
Small Heterodimer Partner Modulates Macrophage Differentiation during Innate Immune Response through the Regulation of Peroxisome Proliferator Activated Receptor Gamma, Mitogen-Activated Protein Kinase, and Nuclear Factor Kappa B Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Chemicals, Plasmids, and Antibodies
2.2. Animal Studies
2.3. Hepatic Cell Isolation and Flow Cytometry Analysis
2.4. Liver Histology and Immunohistochemistry
2.5. Real-Time Quantitative PCR
2.6. Western Blotting and Immunoprecipitation
2.7. Statistical Analysis
3. Results
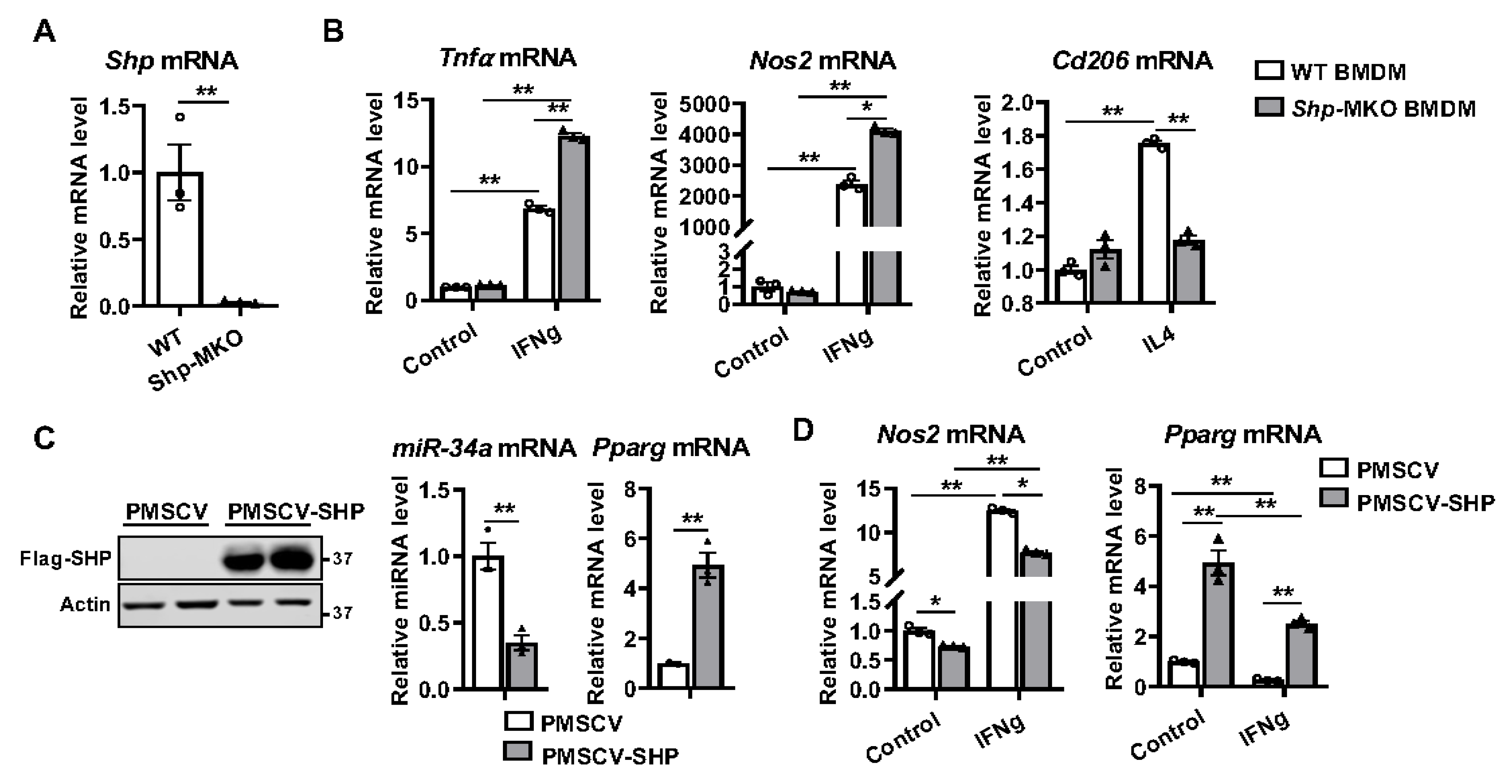
3.1. Macrophage Differentiation Alters Shp mRNA Expression
3.2. Shp Deletion in Macrophages Enhances M1 Polarization but Impairs M2 Differentiation
3.3. SHP Overexpression Increases the Expression of Pparg and Inhibits M1 Differentiation
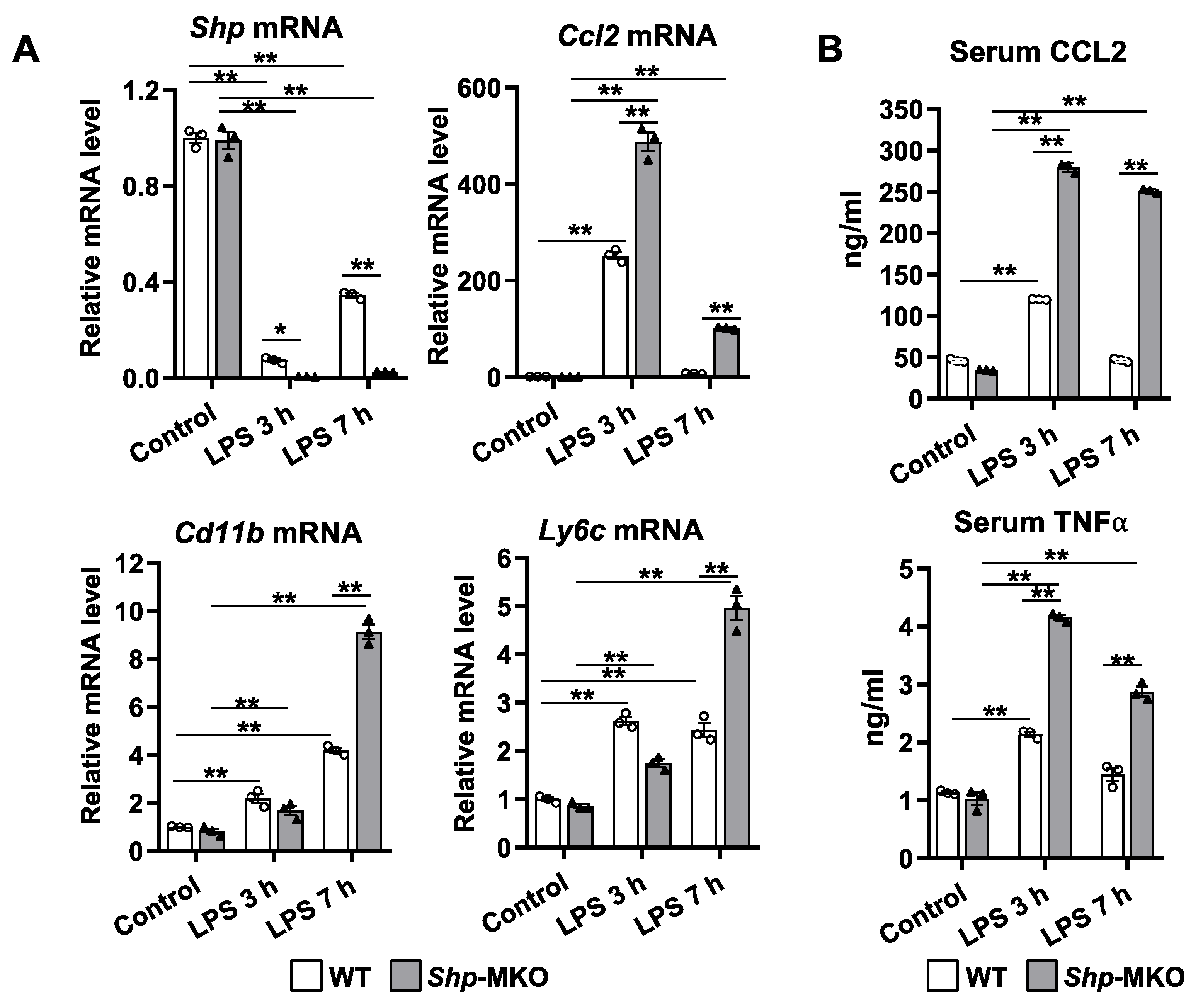
3.4. Shp Knockout Leads to a Persistent Hepatic Infiltration of Proinflammatory Monocytes and M1 Macrophage Differentiation following LPS Challenge
3.5. Shp Deletion in Myeloid Cells Leads to Increased Cytokine Production in Response to LPS Challenge
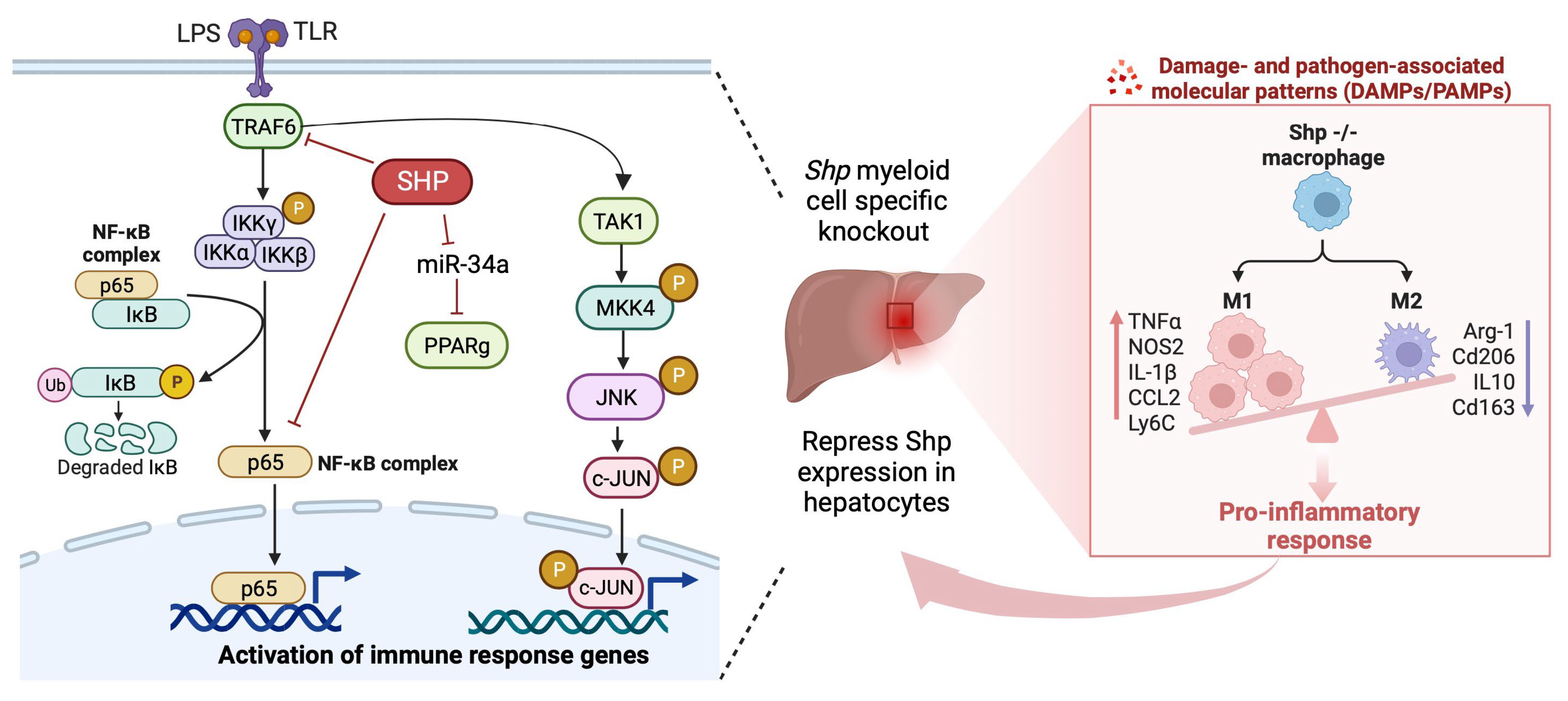
3.6. Shp Deletion Results in Hyperactivation of Both MAPK and NF-κB Signaling Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Gao, B.; Jeong, W.I.; Tian, Z. Liver: An organ with predominant innate immunity. Hepatology 2008, 47, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Rietschel, E.T. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001, 7, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.T. When two is better than one: Macrophages and neutrophils work in concert in innate immunity as complementary and cooperative partners of a myeloid phagocyte system. J. Leukoc. Biol. 2010, 87, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Cai, N.; Zhu, J.; Yang, X.; Liang, H.; Zhang, W. Tumor-associated macrophages in liver cancer: From mechanisms to therapy. Cancer Commun. 2022, 42, 1112–1140. [Google Scholar] [CrossRef]
- Lichtnekert, J.; Kawakami, T.; Parks, W.C.; Duffield, J.S. Changes in macrophage phenotype as the immune response evolves. Curr. Opin. Pharmacol. 2013, 13, 555–564. [Google Scholar] [CrossRef]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Peiseler, M.; Schwabe, R.; Hampe, J.; Kubes, P.; Heikenwalder, M.; Tacke, F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease—Novel insights into cellular communication circuits. J. Hepatol. 2022, 77, 1136–1160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hagedorn, C.H.; Wang, L. Role of nuclear receptor SHP in metabolism and cancer. Biochim. Biophys. Acta 2011, 1812, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Wang, L.; Liu, J.; Saha, P.; Huang, J.; Chan, L.; Spiegelman, B.; Moore, D.D. The orphan nuclear receptor SHP regulates PGC-1alpha expression and energy production in brown adipocytes. Cell Metab. 2005, 2, 227–238. [Google Scholar] [CrossRef]
- Suh, Y.H.; Kim, S.Y.; Lee, H.Y.; Jang, B.C.; Bae, J.H.; Sohn, J.N.; Bae, J.H.; Suh, S.I.; Park, J.W.; Lee, K.U.; et al. Overexpression of short heterodimer partner recovers impaired glucose-stimulated insulin secretion of pancreatic beta-cells overexpressing UCP2. J. Endocrinol. 2004, 183, 133–144. [Google Scholar] [CrossRef]
- Jin, D.; Lu, T.; Ni, M.; Wang, H.; Zhang, J.; Zhong, C.; Shen, C.; Hao, J.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; et al. Farnesoid X Receptor Activation Protects Liver from Ischemia/Reperfusion Injury by Up-Regulating Small Heterodimer Partner in Kupffer Cells. Hepatol. Commun. 2020, 4, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Shahoei, S.H.; Kim, Y.C.; Cler, S.J.; Ma, L.; Anakk, S.; Kemper, J.K.; Nelson, E.R. Small Heterodimer Partner Regulates Dichotomous T Cell Expansion by Macrophages. Endocrinology 2019, 160, 1573–1589. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Yuk, J.M.; Kim, J.J.; Hwang, J.H.; Lee, C.H.; Kim, J.M.; Oh, G.T.; Choi, H.S.; Jo, E.K. Small heterodimer partner-targeting therapy inhibits systemic inflammatory responses through mitochondrial uncoupling protein 2. PLoS ONE 2013, 8, e63435. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.M.; Jin, H.S.; Jo, E.K. Small Heterodimer Partner and Innate Immune Regulation. Endocrinol. Metab. 2016, 31, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Johansson, L.; Bavner, A.; Thomsen, J.S.; Farnegardh, M.; Gustafsson, J.A.; Treuter, E. The orphan nuclear receptor SHP utilizes conserved LXXLL-related motifs for interactions with ligand-activated estrogen receptors. Mol. Cell. Biol. 2000, 20, 1124–1133. [Google Scholar] [CrossRef]
- Yuk, J.M.; Shin, D.M.; Lee, H.M.; Kim, J.J.; Kim, S.W.; Jin, H.S.; Yang, C.S.; Park, K.A.; Chanda, D.; Kim, D.K.; et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat. Immunol. 2011, 12, 742–751. [Google Scholar] [CrossRef]
- Noh, J.R.; Kim, Y.H.; Kim, D.K.; Hwang, J.H.; Kim, K.S.; Choi, D.H.; Lee, S.J.; Lee, H.G.; Lee, T.G.; Weng, H.L.; et al. Small heterodimer partner negatively regulates C-X-C motif chemokine ligand 2 in hepatocytes during liver inflammation. Sci. Rep. 2018, 8, 15222. [Google Scholar] [CrossRef]
- Zou, A.; Magee, N.; Deng, F.; Lehn, S.; Zhong, C.; Zhang, Y. Hepatocyte nuclear receptor SHP suppresses inflammation and fibrosis in a mouse model of nonalcoholic steatohepatitis. J. Biol. Chem. 2018, 293, 8656–8671. [Google Scholar] [CrossRef]
- Magee, N.; Zou, A.; Ghosh, P.; Ahamed, F.; Delker, D.; Zhang, Y. Disruption of hepatic small heterodimer partner induces dissociation of steatosis and inflammation in experimental nonalcoholic steatohepatitis. J. Biol. Chem. 2020, 295, 994–1008. [Google Scholar] [CrossRef]
- Toda, G.; Yamauchi, T.; Kadowaki, T.; Ueki, K. Preparation and culture of bone marrow-derived macrophages from mice for functional analysis. STAR Protoc. 2021, 2, 100246. [Google Scholar] [CrossRef]
- Vrochides, D.; Papanikolaou, V.; Pertoft, H.; Antoniades, A.A.; Heldin, P. Biosynthesis and degradation of hyaluronan by nonparenchymal liver cells during liver regeneration. Hepatology 1996, 23, 1650–1655. [Google Scholar] [CrossRef] [PubMed]
- Magee, N.; Ahamed, F.; Eppler, N.; Jones, E.; Ghosh, P.; He, L.; Zhang, Y. Hepatic transcriptome profiling reveals early signatures associated with disease transition from non-alcoholic steatosis to steatohepatitis. Liver Res. 2022, 6, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Heming, M.; Gran, S.; Jauch, S.L.; Fischer-Riepe, L.; Russo, A.; Klotz, L.; Hermann, S.; Schafers, M.; Roth, J.; Barczyk-Kahlert, K. Peroxisome Proliferator-Activated Receptor-gamma Modulates the Response of Macrophages to Lipopolysaccharide and Glucocorticoids. Front. Immunol. 2018, 9, 893. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.L.; Chow, A.; Spanbroek, R.; Marcelin, G.; Greter, M.; Jakubzick, C.; Bogunovic, M.; Leboeuf, M.; van Rooijen, N.; Habenicht, A.J.; et al. Systemic analysis of PPARgamma in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J. Immunol. 2012, 189, 2614–2624. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Padhye, A.; Sharma, A.; Song, G.; Miao, J.; Mo, Y.Y.; Wang, L.; Kemper, J.K. A pathway involving farnesoid X receptor and small heterodimer partner positively regulates hepatic sirtuin 1 levels via microRNA-34a inhibition. J. Biol. Chem. 2010, 285, 12604–12611. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, Y.; Wu, S.; He, J.; Lou, L.; Ye, W.; Wang, J. microRNA-34a and microRNA-34c promote the activation of human hepatic stellate cells by targeting peroxisome proliferator-activated receptor gamma. Mol. Med. Rep. 2015, 11, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Zarkesh, M.; Tabaei, K.; Akbarzadeh, M.; Daneshafrooz, A.; Zadeh-Vakili, A. Association of miR-34a and miR-143 levels with PPARgamma gene expression in adipose tissues of non-diabetic adults. J. Physiol. Anthropol. 2022, 41, 13. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Z.; Shi, J.; Yu, X.; Li, C.; Liu, J.; Zhang, F.; Chen, H.; Zheng, W. Macrophage polarization toward M1 phenotype through NF-kappaB signaling in patients with Behcet’s disease. Arthritis Res. Ther. 2022, 24, 249. [Google Scholar] [CrossRef]
- Kerneur, C.; Cano, C.E.; Olive, D. Major pathways involved in macrophage polarization in cancer. Front. Immunol. 2022, 13, 1026954. [Google Scholar] [CrossRef]
- Tian, L.; Li, W.; Yang, L.; Chang, N.; Fan, X.; Ji, X.; Xie, J.; Yang, L.; Li, L. Cannabinoid Receptor 1 Participates in Liver Inflammation by Promoting M1 Macrophage Polarization via RhoA/NF-kappaB p65 and ERK1/2 Pathways, Respectively, in Mouse Liver Fibrogenesis. Front. Immunol. 2017, 8, 1214. [Google Scholar] [CrossRef]
- Krenkel, O.; Hundertmark, J.; Abdallah, A.T.; Kohlhepp, M.; Puengel, T.; Roth, T.; Branco, D.P.P.; Mossanen, J.C.; Luedde, T.; Trautwein, C.; et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut 2020, 69, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bonnardel, J.; Haest, B.; Vanderborght, B.; Wagner, C.; Remmerie, A.; Bujko, A.; Martens, L.; Thone, T.; Browaeys, R.; et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 2022, 185, 379–396.e38. [Google Scholar] [CrossRef] [PubMed]
- Vonderlin, J.; Chavakis, T.; Sieweke, M.; Tacke, F. The Multifaceted Roles of Macrophages in NAFLD Pathogenesis. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 1311–1324. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chang, N.; Li, L. Heterogeneity and Function of Kupffer Cells in Liver Injury. Front. Immunol. 2022, 13, 940867. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Koh, Y.K.; Kim, T.H.; Kwon, S.K.; Im, S.S.; Choi, H.S.; Kim, K.S.; Ahn, Y.H. Transcriptional activation of SHP by PPAR-gamma in liver. Biochem. Biophys. Res. Commun. 2007, 360, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Xiang, X.; Xu, D.; Cui, K.; Pang, Y.; Xu, W.; Mai, K.; Ai, Q. LPS Stimulation Induces Small Heterodimer Partner Expression Through the AMPK-NRF2 Pathway in Large Yellow Croaker (Larimichthys crocea). Front. Immunol. 2021, 12, 753681. [Google Scholar] [CrossRef]
- Chung, H.T. SHP gains citizenship of the AMPK kingdom. Cell. Mol. Immunol. 2011, 8, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.Y.; Schuppan, D. AMPK regulates macrophage polarization in adipose tissue inflammation and NASH. J. Hepatol. 2013, 58, 619–621. [Google Scholar] [CrossRef]
- Yao, Q.; Liu, J.; Zhang, Z.; Li, F.; Zhang, C.; Lai, B.; Xiao, L.; Wang, N. Peroxisome proliferator-activated receptor gamma (PPARgamma) induces the gene expression of integrin alpha(V)beta(5) to promote macrophage M2 polarization. J. Biol. Chem. 2018, 293, 16572–16582. [Google Scholar] [CrossRef]
- Chen, J.Y.; Song, C.X.; Lei, S.Y.; Li, J.; Zuo, A.J.; Xu, D.; Guo, Y. CTRP9 induces macrophages polarization into M1 phenotype through activating JNK pathway and enhances VSMCs apoptosis in macrophages and VSMCs co-culture system. Exp. Cell Res. 2020, 395, 112194. [Google Scholar] [CrossRef]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Ehling, J.; Bartneck, M.; Wei, X.; Gremse, F.; Fech, V.; Mockel, D.; Baeck, C.; Hittatiya, K.; Eulberg, D.; Luedde, T.; et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014, 63, 1960–1971. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zhang, J.; Zhang, Y.; Shu, Z.; Xu, P.; He, L.; Yang, C.; Zhang, J.; Wang, H.; Li, Y.; et al. Hepatic recruitment of CD11b+Ly6C+ inflammatory monocytes promotes hepatic ischemia/reperfusion injury. Int. J. Mol. Med. 2018, 41, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Brempelis, K.J.; Crispe, I.N. Infiltrating monocytes in liver injury and repair. Clin. Transl. Immunol. 2016, 5, e113. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Liu, L.; Sun, L.; Fang, P.; Snyder, N.; Saredy, J.; Ji, Y.; Shen, W.; Qin, X.; Wu, Q.; et al. Immunological Feature and Transcriptional Signaling of Ly6C Monocyte Subsets From Transcriptome Analysis in Control and Hyperhomocysteinemic Mice. Front. Immunol. 2021, 12, 632333. [Google Scholar] [CrossRef] [PubMed]
- Mussbacher, M.; Derler, M.; Basilio, J.; Schmid, J.A. NF-kappaB in monocytes and macrophages—An inflammatory master regulator in multitalented immune cells. Front. Immunol. 2023, 14, 1134661. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M. Role of K63-linked polyubiquitination in NF-kappaB signalling: Which ligase catalyzes and what molecule is targeted? J. Biochem. 2012, 151, 115–118. [Google Scholar] [CrossRef]
- McDermott, E.P.; O’Neill, L.A. Ras participates in the activation of p38 MAPK by interleukin-1 by associating with IRAK, IRAK2, TRAF6, and TAK-1. J. Biol. Chem. 2002, 277, 7808–7815. [Google Scholar] [CrossRef]
- Mason, N.J.; Fiore, J.; Kobayashi, T.; Masek, K.S.; Choi, Y.; Hunter, C.A. TRAF6-dependent mitogen-activated protein kinase activation differentially regulates the production of interleukin-12 by macrophages in response to Toxoplasma gondii. Infect. Immun. 2004, 72, 5662–5667. [Google Scholar] [CrossRef]
- Abram, C.L.; Roberge, G.L.; Hu, Y.; Lowell, C.A. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 2014, 408, 89–100. [Google Scholar] [CrossRef]
- Clausen, B.E.; Burkhardt, C.; Reith, W.; Renkawitz, R.; Forster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999, 8, 265–277. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahamed, F.; Eppler, N.; Jones, E.; He, L.; Zhang, Y. Small Heterodimer Partner Modulates Macrophage Differentiation during Innate Immune Response through the Regulation of Peroxisome Proliferator Activated Receptor Gamma, Mitogen-Activated Protein Kinase, and Nuclear Factor Kappa B Pathways. Biomedicines 2023, 11, 2403. https://doi.org/10.3390/biomedicines11092403
Ahamed F, Eppler N, Jones E, He L, Zhang Y. Small Heterodimer Partner Modulates Macrophage Differentiation during Innate Immune Response through the Regulation of Peroxisome Proliferator Activated Receptor Gamma, Mitogen-Activated Protein Kinase, and Nuclear Factor Kappa B Pathways. Biomedicines. 2023; 11(9):2403. https://doi.org/10.3390/biomedicines11092403
Chicago/Turabian StyleAhamed, Forkan, Natalie Eppler, Elizabeth Jones, Lily He, and Yuxia Zhang. 2023. "Small Heterodimer Partner Modulates Macrophage Differentiation during Innate Immune Response through the Regulation of Peroxisome Proliferator Activated Receptor Gamma, Mitogen-Activated Protein Kinase, and Nuclear Factor Kappa B Pathways" Biomedicines 11, no. 9: 2403. https://doi.org/10.3390/biomedicines11092403