
The Human Soluble NKG2D Ligand Differentially Impacts Tumorigenicity and Progression in Temporal and Model-Dependent Modes
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Mice and Cell Lines
2.2. In Vivo Experiments
2.3. Flow Cytometry Analysis
2.4. Statistics
3. Results
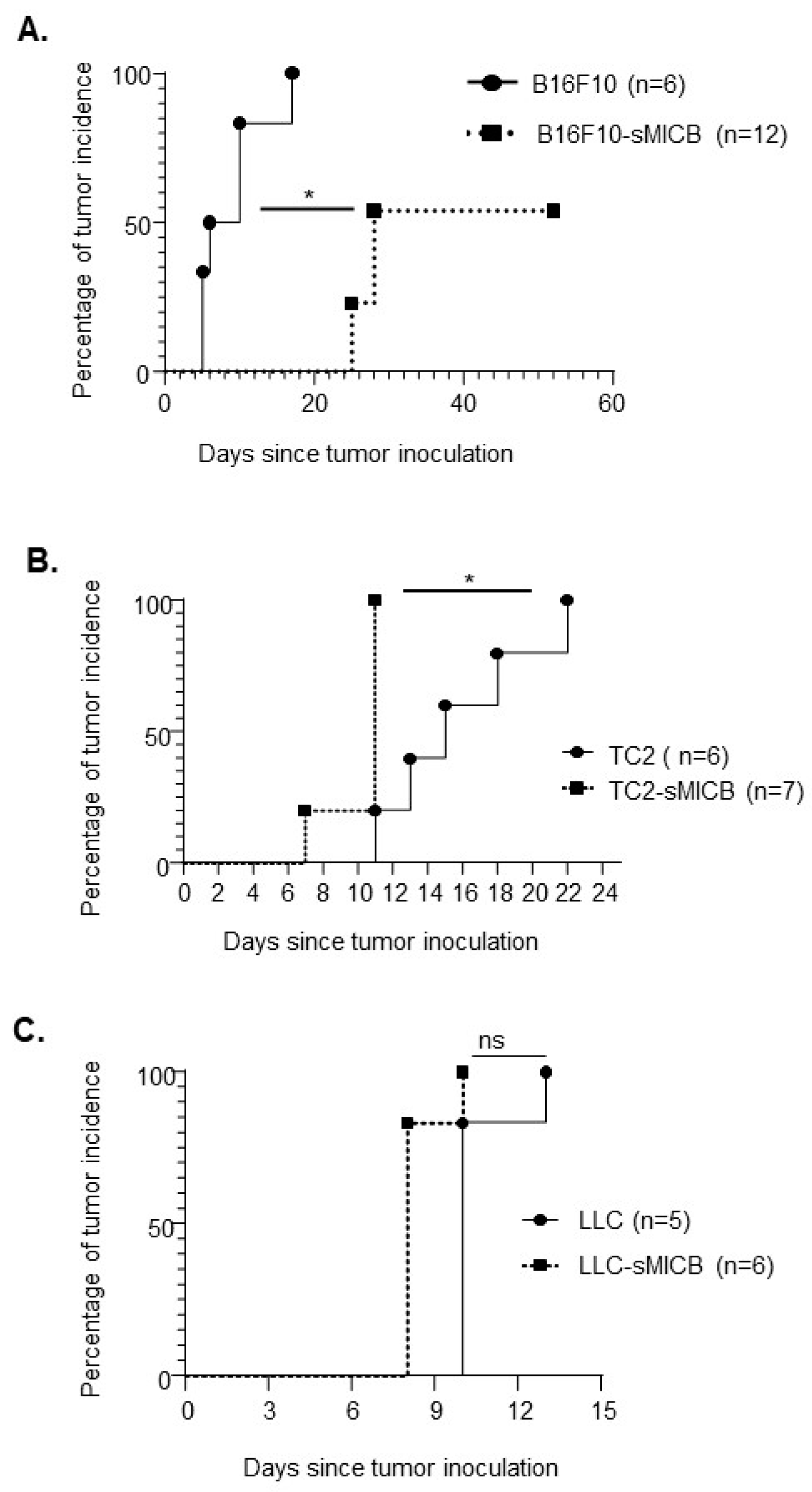
3.1. Model-Dependent Impact of Soluble Human NKG2D Ligand on Tumor Establishment
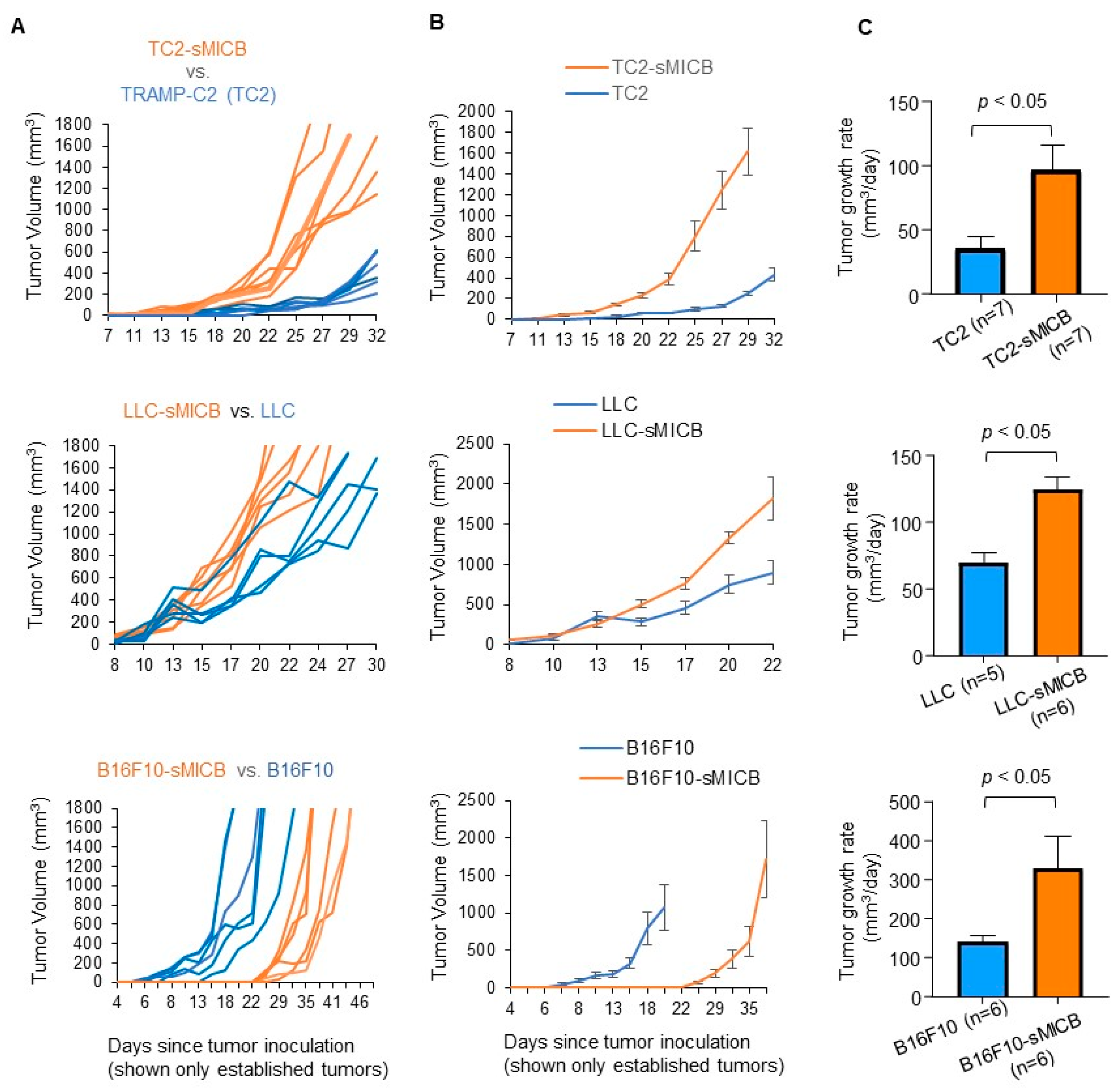
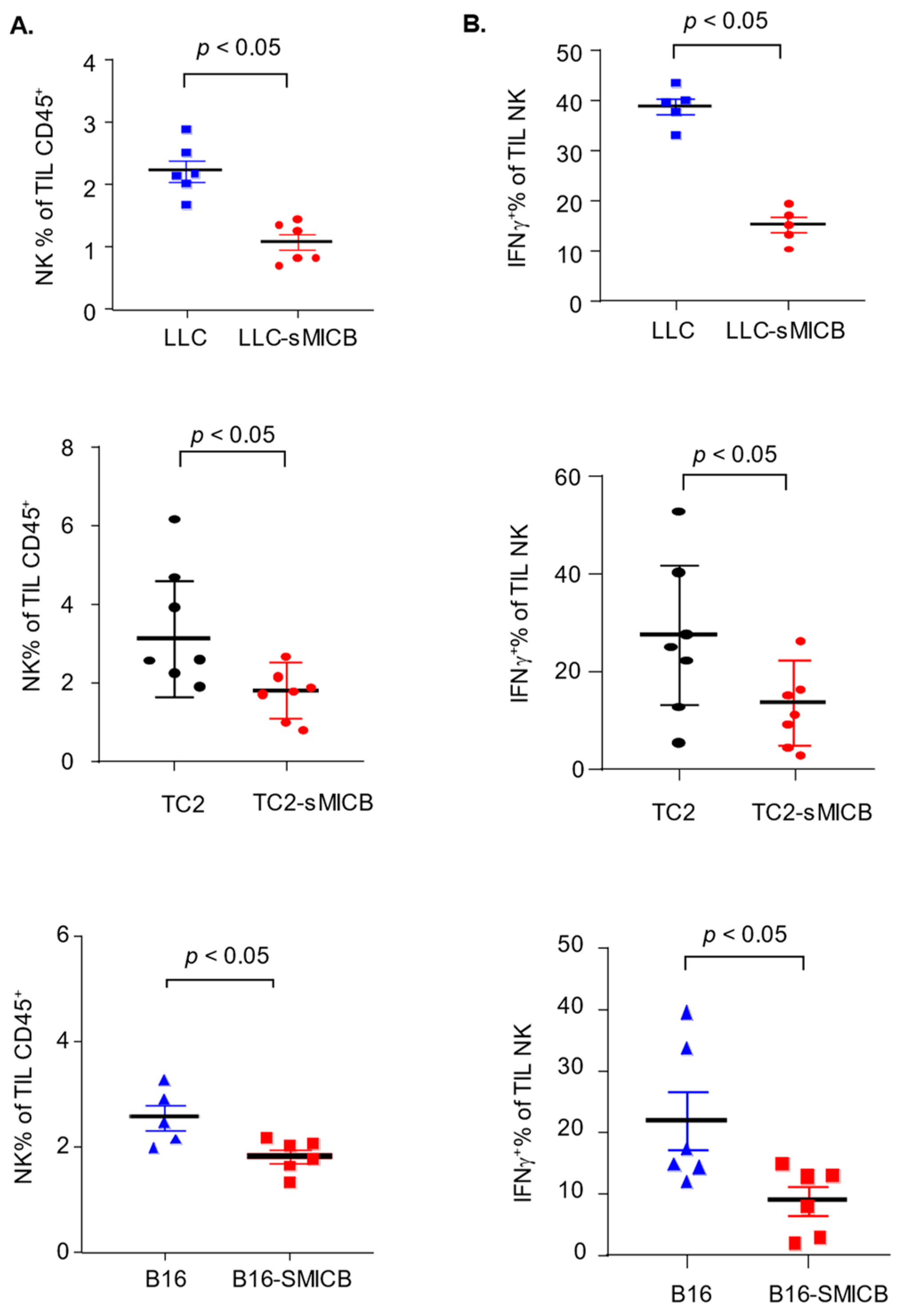
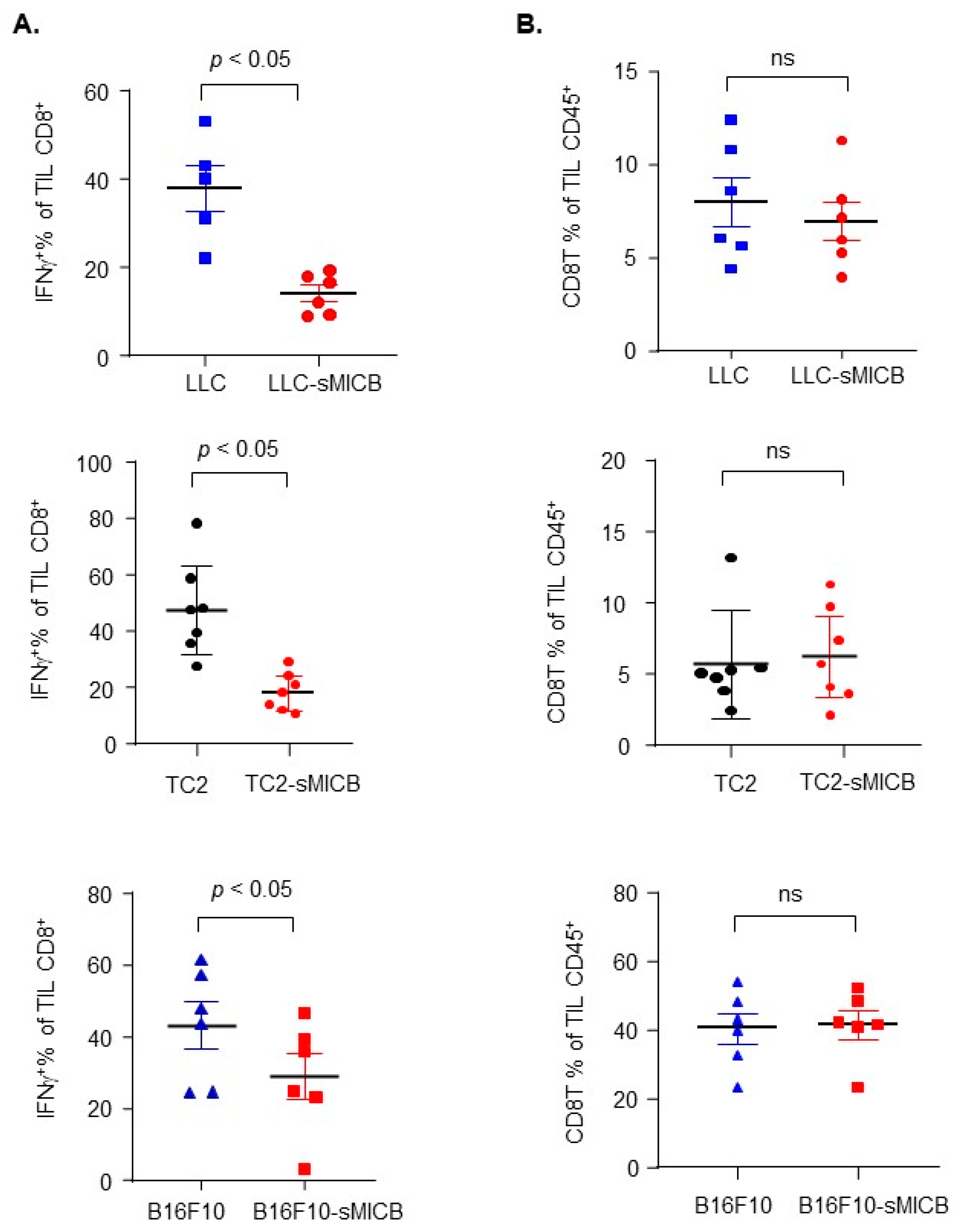
3.2. Model-Independent Impact of sMIC on the Growth of Established Tumors and on the Function of Tumoral Effector Cells
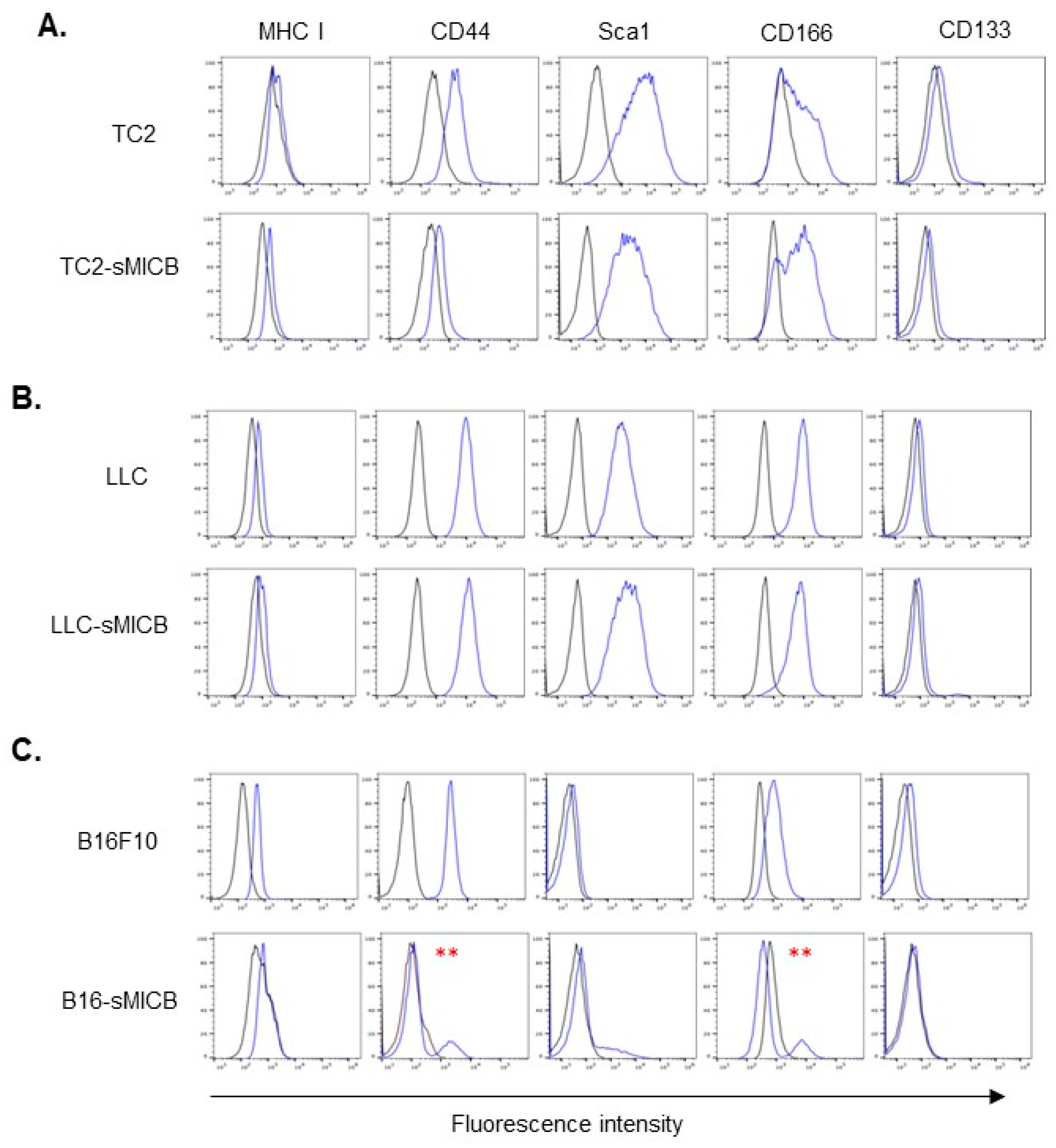
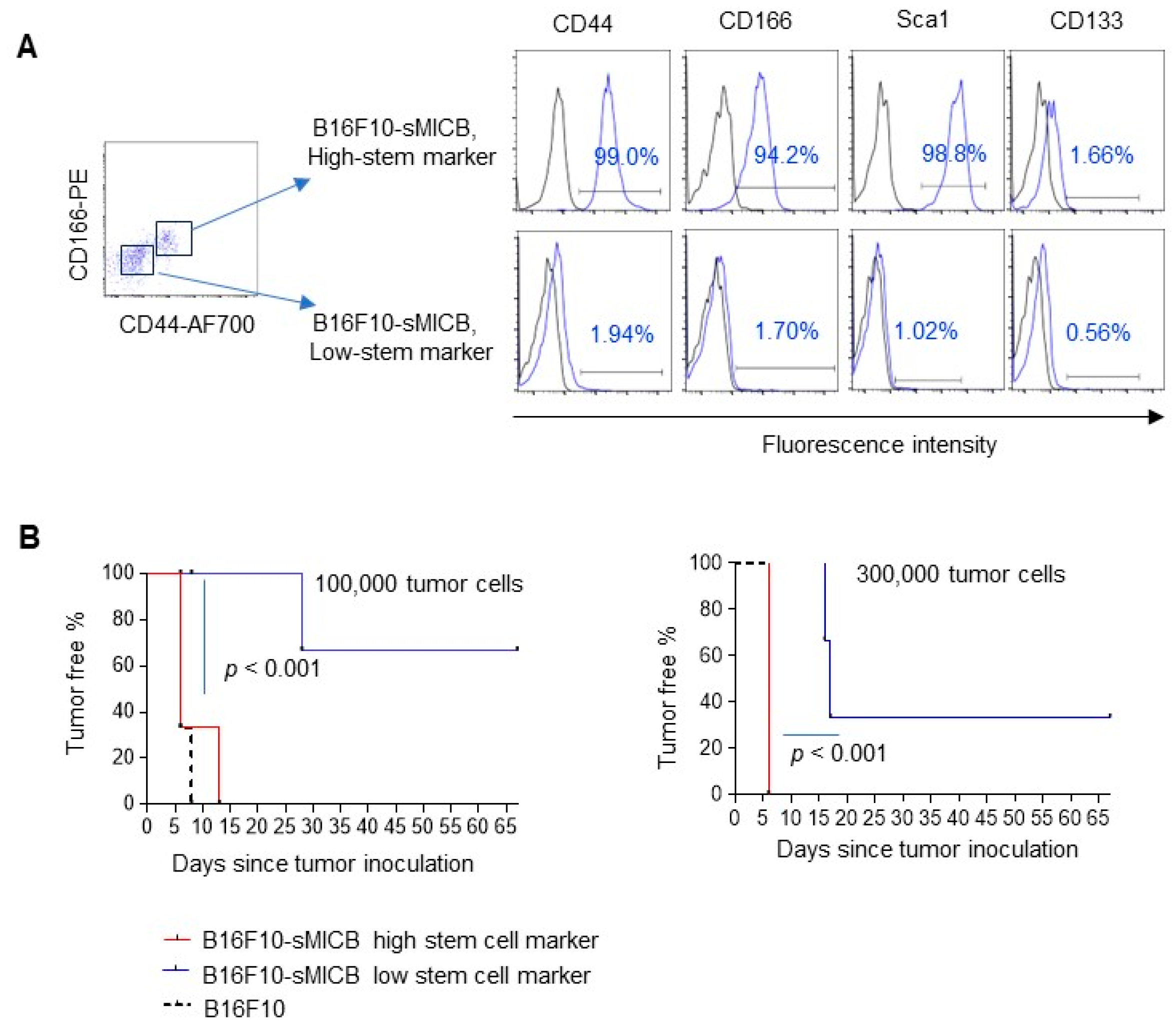
3.3. Tumor Cell Stem-like Property Rather Than sMIC Expression Determines the Ability of In Vivo Tumor Establishment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 2002, 17, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 2003, 3, 781–790. [Google Scholar] [CrossRef]
- Roberts, A.I.; Lee, L.; Schwarz, E.; Groh, V.; Spies, T.; Ebert, E.C.; Jabri, B. NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J. Immunol. 2001, 167, 5527–5530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Basher, F.; Wu, J.D. NKG2D Ligands in Tumor Immunity: Two Sides of a Coin. Front. Immunol. 2015, 6, 97. [Google Scholar] [CrossRef]
- Groh, V.; Rhinehart, R.; Randolph-Habecker, J.; Topp, M.S.; Riddell, S.R.; Spies, T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat. Immunol. 2001, 2, 255–260. [Google Scholar] [CrossRef]
- Dhar, P.; Wu, J.D. NKG2D and its ligands in cancer. Curr. Opin. Immunol. 2018, 51, 55–61. [Google Scholar] [CrossRef]
- Frazao, A.; Rethacker, L.; Messaoudene, M.; Avril, M.F.; Toubert, A.; Dulphy, N.; Caignard, A. NKG2D/NKG2-Ligand Pathway Offers New Opportunities in Cancer Treatment. Front. Immunol. 2019, 10, 661. [Google Scholar] [CrossRef]
- Fuertes, M.B.; Domaica, C.I.; Zwirner, N.W. Leveraging NKG2D Ligands in Immuno-Oncology. Front. Immunol. 2021, 12, 713158. [Google Scholar] [CrossRef]
- Groh, V.; Rhinehart, R.; Secrist, H.; Bauer, S.; Grabstein, K.H.; Spies, T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc. Natl. Acad. Sci. USA 1999, 96, 6879–6884. [Google Scholar] [CrossRef]
- Groh, V.; Steinle, A.; Bauer, S.; Spies, T. Recognition of stress-induced MHC molecules by intestinal epithelial Gammadelta T cells. Science 1998, 279, 1737–1740. [Google Scholar] [CrossRef]
- Lopez-Soto, A.; Huergo-Zapico, L.; Acebes-Huerta, A.; Villa-Alvarez, M.; Gonzalez, S. NKG2D signaling in cancer immunosurveillance. Int. J. Cancer 2015, 136, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, E.; Koch, J.; Cerwenka, A.; Steinle, A. New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunology 2013, 2, e26097. [Google Scholar] [CrossRef] [PubMed]
- Cerwenka, A.; Baron, J.L.; Lanier, L.L. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 11521–11526. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef]
- Chen, J.; Xu, H.; Zhu, X.X. Abnormal expression levels of sMICA and NKG2D are correlated with poor prognosis in pancreatic cancer. Ther. Clin. Risk Manag. 2016, 12, 11–18. [Google Scholar] [CrossRef]
- Arai, J.; Otoyama, Y.; Fujita, K.I.; Goto, K.; Tojo, M.; Katagiri, A.; Nozawa, H.; Kubota, Y.; Takahashi, T.; Ishida, H.; et al. Baseline soluble MICA levels act as a predictive biomarker for the efficacy of regorafenib treatment in colorectal cancer. BMC Cancer 2022, 22, 428. [Google Scholar] [CrossRef]
- Zingoni, A.; Vulpis, E.; Cecere, F.; Amendola, M.G.; Fuerst, D.; Saribekyan, T.; Achour, A.; Sandalova, T.; Nardone, I.; Peri, A.; et al. MICA-129 Dimorphism and Soluble MICA Are Associated with the Progression of Multiple Myeloma. Front. Immunol. 2018, 9, 926. [Google Scholar] [CrossRef]
- Rebmann, V.; Schutt, P.; Brandhorst, D.; Opalka, B.; Moritz, T.; Nowrousian, M.R.; Grosse-Wilde, H. Soluble MICA as an independent prognostic factor for the overall survival and progression-free survival of multiple myeloma patients. Clin. Immunol. 2007, 123, 114–120. [Google Scholar] [CrossRef]
- Jinushi, M.; Vanneman, M.; Munshi, N.C.; Tai, Y.T.; Prabhala, R.H.; Ritz, J.; Neuberg, D.; Anderson, K.C.; Carrasco, D.R.; Dranoff, G. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc. Natl. Acad. Sci. USA 2008, 105, 1285–1290. [Google Scholar] [CrossRef]
- Wu, B.J.; Li, W.P.; Qian, C.; Ding, W.; Zhou, Z.W.; Jiang, H. Serum soluble MICB (sMICB) correlates with disease progression and survival in melanoma patients. Tumour Biol. 2013, 34, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Nuckel, H.; Switala, M.; Sellmann, L.; Horn, P.A.; Durig, J.; Duhrsen, U.; Kuppers, R.; Grosse-Wilde, H.; Rebmann, V. The prognostic significance of soluble NKG2D ligands in B-cell chronic lymphocytic leukemia. Leukemia 2010, 24, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Maccalli, C.; Giannarelli, D.; Chiarucci, C.; Cutaia, O.; Giacobini, G.; Hendrickx, W.; Amato, G.; Annesi, D.; Bedognetti, D.; Altomonte, M.; et al. Soluble NKG2D ligands are biomarkers associated with the clinical outcome to immune checkpoint blockade therapy of metastatic melanoma patients. Oncoimmunology 2017, 6, e1323618. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Yi Lo, P.H.; Sawai, H.; Kato, N.; Takahashi, A.; Deng, Z.; Urabe, Y.; Mbarek, H.; Tokunaga, K.; Tanaka, Y.; et al. Soluble MICA and a MICA variation as possible prognostic biomarkers for HBV-induced hepatocellular carcinoma. PLoS ONE 2012, 7, e44743. [Google Scholar] [CrossRef] [PubMed]
- Ferrari de Andrade, L.; Kumar, S.; Luoma, A.M.; Ito, Y.; Alves da Silva, P.H.; Pan, D.; Pyrdol, J.W.; Yoon, C.H.; Wucherpfennig, K.W. Inhibition of MICA and MICB Shedding Elicits NK-Cell-Mediated Immunity against Tumors Resistant to Cytotoxic T Cells. Cancer Immunol. Res. 2020, 8, 769–780. [Google Scholar] [CrossRef]
- Ferrari de Andrade, L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef]
- Basher, F.; Dhar, P.; Wang, X.; Wainwright, D.A.; Zhang, B.; Sosman, J.; Ji, Z.; Wu, J.D. Antibody targeting tumor-derived soluble NKG2D ligand sMIC reprograms NK cell homeostatic survival and function and enhances melanoma response to PDL1 blockade therapy. J. Hematol. Oncol. 2020, 13, 74. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, J.; Liu, D.; Li, G.; Staveley-O’Carroll, K.F.; Li, Z.; Wu, J.D. Nonblocking Monoclonal Antibody Targeting Soluble MIC Revamps Endogenous Innate and Adaptive Antitumor Responses and Eliminates Primary and Metastatic Tumors. Clin. Cancer Res. 2015, 21, 4819–4830. [Google Scholar] [CrossRef]
- Zhang, J.; Larrocha, P.S.; Zhang, B.; Wainwright, D.; Dhar, P.; Wu, J.D. Antibody targeting tumor-derived soluble NKG2D ligand sMIC provides dual co-stimulation of CD8 T cells and enables sMIC+ tumors respond to PD1/PD-L1 blockade therapy. J. Immunother. Cancer 2019, 7, 223. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, D.; Li, G.; Staveley-O’Carroll, K.F.; Graff, J.N.; Li, Z.; Wu, J.D. Antibody-mediated neutralization of soluble MIC significantly enhances CTLA4 blockade therapy. Sci. Adv. 2017, 3, e1602133. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Gowen, B.G.; Zhang, L.; Wang, L.; Lau, S.; Iannello, A.; Xu, J.; Rovis, T.L.; Xiong, N.; Raulet, D.H. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science 2015, 348, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, M.; Steinle, A. The NKG2D axis: An emerging target in cancer immunotherapy. Expert Opin. Ther. Targets 2019, 23, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, D.; Mandelboim, O. NKG2D Ligands-Critical Targets for Cancer Immune Escape and Therapy. Front. Immunol. 2018, 9, 2040. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.D.; Atteridge, C.L.; Wang, X.; Seya, T.; Plymate, S.R. Obstructing shedding of the immunostimulatory MHC class I chain-related gene B prevents tumor formation. Clin. Cancer Res. 2009, 15, 632–640. [Google Scholar] [CrossRef]
- Liu, G.; Lu, S.; Wang, X.; Page, S.T.; Higano, C.S.; Plymate, S.R.; Greenberg, N.M.; Sun, S.; Li, Z.; Wu, J.D. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. J. Clin. Investig. 2013, 123, 4410–4422. [Google Scholar] [CrossRef]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One”. Front. Immunol. 2018, 9, 476. [Google Scholar] [CrossRef]
- Nausch, N.; Cerwenka, A. NKG2D ligands in tumor immunity. Oncogene 2008, 27, 5944–5958. [Google Scholar] [CrossRef]
- Berkley, A.M.; Toy, E.; Cook, R.; Ye, Z.; Grogan, J.; Schartner, J.; Kim, J. Immunosuppressive effects of sMIC abrogate immunotherapy efficacy in a mouse model of breast cancer. J. Immunol. 2018, 200 (Suppl. S1), 57.42. [Google Scholar] [CrossRef]
- Dhar, P.; Basher, F.; Ji, Z.; Huang, L.; Qin, S.; Wainwright, D.A.; Robinson, J.; Hagler, S.; Zhou, J.; MacKay, S.; et al. Tumor-derived NKG2D ligand sMIC reprograms NK cells to an inflammatory phenotype through CBM signalosome activation. Commun. Biol. 2021, 4, 905. [Google Scholar] [CrossRef]
- Lopez-Soto, A.; Gonzalez, S.; Galluzzi, L. Soluble NKG2D ligands limit the efficacy of immune checkpoint blockade. Oncoimmunology 2017, 6, e1346766. [Google Scholar] [CrossRef]
- Bajaj, J.; Diaz, E.; Reya, T. Stem cells in cancer initiation and progression. J. Cell Biol. 2020, 219, e201911053. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Ganesh, K. Metastasis-Initiating Cells and Ecosystems. Cancer Discov. 2021, 11, 971–994. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Chen, X.; Liang, R.; Lin, H.; Chen, K.; Chen, L.; Tian, G.; Zhu, X. CD166 promotes cancer stem cell-like phenotype via the EGFR/ERK1/2 pathway in the nasopharyngeal carcinoma cell line CNE-2R. Life Sci. 2021, 267, 118983. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Yang, X.; Wang, L.; Clark, D.; Zuo, H.; Ye, D.; Chen, W.; Zhang, P. Plasma membrane proteomics of tumor spheres identify CD166 as a novel marker for cancer stem-like cells in head and neck squamous cell carcinoma. Mol. Cell Proteom. 2013, 12, 3271–3284. [Google Scholar] [CrossRef]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef]
- Swart, G.W.; Lunter, P.C.; Kilsdonk, J.W.; Kempen, L.C. Activated leukocyte cell adhesion molecule (ALCAM/CD166): Signaling at the divide of melanoma cell clustering and cell migration? Cancer Metastasis Rev. 2005, 24, 223–236. [Google Scholar] [CrossRef]
- Darvishi, B.; Boroumandieh, S.; Majidzadeh, A.K.; Salehi, M.; Jafari, F.; Farahmand, L. The role of activated leukocyte cell adhesion molecule (ALCAM) in cancer progression, invasion, metastasis and recurrence: A novel cancer stem cell marker and tumor-specific prognostic marker. Exp. Mol. Pathol. 2020, 115, 104443. [Google Scholar] [CrossRef]
- Ferragut, F.; Vachetta, V.S.; Troncoso, M.F.; Rabinovich, G.A.; Elola, M.T. ALCAM/CD166: A pleiotropic mediator of cell adhesion, stemness and cancer progression. Cytokine Growth Factor Rev. 2021, 61, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Mohammad, K.S.; Wu, H.; Crean, C.; Poteat, B.; Cheng, Y.; Cardoso, A.A.; Machal, C.; Hanenberg, H.; Abonour, R.; et al. Cell Adhesion Molecule CD166 Drives Malignant Progression and Osteolytic Disease in Multiple Myeloma. Cancer Res. 2016, 76, 6901–6910. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Dai, Z.; Reeves, R.S.; Caballero-Benitez, A.; Duran, K.L.; Delrow, J.J.; Porter, P.L.; Spies, T.; Groh, V. Autonomous stimulation of cancer cell plasticity by the human NKG2D lymphocyte receptor coexpressed with its ligands on cancer cells. PLoS ONE 2014, 9, e108942. [Google Scholar] [CrossRef] [PubMed]
- Benitez, A.C.; Dai, Z.; Mann, H.H.; Reeves, R.S.; Margineantu, D.H.; Gooley, T.A.; Groh, V.; Spies, T. Expression, signaling proficiency, and stimulatory function of the NKG2D lymphocyte receptor in human cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4081–4086. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Caballero-Benitez, A.; Gewe, M.M.; Jenkins, I.C.; Drescher, C.W.; Strong, R.K.; Spies, T.; Groh, V. Control of Tumor Initiation by NKG2D Naturally Expressed on Ovarian Cancer Cells. Neoplasia 2017, 19, 471–482. [Google Scholar] [CrossRef]
- Jinushi, M.; Hodi, F.S.; Dranoff, G. Therapy-induced antibodies to MHC class I chain-related protein A antagonize immune suppression and stimulate antitumor cytotoxicity. Proc. Natl. Acad. Sci. USA 2006, 103, 9190–9195. [Google Scholar] [CrossRef]
- Barrow, A.D.; Edeling, M.A.; Trifonov, V.; Luo, J.; Goyal, P.; Bohl, B.; Bando, J.K.; Kim, A.H.; Walker, J.; Andahazy, M.; et al. Natural Killer Cells Control Tumor Growth by Sensing a Growth Factor. Cell 2018, 172, 534–548.e19. [Google Scholar] [CrossRef]
- El Costa, H.; Casemayou, A.; Aguerre-Girr, M.; Rabot, M.; Berrebi, A.; Parant, O.; Clouet-Delannoy, M.; Lombardelli, L.; Jabrane-Ferrat, N.; Rukavina, D.; et al. Critical and differential roles of NKp46- and NKp30-activating receptors expressed by uterine NK cells in early pregnancy. J. Immunol. 2008, 181, 3009–3017. [Google Scholar] [CrossRef]
- Coudert, J.D.; Zimmer, J.; Tomasello, E.; Cebecauer, M.; Colonna, M.; Vivier, E.; Held, W. Altered NKG2D function in NK cells induced by chronic exposure to NKG2D ligand-expressing tumor cells. Blood 2005, 106, 1711–1717. [Google Scholar] [CrossRef]
- Hanaoka, N.; Jabri, B.; Dai, Z.; Ciszewski, C.; Stevens, A.M.; Yee, C.; Nakakuma, H.; Spies, T.; Groh, V. NKG2D initiates caspase-mediated CD3zeta degradation and lymphocyte receptor impairments associated with human cancer and autoimmune disease. J. Immunol. 2010, 185, 5732–5742. [Google Scholar] [CrossRef]
- Rajasekaran, K.; Kumar, P.; Schuldt, K.M.; Peterson, E.J.; Vanhaesebroeck, B.; Dixit, V.; Thakar, M.S.; Malarkannan, S. Signaling by Fyn-ADAP via the Carma1-Bcl-10-MAP3K7 signalosome exclusively regulates inflammatory cytokine production in NK cells. Nat. Immunol. 2013, 14, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, N.; Yu, Y.; Zhou, L.; Niu, C.; Liu, Y.; Tian, H.; Lv, Z.; Han, F.; Cui, J. Prognostic value of MICA/B in cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 96384–96395. [Google Scholar] [CrossRef] [PubMed]
- Holdenrieder, S.; Stieber, P.; Peterfi, A.; Nagel, D.; Steinle, A.; Salih, H.R. Soluble MICA in malignant diseases. Int. J. Cancer 2006, 118, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Holdenrieder, S.; Stieber, P.; Peterfi, A.; Nagel, D.; Steinle, A.; Salih, H.R. Soluble MICB in malignant diseases: Analysis of diagnostic significance and correlation with soluble MICA. Cancer Immunol. Immunother. 2006, 55, 1584–1589. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serritella, A.V.; Saenz-Lopez Larrocha, P.; Dhar, P.; Liu, S.; Medd, M.M.; Jia, S.; Cao, Q.; Wu, J.D. The Human Soluble NKG2D Ligand Differentially Impacts Tumorigenicity and Progression in Temporal and Model-Dependent Modes. Biomedicines 2024, 12, 196. https://doi.org/10.3390/biomedicines12010196
Serritella AV, Saenz-Lopez Larrocha P, Dhar P, Liu S, Medd MM, Jia S, Cao Q, Wu JD. The Human Soluble NKG2D Ligand Differentially Impacts Tumorigenicity and Progression in Temporal and Model-Dependent Modes. Biomedicines. 2024; 12(1):196. https://doi.org/10.3390/biomedicines12010196
Chicago/Turabian StyleSerritella, Anthony V., Pablo Saenz-Lopez Larrocha, Payal Dhar, Sizhe Liu, Milan M. Medd, Shengxian Jia, Qi Cao, and Jennifer D. Wu. 2024. "The Human Soluble NKG2D Ligand Differentially Impacts Tumorigenicity and Progression in Temporal and Model-Dependent Modes" Biomedicines 12, no. 1: 196. https://doi.org/10.3390/biomedicines12010196