pathol. Res. Pract. 2021, 227, 153621. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Pérez, S.; Cabrera, E.; Salido, E.; Lim, M.; Reid, L.; Lakhani, S.R.; Khanna, K.K.; Saunus, J.M.; Freire, R. DUB3 and USP7 de-ubiquitinating enzymes control replication inhibitor Geminin: Molecular characterization and associations with breast cancer. Oncogene 2017, 36, 4802–4809. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Liao, Y.; Huang, C.; Liu, Y.; He, J.; Shao, Z.; Jiang, L.; Dou, Q.P.; Liu, J.; Huang, H. Deubiquitination and stabilization of estrogen receptor alpha by ubiquitin-specific protease 7 promotes breast tumorigenesis. Cancer Lett. 2019, 465, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Valles, G.J.; Bezsonova, I.; Woodgate, R.; Ashton, N.W. USP7 Is a Master Regulator of Genome Stability. Front. Cell Dev. Biol. 2020, 8, 717. [Google Scholar] [CrossRef] [PubMed]
- Galarreta, A.; Valledor, P.; Ubieto-Capella, P.; Lafarga, V.; Zarzuela, E.; Muñoz, J.; Malumbres, M.; Lecona, E.; Fernandez-Capetillo, O. USP7 limits CDK1 activity throughout the cell cycle. EMBO J. 2021, 40, e99692. [Google Scholar] [CrossRef] [PubMed]
- Lecona, E.; Rodriguez-Acebes, S.; Specks, J.; Lopez-Contreras, A.J.; Ruppen, I.; Murga, M.; Muñoz, J.; Mendez, J.; Fernandez-Capetillo, O. USP7 is a SUMO deubiquitinase essential for DNA replication. Nat. Struct. Mol. Biol. 2016, 23, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, M.; Nguyen, T.; Gallo, D.; Luthra, N.; Brown, G.W.; Saridakis, V.; Frappier, L. A role for USP7 in DNA replication. Mol. Cell. Biol. 2014, 34, 132–145. [Google Scholar] [CrossRef] [PubMed]
- McGarry, T.J.; Kirschner, M.W. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 1998, 93, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Vega, I.A.-D.; Martín, Y.; Smits, V.A. USP7 controls Chk1 protein stability by direct deubiquitination. Cell Cycle 2014, 13, 3921–3926. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Ma, S.; Shan, L.; Wang, Y.; Wang, Y.; Cao, C.; Liu, B.; Yang, C.; Wang, L.; Tian, S.; et al. Ubiquitin-specific protease 7 sustains DNA damage response and promotes cervical carcinogenesis. J. Clin. Investig. 2018, 128, 4280–4296. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ma, S.; Song, N.; Li, X.; Liu, L.; Yang, S.; Ding, X.; Shan, L.; Zhou, X.; Su, D.; et al. Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J. Clin. Investig. 2016, 126, 2205–2220. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Zhu, Y.; Wang, J.; Wang, T.; Wang, X.; Yan, L. Effects of USP7 on radiation sensitivity through p53 pathway in laryngeal squamous cell carcinoma. Transl. Oncol. 2022, 22, 101466. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Marchenko, N.; Palacios, G.; Moll, U.M. A role of HAUSP in tumor suppression in a human colon carcinoma xenograft model. Cell Cycle 2008, 7, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Molkentine, D.P.; Molkentine, J.M.; Bridges, K.A.; Valdecanas, D.R.; Dhawan, A.; Bahri, R.; Hefner, A.J.; Kumar, M.; Yang, L.; Abdelhakiem, M.; et al. p16 Represses DNA Damage Repair via a Novel Ubiquitin-Dependent Signaling Cascade. Cancer Res. 2022, 82, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Eklund, A.C.; Li, Q.; McClelland, S.E.; Endesfelder, D.; Tan, P.; Tan, I.B.; Richardson, A.L.; Szallasi, Z.; Swanton, C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011, 71, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.I.; Guedes, R.A.; Salvador, J.A.R. Highlights in USP7 inhibitors for cancer treatment. Front. Chem. 2022, 10, 1005727. [Google Scholar] [CrossRef]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 2014, 16, 864–875. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, Y.; Wang, T.; Zhang, J.; Zhou, Z.; Sun, Y.; Wang, S.; Shi, Y.; Luan, X.; Zhang, Y.; et al. The USP7 Inhibitor P5091 Induces Cell Death in Ovarian Cancers with Different P53 Status. Cell Physiol. Biochem. 2017, 43, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Naipal, K.A.; Verkaik, N.S.; Ameziane, N.; van Deurzen, C.H.; ter Brugge, P.; Meijers, M.; Sieuwerts, A.M.; Martens, J.W.; O’Connor, M.J.; Vrieling, H.; et al. Functional ex vivo assay to select homologous recombination-deficient breast tumors for PARP inhibitor treatment. Clin. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, Y.; Hyodo, M.; Suzuki, M.; Tanaka, Y.; Horikoshi, Y.; Murakami, Y.; Torigoe, H.; Mano, H.; Tashiro, S.; Yoshioka, K.-I. Replication-stress-associated DSBs induced by ionizing radiation risk genomic destabilization and associated clonal evolution. iScience 2021, 24, 102313. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Köcher, S.; Rieckmann, T.; Rohaly, G.; Mansour, W.Y.; Dikomey, E.; Dornreiter, I.; Dahm-Daphi, J. Radiation-induced double-strand breaks require ATM but not Artemis for homologous recombination during S-phase. Nucleic Acids Res. 2012, 40, 8336–8347. [Google Scholar] [CrossRef]
- Zhao, G.-Y.; Lin, Z.-W.; Lu, C.-L.; Gu, J.; Yuan, Y.-F.; Xu, F.-K.; Liu, R.-H.; Ge, D.; Ding, J.-Y. USP7 overexpression predicts a poor prognosis in lung squamous cell carcinoma and large cell carcinoma. Tumour. Biol. 2015, 36, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Giovinazzi, S.; Morozov, V.M.; Summers, M.K.; Reinhold, W.C.; Ishov, A.M. USP7 and Daxx regulate mitosis progression and taxane sensitivity by affecting stability of Aurora-A kinase. Cell Death Differ. 2013, 20, 721–731. [Google Scholar] [CrossRef]
- Peng, Y.; Liu, Y.; Gao, Y.; Yuan, B.; Qi, X.; Fu, Y.; Zhu, Q.; Cao, T.; Zhang, S.; Yin, L.; et al. USP7 is a novel Deubiquitinase sustaining PLK1 protein stability and regulating chromosome alignment in mitosis. J. Exp. Clin. Cancer Res. 2019, 38, 468. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Jiang, Y.; Ng, H.H.W.; Man, E.P.S.; Chen, J.; Khoo, U.-S.; Gong, Q.; Huen, M.S.Y. Dual-utility NLS drives RNF169-dependent DNA damage responses. Proc. Natl. Acad. Sci. USA 2017, 114, E2872–E2881. [Google Scholar] [CrossRef] [PubMed]
- Agathanggelou, A.; Smith, E.; Davies, N.J.; Kwok, M.; Zlatanou, A.; Oldreive, C.E.; Mao, J.; Da Costa, D.; Yadollahi, S.; Perry, T.; et al. USP7 inhibition alters homologous recombination repair and targets CLL cells independently of ATM/p53 functional status. Blood 2017, 130, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Malapelle, U.; Morra, F.; Ilardi, G.; Visconti, R.; Merolla, F.; Cerrato, A.; Napolitano, V.; Monaco, R.; Guggino, G.; Monaco, G.; et al. USP7 inhibitors, downregulating CCDC6, sensitize lung neuroendocrine cancer cells to PARP-inhibitor drugs. Lung Cancer 2017, 107, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, J.; Wang, Y.; Liu, X.; Yang, X.; Liao, Z.; Deng, S.; Deng, Y.; Zhou, Z.; Tian, Y.; et al. Deubiquitinase USP7 stabilizes KDM5B and promotes tumor progression and cisplatin resistance in nasopharyngeal carcinoma through the ZBTB16/TOP2A axis. Cell Death Differ. 2024, 31, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Grunblatt, E.; Wu, N.; Zhang, H.; Liu, X.; Norton, J.P.; Ohol, Y.; Leger, P.; Hiatt, J.B.; Eastwood, E.C.; Thomas, R.; et al. MYCN drives chemoresistance in small cell lung cancer while USP7 inhibition can restore chemosensitivity. Genes Dev. 2020, 34, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Sharma, N.; He, J.; Wani, G.; Wani, A.A. USP7 deubiquitinase promotes ubiquitin-dependent DNA damage signaling by stabilizing RNF168. Cell Cycle 2015, 14, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.G.S.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef] [PubMed]
- An, T.; Gong, Y.; Li, X.; Kong, L.; Ma, P.; Gong, L.; Zhu, H.; Yu, C.; Liu, J.; Zhou, H.; et al. USP7 inhibitor P5091 inhibits Wnt signaling and colorectal tumor growth. Biochem. Pharmacol. 2017, 131, 29–39. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vogt, M.; Classen, S.; Krause, A.K.; Peter, N.-J.; Petersen, C.; Rothkamm, K.; Borgmann, K.; Meyer, F. USP7 Deregulation Impairs S Phase Specific DNA Repair after Irradiation in Breast Cancer Cells. Biomedicines 2024, 12, 762. https://doi.org/10.3390/biomedicines12040762
Vogt M, Classen S, Krause AK, Peter N-J, Petersen C, Rothkamm K, Borgmann K, Meyer F. USP7 Deregulation Impairs S Phase Specific DNA Repair after Irradiation in Breast Cancer Cells. Biomedicines. 2024; 12(4):762. https://doi.org/10.3390/biomedicines12040762
Chicago/Turabian StyleVogt, Marie, Sandra Classen, Ann Kristin Krause, Nadja-Juanita Peter, Cordula Petersen, Kai Rothkamm, Kerstin Borgmann, and Felix Meyer. 2024. "USP7 Deregulation Impairs S Phase Specific DNA Repair after Irradiation in Breast Cancer Cells" Biomedicines 12, no. 4: 762. https://doi.org/10.3390/biomedicines12040762