
Insight into Genetic Mutations of SZT2: Is It a Syndrome?

 , , and
, , and
Abstract
:1. Introduction
2. Case Report
2.1. Proband I
2.2. Proband II
3. Materials and Methods
3.1. Search Strategy
3.2. Sample Collections
4. Results
4.1. Literature Review
4.2. Baseline, Demographic, and Clinical Characteristics
4.3. Seizure Semiology
4.4. Neuroimaging and Electrophysiological Characteristics
4.5. Management and Therapeutic Intervention
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meyerhoff, J.; Muhie, S.; Chakraborty, N.; Naidu, L.; Sowe, B.; Hammamieh, R.; Jett, M.; Gautam, A. Microdissection of Mouse Brain into Functionally and Anatomically Different Regions. J. Vis. Exp. 2021, 2021, e61941. [Google Scholar] [CrossRef]
- Kasperavičiūtė, D.; Catarino, C.B.; Heinzen, E.L.; Depondt, C.; Cavalleri, G.L.; Caboclo, L.O.; Tate, S.K.; Jamnadas-Khoda, J.; Chinthapalli, K.; Clayton, L.M.; et al. Common genetic variation and susceptibility to partial epilepsies: A genome-wide association study. Brain 2010, 133, 2136–2147. [Google Scholar] [CrossRef] [PubMed]
- Trivisano, M.; Rivera, M.; Terracciano, A.; Ciolfi, A.; Napolitano, A.; Pepi, C.; Calabrese, C.; Digilio, M.C.; Tartaglia, M.; Curatolo, P.; et al. Developmental and epileptic encephalopathy due to SZT2 genomic variants: Emerging features of a syndromic condition. Epilepsy Behav. 2020, 108, 107097. [Google Scholar] [CrossRef]
- Trivisano, M.; Specchio, N. What are the epileptic encephalopathies? Curr. Opin. Neurol. 2020, 33, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Rochtus, A.; Olson, H.E.; Smith, L.; Keith, L.G.; El Achkar, C.; Taylor, A.; Mahida, S.; Park, M.; Kelly, M.; Shain, C.; et al. Genetic diagnoses in epilepsy: The impact of dynamic exome analysis in a pediatric cohort. Epilepsia 2020, 61, 249. [Google Scholar] [CrossRef]
- Frankel, W.N.; Yang, Y.; Mahaffey, C.L.; Beyer, B.J.; O’Brien, T.P. Szt2, a novel gene for seizure threshold in mice. Genes Brain Behav. 2009, 8, 568–576. [Google Scholar] [CrossRef]
- Naseer, M.I.; Alwasiyah, M.K.; Abdulkareem, A.A.; Bajammal, R.A.; Trujillo, C.; Abu-Elmagd, M.; Alam Jafri, M.; Chaudhary, A.G.; Al-Qahtani, M.H. A novel homozygous mutation in SZT2 gene in Saudi family with developmental delay, macrocephaly and epilepsy. Genes Genom. 2018, 401, 149–155. [Google Scholar] [CrossRef]
- Basel-Vanagaite, L.; Hershkovitz, T.; Heyman, E.; Raspall-Chaure, M.; Kakar, N.; Smirin-Yosef, P.; Vila-Pueyo, M.; Kornreich, L.; Thiele, H.; Bode, H.; et al. Biallelic SZT2 mutations cause infantile encephalopathy with epilepsy and dysmorphic corpus callosum. Am. J. Hum. Genet. 2013, 93, 524–529. [Google Scholar] [CrossRef]
- Kariminejad, A.; Yazdan, H.; Rahimian, E.; Kalhor, Z.; Fattahi, Z.; Zonooz, M.F.; Najmabadi, H.; Ashrafi, M. SZT2 mutation in a boy with intellectual disability, seizures and autistic features. Eur. J. Med. Genet. 2019, 62, 103556. [Google Scholar] [CrossRef]
- Toutzaris, D.; Lewerenz, J.; Albrecht, P.; Jensen, L.T.; Letz, J.; Geerts, A.; Golz, S.; Methner, A. A novel giant peroxisomal superoxide dismutase motif-containing protein. Free Radic. Biol. Med. 2010, 48, 811–820. [Google Scholar] [CrossRef] [PubMed]
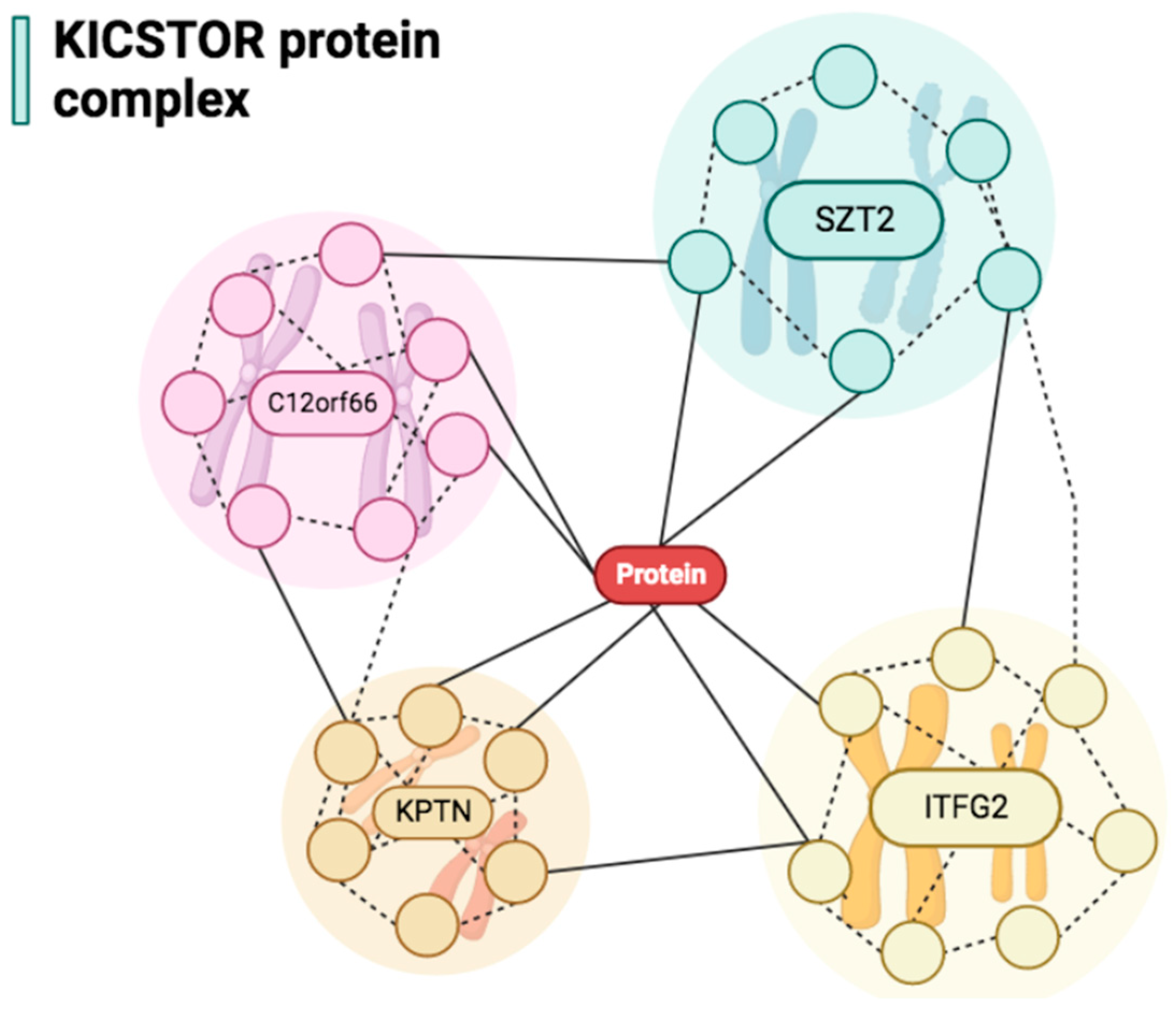
- Wolfson, R.L.; Chantranupong, L.; Wyant, G.A.; Gu, X.; Orozco, J.M.; Shen, K.; Condon, K.J.; Petri, S.; Kedir, J.; Scaria, S.M.; et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 2017, 543, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Yin, N.; Li, M.O. SZT2 dictates GATOR control of mTORC1 signalling. Nature 2017, 543, 433–437. [Google Scholar] [CrossRef]
- Sakai, Y.; Kassai, H.; Nakayama, H.; Fukaya, M.; Maeda, T.; Nakao, K.; Hashimoto, K.; Sakagami, H.; Kano, M.; Aiba, A. Hyperactivation of mTORC1 disrupts cellular homeostasis in cerebellar Purkinje cells. Sci. Rep. 2019, 9, 2799. [Google Scholar] [CrossRef]
- Cattelani, C.; Lesiak, D.; Liebscher, G.; Singer, I.I.; Stasyk, T.; Wallnöfer, M.H.; Heberle, A.M.; Corti, C.; Hess, M.W.; Pfaller, K.; et al. The SZT2 Interactome Unravels New Functions of the KICSTOR Complex. Cells 2021, 10, 2711. [Google Scholar] [CrossRef] [PubMed]
- Unni, N.; Arteaga, C.L. Is Dual mTORC1 and mTORC2 Therapeutic Blockade Clinically Feasible in Cancer? JAMA Oncol. 2019, 5, 1564–1565. [Google Scholar] [CrossRef]
- Venkatesan, C.; Angle, B.; Millichap, J.J. Early-life epileptic encephalopathy secondary to SZT2 pathogenic recessive variants. Epileptic Disord. 2016, 18, 195–200. [Google Scholar] [CrossRef]
- Baple, E.L.; Maroofian, R.; Chioza, B.A.; Izadi, M.; Cross, H.E.; Al-Turki, S.; Barwick, K.; Skrzypiec, A.; Pawlak, R.; Wagner, K.; et al. Mutations in KPTN cause macrocephaly, neurodevelopmental delay, and seizures. Am. J. Hum. Genet. 2013, 94, 87–94. [Google Scholar] [CrossRef]
- Falcone, M.; Yariz, K.O.; Ross, D.B.; Foster, J.; Menendez, I.; Tekin, M. An amino acid deletion in SZT2 in a family with non-syndromic intellectual disability. PLoS ONE 2013, 8, e82810. [Google Scholar] [CrossRef]
- Tsuchida, N.; Nakashima, M.; Miyauchi, A.; Yoshitomi, S.; Kimizu, T.; Ganesan, V.; Teik, K.; Ch’Ng, G.-S.; Kato, M.; Mizuguchi, T.; et al. Novel biallelic SZT2 mutations in 3 cases of early-onset epileptic encephalopathy. Clin. Genet. 2018, 93, 266–274. [Google Scholar] [CrossRef]
- Imaizumi, T.; Kumakura, A.; Yamamoto-Shimojima, K.; Ondo, Y.; Yamamoto, T. Identification of a rare homozygous SZT2 variant due to uniparental disomy in a patient with a neurodevelopmental disorder. Intractable Rare Dis. Res. 2018, 7, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Domingues, F.S.; König, E.; Schwienbacher, C.; Volpato, C.B.; Picard, A.; Cantaloni, C.; Mascalzoni, D.; Lackner, P.; Heimbach, A.; Hoffmann, P.; et al. Compound heterozygous SZT2 mutations in two siblings with early-onset epilepsy, intellectual disability and macrocephaly. Seizure 2019, 66, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Takahashi, S.; Kuroda, M.; Takeguchi, R.; Suzuki, N.; Makita, Y.; Narumi-Kishimoto, Y.; Kaname, T. Biallelic SZT2 variants in a child with developmental and epileptic encephalopathy. Epileptic Disord. 2020, 22, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Cherian, A.; Divya, K.P.; Pavuluri, H.; Thomas, B. The Dysfunctional Gangway: SZT2-associated Epilepsy with Thick Corpus Callosum. J. Pediatr. Neurosci. 2021, 16, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yang, L.M.; Liao, H.M.; Fang, H.J.; Ning, Z.S.; Liao, C.S.; Wu, L.W. Genetic analysis of developmental and epileptic encephalopathy caused by novel biallelic SZT2 gene mutations in three Chinese Han infants: A case series and literature review. Neurol Sci. 2022, 43, 5039–5048. [Google Scholar] [CrossRef]
- Nakamura, Y.; Togawa, Y.; Okuno, Y.; Muramatsu, H.; Nakabayashi, K.; Kuroki, Y.; Ieda, D.; Hori, I.; Negishi, Y.; Togawa, T.; et al. Biallelic mutations in SZT2 cause a discernible clinical entity with epilepsy, developmental delay, macrocephaly and a dysmorphic corpus callosum. Brain Dev. 2018, 40, 134–139. [Google Scholar] [CrossRef]
- Pizzino, A.; Whitehead, M.; Rasekh, P.S.; Murphy, J.; Helman, G.; Bloom, M.; Evans, S.H.; Murnick, J.G.; Conry, J.; Taft, R.J.; et al. Mutations in SZT2 result in early-onset epileptic encephalopathy and leukoencephalopathy. Am. J. Med. Genet Part A 2018, 176, 1443–1448. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Gropman, A.; Brantner, C.A.; Chiaramello, A. Novel metabolic signatures of compound heterozygous Szt2 variants in a case of early-onset of epileptic encephalopathy. Clin. Case Rep. 2018, 6, 2376. [Google Scholar] [CrossRef]
- Iodice, A.; Spagnoli, C.; Frattini, D.; Salerno, G.G.; Rizzi, S.; Fusco, C. Biallelic SZT2 mutation with early onset of focal status epilepticus: Useful diagnostic clues other than epilepsy, intellectual disability and macrocephaly. Seizure 2019, 69, 296–297. [Google Scholar] [CrossRef]
- Sun, X.; Zhong, X.; Li, T. Novel SZT2 mutations in three patients with developmental and epileptic encephalopathies. Mol. Genet. Genom. Med. 2019, 7, e926. [Google Scholar] [CrossRef]
- Green, B.N.; Johnson, C.D.; Adams, A. Writing narrative literature reviews for peer-reviewed journals: Secrets of the trade. J. Chiropr. Med. 2006, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Riley, D.S.; Barber, M.S.; Kienle, G.S.; Aronson, J.K.; von Schoen-Angerer, T.; Tugwell, P.; Kiene, H.; Helfand, M.; Altman, D.G.; Sox, H.; et al. CARE guidelines for case reports: Explanation and elaboration document. J. Clin. Epidemiol. 2017, 89, 218–235. [Google Scholar] [CrossRef]
- Vanderver, A.; Simons, C.; Helman, G.; Crawford, J.; Wolf, N.I.; Bernard, G.; Pizzino, A.; Schmidt, J.L.; Takanohashi, A.; Miller, D.; et al. Whole exome sequencing in patients with white matter abnormalities. Ann. Neurol. 2016, 79, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- El Halabi, T.; Dirani, M.; Hotait, M.; Nasreddine, W.; Beydoun, A. A novel possible familial cause of epilepsy of infancy with migrating focal seizures related to SZT2 gene variant. Epilepsia Open 2021, 6, 73–78. [Google Scholar] [CrossRef]
- Shao, D.; Villet, O.; Zhang, Z.; Choi, S.W.; Yan, J.; Ritterhoff, J.; Gu, H.; Djukovic, D.; Christodoulou, D.; Kolwicz, S.C.; et al. Glucose promotes cell growth by suppressing branched-chain amino acid degradation. Nat. Commun. 2018, 9, 2935. [Google Scholar] [CrossRef]
- Yeung, K.S.; Tso, W.W.Y.; Ip, J.J.K.; Mak, C.C.Y.; Leung, G.K.C.; Tsang, M.H.Y.; Ying, D.; Pei, S.L.C.; Lee, S.L.; Yang, W.; et al. Identification of mutations in the PI3K-AKT-mTOR signalling pathway in patients with macrocephaly and developmental delay and/or autism. Mol. Autism. 2017, 8, 66. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
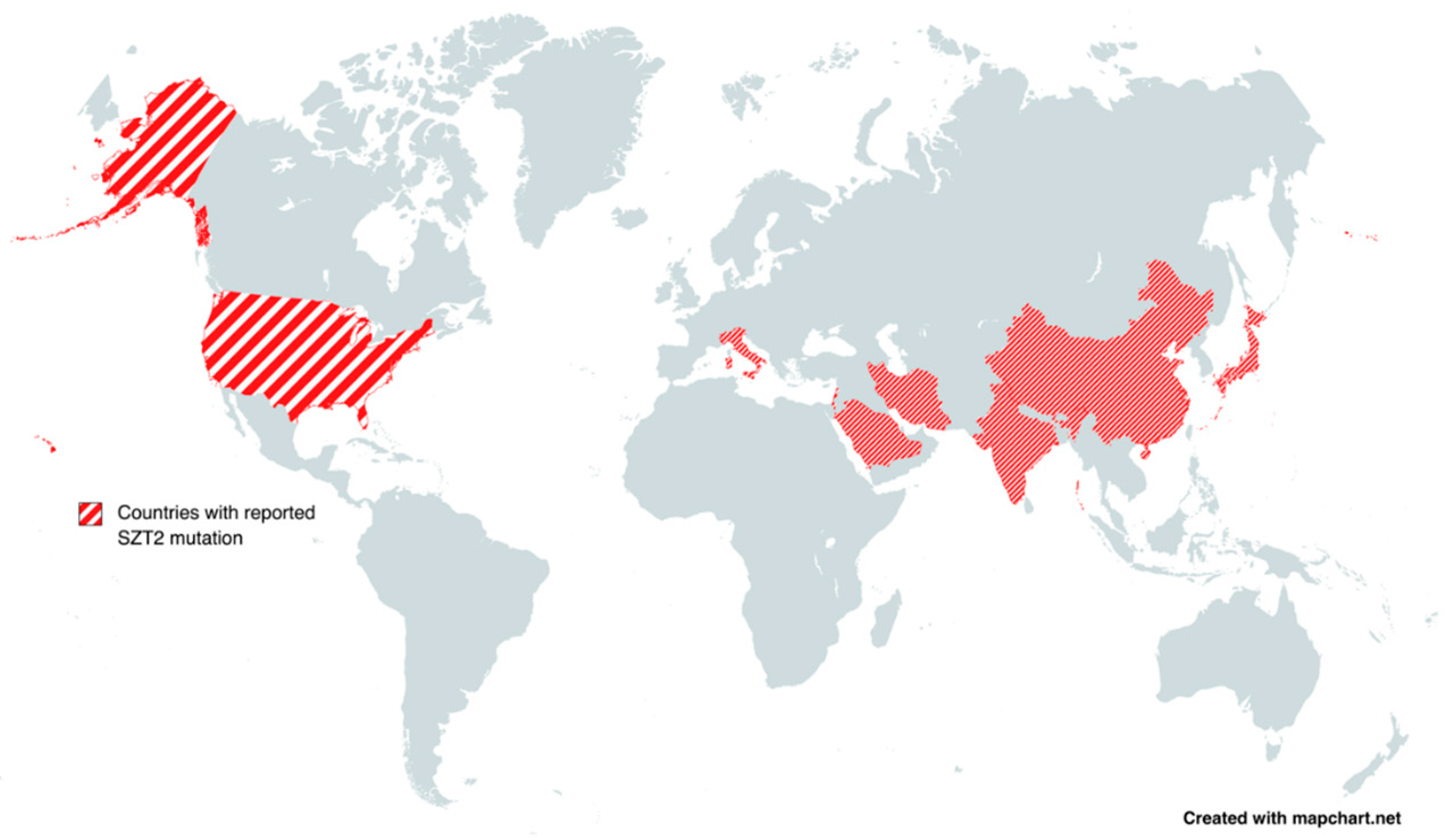
| Patient No. | Authors | Age | Sex | Reported Country | Facial Dysmorphism | Seizures | Intellectual Disability | Autism | Developmental Delay | Hypotonia |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Basel-Vanagaite et al., 2013 [9] | 10 y | F | Israel | + | + | NM | NM | + (severe) | + |
| 2 | Basel-Vanagaite et al., 2013 [9] | 9 y | M | Israel | + | + | NM | NM | + (severe) | + |
| 3 | Falcone et al., 2013 (patient 1) [19] | 18 y | M | USA | — | — | + | — | + | — |
| 4 | Falcone et al., 2013 (patient 2) [19] | 10 y | M | USA | — | — | + | — | + | — |
| 5 | Falcone et al., 2013 (patient 3) [19] | 7 y | M | USA | — | — | — | — | + (only speech) | — |
| 6 | Venkatesan et al., 2016 [17] | 3 y | M | USA | + | + | NM | NM | + | + |
| 7 | Vanderver et al., 2016 [33] | 7 y | F | USA | + | + | NM | NM | + | NM |
| 8 | Tsuchida et al., 2018 (patient 1) [20] | 4 y | F | Japan | + | + | + | — | + (severe) | + |
| 9 | Tsuchida et al., 2018 (patient 2) [20] | 2 y | M | Japan | + | + | + | — | + (severe) | + |
| 10 | Tsuchida et al., 2018 (patient 3) [20] | 5 y | F | Japan | + | + | + | — | + (severe) | + |
| 11 | Nakamura et al., 2018 [26] | 4 y | F | Japan | + | + | NM | NM | + | NM |
| 12 | Pizzino et al., 2018 [27] | 20 m | F | USA | + | + | NM | NM | + | NM |
| 13 | Naseer et al., 2018 (patient 1) [8] | 8 y | F | Saudi Arabia | — | + | NM | NM | + | + |
| 14 | Naseer et al., 2018 (patient 2) [8] | 5 y | F | Saudi Arabia | — | + | NM | NM | + | + |
| 15 | Kariminejad et al., 2019 [10] | 6 y | M | Iran | + (prominent forehead) | + | NM | + | + | — |
| 16 | Imaizumi et al., 2018 [21] | 15 y | M | Japan | — | + | NM | + | + | — |
| 17 | Uittenbogaard et al., 2018 [28] | 4 y | M | USA | — | + | NM | NM | + | + |
| 18 | Domingues et al., 2019 (patient 1) [22] | 29 y | M | Italy | + | + | + | + | + | — |
| 19 | Domingues et al., 2019 (patient 2) [22] | 19 y | M | Italy | + | + | + | + | + | — |
| 20 | Iodice et al., 2019 [29] | 6 y | F | Ukraine | + | + | + | NM | + (psychomotor) | + (severe) |
| 21 | Sun et al., 2019 (patient 1) [30] | 10 m | M | China | + | + | NM | NM | + | + |
| 22 | Sun et al., 2019 (patient 2) [30] | 18 m | F | China | + | + | NM | NM | + | + |
| 23 | Sun et al., 2019 (patient 3) [30] | 17 m | M | China | + | + | NM | NM | + | + |
| 24 | Trivisano et al., 2020 (patient 1) [3] | 11 y | F | Italy | + | + | + | — | — | — |
| 25 | Trivisano et al., 2020 (patient 1) [3] | 6 y | F | Italy | + | + | + | — | — | — |
| 26 | Tanaka et al., 2020 [23] | 4 y | F | Japan | + | + | NM | NM | + (walk and speech) | + |
| 27 | El Halabi et al., 2021 (patient 1) [34] | 11 m | F | Lebanon | + | + | — | — | + | — |
| 28 | El Halabi et al., 2021 (patient 2) [34] | 19 m | F | Lebanon | + | + | — | — | + | — |
| 29 | Cherian et al., 2021 [24] | 3 y | F | India | + | + | — | — | + | + (truncal) |
| 30 | Yang et al., 2022 (patient 1) [25] | NM | NM | China | NM | NM | NM | NM | NM | NM |
| 31 | Yang et al., 2022 (patient 2) [25] | NM | NM | China | NM | NM | NM | NM | NM | NM |
| 32 | Yang et al., 2022 (patient 3) [25] | NM | NM | China | NM | NM | NM | NM | NM | NM |
| Patient No. | Authors | Consanguinity | Parents Origin | Genetic Mutation | Patient Allele | Paternal Allele | Maternal Allele |
|---|---|---|---|---|---|---|---|
| 1 | Basel-Vanagaite et al., 2013 [9] | — | Iraqi–Jewish | Homozygous mutation in the SZT2 gene | Homozygous for paternally and maternally inherited variant at c.73C<T | c.73C>T p.Arg25* | c.73C>T p.Arg25* |
| 2 | Basel-Vanagaite et al., 2013 [9] | — | Spanish | Heterozygous mutation in the SZT2 gene | Compound of heterozygous for paternally inherited variant at c.1496G>T Maternally inherited variant at c.2092C>T (p.Gln698*) | c.1496G>T p.Gly412Alafs*86 | c.2092C>T (p.Gln698*) |
| 3 | Falcone et al., 2013 (patient 1) [19] | + (first degree) | Southern Italy | Homozygous mutation in the SZT2 gene | Homozygous for paternally and maternally inherited variant at c.4202_4204delTTC | c.4202_4204delTTC (p.Phe1401del) | c.4202_4204delTTC (p.Phe1401del) |
| 4 | Falcone et al., 2013 (patient 2) [19] | + (first degree) | Southern Italy | Homozygous mutation in the SZT2 gene | Homozygous for paternally and maternally inherited variant at c.4202_4204delTTC | c.4202_4204delTTC (p.Phe1401del) | c.4202_4204delTTC (p.Phe1401del) |
| 5 | Falcone et al., 2013 (patient 3) [19] | + (first degree) | Southern Italy | Homozygous mutation in the SZT2 gene | Homozygous for paternally and maternally inherited variant at c.4202_4204delTTC | c.4202_4204delTTC (p.Phe1401del) | c.4202_4204delTTC (p.Phe1401del) |
| 6 | Venkatesan et al., 2016 [17] | NM | NM | Heterozygous mutation in the SZT2 gene | Compound of Heterozygous for Paternally inherited variant at c.3509_3512delCAGA (p.T1170RfsX22) Maternally inherited variant at c.9703 C>T (p.R3235X) | c.3509_3512delCAGA (p.T1170RfsX22) | c.9703C>T (p.R3235X) |
| 7 | Vanderver et al., 2016 [33] | NM | NM | SZT2 gene | NM | c.5499del (p. Phe1834Serfs*47) | c.6916G>A (p. Gly2306Arg) |
| 8 | Tsuchida et al., 2018 (patient 1) [20] | — | Japanese | Heterozygous mutation in the SZT2 gene | Compound of heterozygous for paternally inherited variant at c.3700_3716del:p.(Asn1234Alafs*35) Maternally inherited variant at c.5482del:p.(Gly1829Valfs*52) | c.3700_3716del (p.Asn1234Alafs*35) | c.5482del (p.Gly1829Valfs*52) |
| 9 | Tsuchida et al., 2018 (patient 2) [20] | — | Japanese | Heterozygous mutation in the SZT2 gene | Compound of heterozygous for paternally inherited variant at c.3947dup:p.(Glu1317Glyfs*4) Maternally inherited variant at c.2929+1G>A: p.(Leu939Aspfs*19) | c.3947dup (p.Glu1317Glyfs*4) | c.2929+1G>A (p.Leu939Aspfs*19) |
| 10 | Tsuchida et al., 2018 (patient 3) [20] | + | Malaysian | Heterozygous mutation in the SZT2 gene | Compound of heterozygous for paternally inherited variant at c.7303C>T:p.(Arg2435Trp) Maternally inherited variant at c.8162C>G:p.(Ser2721Cys) | c.7303C>T (p.Arg2435Trp) | c.8162C>G (p.Ser2721Cys) |
| 11 | Nakamura et al., 2018 [26] | — | Japanese | Heterozygous mutation in the SZT2 gene | Compound heterozygous for two paternally inherited variants at c.4181C>T (p.Pro1394Leu) and c.2930-17_2930-3delinsCTCGTG Maternally inherited variant at c.8596dup (p.Tyr2866Leufs*42) | c.4181C>T (p.Pro1394Leu) and c.2930-17_2930-3delinsCTCGTG | c.8596dup (p.Tyr2866Leufs*42) |
| 12 | Pizzino et al., 2018 [27] | NM | NM | Heterozygous mutation in the SZT2 gene | Compound heterozygous for paternally inherited variant at c.5499delC (p.Phe1834Serfs*47) Maternally inherited variant at c.6916G>A (p.Gly2306Arg) | — | c.6916G>A (p.Gly2306Arg) |
| 13 | Naseer et al., 2018 (patient 1) [8] | + (first degree) | Saudi Arabian | Homozygous mutation in the SZT2 gene | Homozygous for paternally and maternally inherited variant at c.9368G>A | c.9368G>A (p.Gly3123Glu) | c.9368G>A (p.Gly3123Glu) |
| 14 | Naseer et al., 2018 (patient 2) [8] | + (first degree) | Saudi Arabian | Homozygous mutation in the SZT2 gene | Homozygous for paternally and maternally inherited variant at c.9368G>A | c.9368G>A (p.Gly3123Glu) | c.9368G>A (p.Gly3123Glu) |
| 15 | Kariminejad et al., 2019 [10] | + | NM | Homozygous mutation in the SZT2 gene | Homozygous for paternally and maternally inherited variant at NM_015284.3: c.7442G>A (p.Cys2481Tyr) | c.7442G>A (p.Cys2481Tyr) | c.7442G>A (p.Cys2481Tyr) |
| 16 | Imaizumi et al., 2018 [21] | — | Japanese | Homozygous mutation in the SZT2 gene | Homozygous for paternally and maternally inherited variant at c.6553C>T (p.Arg2185Trp) | c.6553C>T (p.Arg2185Trp) | c.6553C>T (p.Arg2185Trp) |
| 17 | Uittenbogaard et al., 2018 [28] | NM | NM | Heterozygous mutation in the SZT2 gene | Compound of heterozygous for paternally inherited variant at c.5949_5951del TGT (p.Val1984del) Maternally inherited variant at c.5174C>T (p.A1725V) | c.5949_5951del TGT (p.Val1984del) | c.5174 C>T (p.A1725V) |
| 18 | Domingues et al., 2019 (patient 1) [22] | — | Caucasian | Heterozygous mutation in the SZT2 gene | Compound of heterozygous for paternally inherited variant at c.6553C>T (p.Arg2185Trp) Maternally inherited variant at c.498G>T (p.Gln166His) | c.6553C>T (p.Arg2185Trp) | c.498G>T (p.Gln166His) |
| 19 | Domingues et al., 2019 (patient 2) [22] | — | Caucasian | Heterozygous mutation in the SZT2 gene | Compound of heterozygous for paternally inherited variant at c.6553C>T (p.Arg2185Trp) Maternally inherited variant at c.498G>T (p.Gln166His) | c.6553C>T (p.Arg2185Trp) | c.498G>T (p.Gln166His) |
| 20 | Iodice et al., 2019 [29] | NM | Ukrainian | Heterozygous mutation in the SZT2 gene | Compound heterozygous for paternally inherited variant at c.3632G>A (p.Arg1211Gln) Maternally inherited variant at c8435delC (p.Ser2812Leufs*41) | c.3632GNA (p.Arg1211Gln) | c8435delC (p. Ser2812Leufs*41) |
| 21 | Sun et al., 2019 (patient 1) [30] | — | NM | Heterozygous mutation in the SZT2 gene | Compound heterozygous for paternally inherited variant at c.1626 + 1G>A: splicing Maternally inherited variant at c.5772dupA (p.C1924 fs) | c.1626 + 1G>A: splicing | c.5772dupA (p.C1924 fs) |
| 22 | Sun et al., 2019 (patient 2) [30] | — | NM | Heterozygous mutation in the SZT2 gene | Compound heterozygous for paternally inherited variant at c.1626 + 1G>A: splicing Maternally inherited variant at c.5772dupA (p.C1924 fs) | c.1626 + 1G>A: splicing | c.5772dupA (p.C1924 fs) |
| 23 | Sun et al., 2019 (patient 3) [30] | — | Chinese | Heterozygous mutation in the SZT2 gene | Compound heterozygous for paternally inherited variant at c.4209C>A (p. C1403X,1973) Maternally inherited variant at c.7307_7308insG (p.A2436fs*22) | c.4209C>A (p.C1403X,1973) | c.7307_7308insG (p.A2436fs*22) |
| 24 | Trivisano et al., 2020 (patient 1) [3] | + | Southern Italy | Homozygous mutation in the SZT2 gene | Homozygous for paternally and maternally inherited variant at c.7825TNG;p.(Trp2609Gly) | c.7825T>G p.(Trp2609Gly) | c.7825T>G p.(Trp2609Gly) |
| 25 | Trivisano et al., 2020 (patient 1) [3] | + | Southern Italy | Homozygous mutation in the SZT2 gene | Homozygous for paternally and maternally inherited variant at c.7825TNG;p.(Trp2609Gly) | c.7825T>G p.(Trp2609Gly) | c.7825T>G p.(Trp2609Gly) |
| 26 | Tanaka et al., 2020 [23] | — | NM | Heterozygous mutation in the SZT2 gene | Compound heterozygous for paternally inherited variant at c.2798C>T (p.Ser933Phe) Maternally inherited variant at c.4549C>T (p.Arg1517Trp) | c.2798C>T (p.Ser933Phe) | c.4549C>T (p.Arg1517Trp) |
| 27 | El Halabi et al., 2021 (patient 1) [34] | + (first degree) | NM | Homozygous mutation in the SZT2 gene | Homozygous for Paternally and maternally inherited variant at c.82C>T (p.Arg28*) | c.82C>T (p.Arg28*) | c.82C>T (p.Arg28*) |
| 28 | El Halabi et al., 2021 (patient 2) [34] | + (first degree) | NM | Homozygous mutation in the SZT2 gene | Homozygous for paternally and maternally inherited variant at c.82C > T p. (Arg28*) | c.82C>T (p.Arg28*) | c.82C>T (p.Arg28*) |
| 29 | Cherian et al., 2021 [24] | — | NM | Homozygous mutation in the SZT2 gene | Homozygous for paternally and maternally inherited variant at (p.Asp2440ArgfsTer18) | p.Asp2440ArgfsTer18 | p. Asp2440ArgfsTer18 |
| 30 | Yang et al., 2022 (patient 1) [25] | NM | NM | Heterozygous mutation in the SZT2 gene | Compound heterozygous for inherited variants at c.2887A>G/c.7970G>A | NM | NM |
| 31 | Yang et al., 2022 (patient 2) [25] | NM | NM | Heterozygous mutation in the SZT2 gene | Compound heterozygous for inherited variants at c.3508A>G/c.7936C>T | NM | NM |
| 32 | Yang et al., 2022 (patient 3) [25] | NM | NM | Heterozygous mutation in the SZT2 gene | Compound heterozygous for inherited variants at c.2489G>T/c.8640_8641insC | NM | NM |
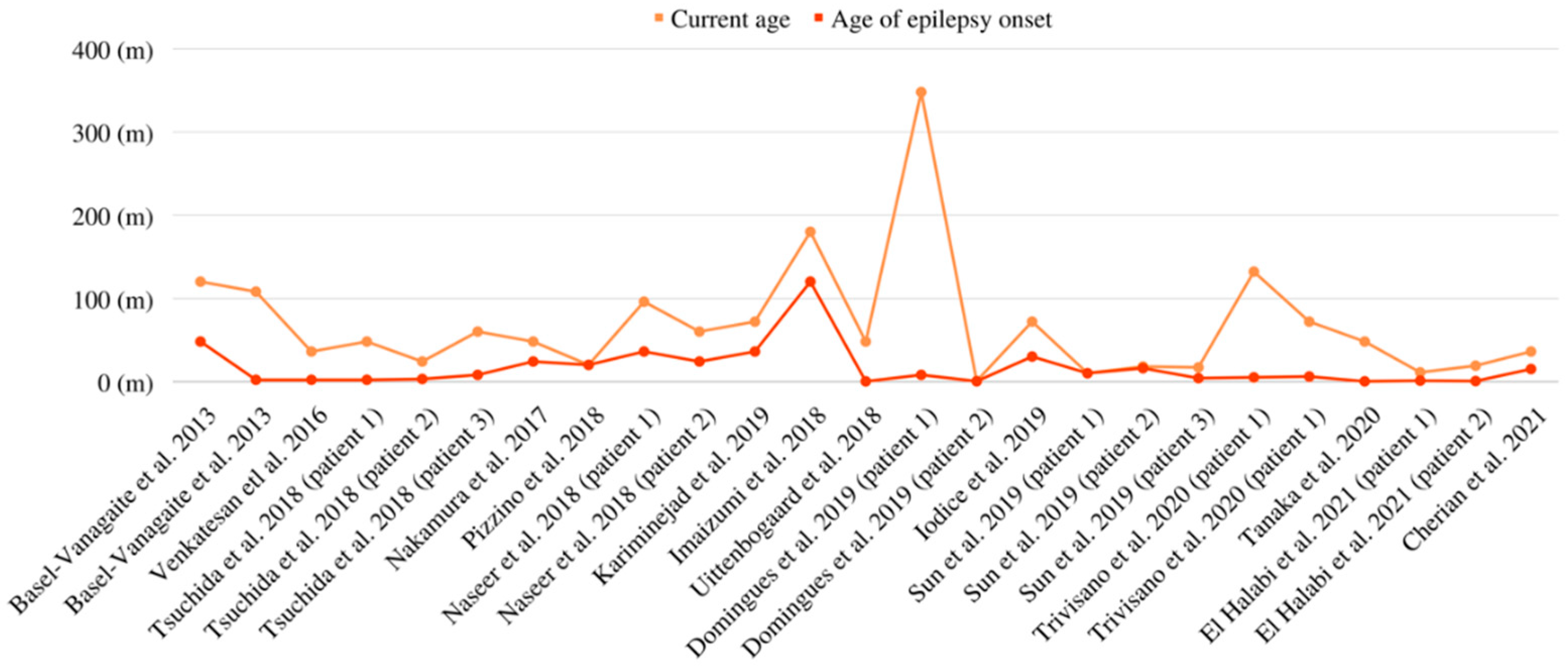
| Patient No. | Authors | Epilepsy Onset | Seizure Semiology | EEG Findings | MRI Findings | Management |
|---|---|---|---|---|---|---|
| 1 | Basel-Vanagaite et al., 2013 [9] | 4 y | Focal to bilateral tonic-clonic seizures | Generalized epileptic discharges | Short and thick CC and persistent CSP | NM |
| 2 | Basel-Vanagaite et al., 2013 [9] | 2 m | Generalized motor tonic-clonic seizures | Slow background and multifocal spikes in either hemisphere | Short and thick CC and persistent CSP | NM |
| 3 | Falcone et al., 2013 (patient 1) [19] | NM | NM | Normal | Normal | NM |
| 4 | Falcone et al., 2013 (patient 2) [19] | NM | NM | Normal | Normal | NM |
| 5 | Falcone et al., 2013 (patient 3) [19] | NM | NM | NM | Normal | NM |
| 6 | Venkatesan et al., 2016 [17] | 2 m | Generalized motor tonic-clonic seizures | Multifocal epileptiform discharges consistent with a diffuse epileptogenic encephalopathy | Right periventricular heterotopia | Phenobarbital (5.8 mg/kg/day), Levetiracetam (56.1 mg/kg/day), Pyridoxine (17 mg/kg/day), Topiramate (2.8 mg/kg/day), Lamotrigine (4.9 mg/kg/day), and Divalproex (17.4 mg/kg/day) |
| 7 | Vanderver et al., 2016 [33] | NM | NM | NM | Deficits in myelination, mild cerebellar atrophy, and volume loss of the CC and FL | NM |
| 8 | Tsuchida et al., 2018 (patient 1) [20] | 2 m | Focal to bilateral tonic-clonic seizures | Slow, 4–5 Hz background activity with spike waves in the left frontocentral areas | Short and thick CC | Phenobarbital, Phenytoin, and potassium bromide |
| 9 | Tsuchida et al., 2018 (patient 2) [20] | 3 m | Seizures presented as either with cyanosis and stopping motion, or as adversive seizures with fencing posture | Rhythmic fast waves, low amplitude fast waves, rhythmic slow waves, bursts of spikes, and sharp waves | Diffuse brain atrophy, thinned corpus callosum, and a persistent CSP | Valproic acid, Clobazam, Phenobarbital, Carbamazepine, Phenytoin, Zonisamide, Topiramate, and Levetiracetam |
| 10 | Tsuchida et al., 2018 (patient 3) [20] | 8 m | Tonic spasm and up-rolling of the eyeballs | Intermittent epileptic discharges over both posterior quadrants. These involved all regions, except the central and anterior areas with poorly organized background | Dilated lateral and third ventricles, and a thin corpus callosum | Clobazam |
| 11 | Nakamura et al., 2018 [26] | 2 y | Focal to bilateral tonic-clonic seizures | Focal epileptic discharges and background slowing | Short and thick CC | Valproate |
| 12 | Pizzino et al., 2018 [27] | 20 m | Focal to bilateral tonic-clonic seizures | Multifocal epileptiform discharges | Increasing size and loss of normal posterior pituitary signal | Lamotrigine, Levetiracetam, Phenytoin, Phenobarbital, Topiramate, Rufinamide, Midazolam, and ketogenic diet |
| 13 | Naseer et al., 2018 (patient 1) [8] | 3 y | NM | Background of predominately beta wave rhythm. Paroxysmal epileptic discharge, mainly on the right central hemisphere | Prominent extra-axial cerebrospinal fluid space with wide Sylvian fissure | NM |
| 14 | Naseer et al., 2018 (patient 2) [8] | 2 y | NM | Background of predominately beta wave rhythm. Paroxysmal epileptic discharge, mainly on the right central hemisphere | Prominent extra-axial cerebrospinal fluid space with wide Sylvian fissure | NM |
| 15 | Kariminejad et al., 2019 [10] | 3 y | Tonic-clonic seizures | Normal | Normal | Diazepam (IM injection), Phenobarbital (90 mg/bd), Sodium valproate (400 mg/bd), and Risperidone (5 mg/daily) |
| 16 | Imaizumi et al., 2018 [21] | 10 y | Complex partial seizures | Multifocal spikes or spikes and waves, predominantly in the frontal lobes | Normal | Unspecified antiepileptic medications |
| 17 | Uittenbogaard et al., 2018 [28] | 1 d | Transient seizures following a hypoxic ischemic event | Excessive left and right frontal sharp wave discharges were indicated | The involvement of the globus pallidum | Phenobarbital and Levetiracetam |
| 18 | Domingues et al., 2019 (patient 1) [22] | 8 m | Focal motor to bilateral tonic-clonic seizures | NM | Normal | Sodium valproate, Vigabatrin, Valproate, and Lacosamide |
| 19 | Domingues et al., 2019 (patient 2) [22] | 4 d | Focal and focal-to-generalized tonic seizures | Sub-continuous bifrontal sharp waves and multifocal epileptiform discharges | Right frontal polymicrogyria | Phenytoin, Carbamazepine, Clobazam, Topiramate, Phenytoin, Zonisamide, Rufinamide, Lacosamide, and ACTH |
| 20 | Iodice et al., 2019 [29] | 30 m | Cluster of afebrile focal-clonic seizures | Epileptic encephalopathy with a Lennox–Gastaut-like pattern | Thick and short CC and right hippocampal atrophy | Levetiracetam, Valproic acid, Rufinamide, and Clobazam |
| 21 | Sun et al., 2019 (patient 1) [30] | 10 m | Focal tonic or clonic seizures; status epilepticus | Slow background, epileptic discharges originating from L medial temporal area on ictal EEG | Enlarged ventricle and delayed myelination in the terminal zone | Levetiracetam, Oxcarbazepine, and Phenobarbital |
| 22 | Sun et al., 2019 (patient 2) [30] | 16 m | Focal or generalized tonic seizures; status epilepticus | Slow background, multifocal discharges on interictal EEG, epileptic discharges originating from L temporal area on ictal EEG | Normal | Valproic acid, Nitrazepam, and Topiramate |
| 23 | Sun et al., 2019 (patient 3) [30] | 4 m | Focal or generalized tonic seizure; status epilepticus | Slow background, multifocal discharges with partial generalization | Subependymal nodules, shortened corpus callosum, enlarged ventricles, and widened cavum septum pellucidum | Levetiracetam, Topiramate, Clonazepam, and Valproic acid |
| 24 | Trivisano et al., 2020 (patient 1) [3] | 5 m | Focal to bilateral tonic-clonic seizures, at times with secondary generalization | Bilateral temporal theta waves and rare spikes in right temporal | Thick CC abnormally shaped | Phenobarbital and Carbamazepine |
| 25 | Trivisano et al., 2020 (patient 1) [3] | 6 m | Focal to bilateral tonic-clonic seizures, at times with secondary generalization during sleep | Altered background, left temporal, right C–P spikes, and slow waves | CC abnormally shaped | Carbamazepine, Levetiracetam, Valproate, Phenobarbital, Clobazam, Oxcarbazepine, Rufinamide, and Lacosamide |
| 26 | Tanaka et al., 2020 [23] | 4 d | Focal seizures followed by tonic or clonic seizures affecting one or both limbs on the left side; occasionally, bilateral tonic-clonic seizures | Multifocal epileptiform discharges | Thin CC, persistent CSP, and atrophy | Phenobarbital, Midazolam, and Carbamazepine (5 mg/kg/day) |
| 27 | El Halabi et al., 2021 (patient 1) [34] | 1 m | Staring, blinking, and flushing | Multifocal independent spikes | Short CC of normal thickness and persistent CSP | Phenytoin, Carbamazepine, Oxcarbazepine, Levetiracetam, Clonazepam, Valproate, potassium, bromide, Biotin or vitamin B7, Pyridoxine, and Folinic acid |
| 28 | El Halabi et al., 2021 (patient 2) [34] | 2 w | Behavioral arrest, increased tone, grimacing, and flushing | Generalized slowing, multifocal independent spikes with ictal activity originating independently from the right frontotemporal, left or bilateral temporal, and on occasions, the ictal discharge propagated from one hemisphere to the other, hypsarrhythmia | Persistent CSP with a CC of normal length and thickness | Levetiracetam, Valproate, Phenobarbital, Clonazepam, Lacosamide, ACTH, potassium bromide, Biotin or vitamin B7, Pyridoxine, and Folinic acid |
| 29 | Cherian et al., 2021 [24] | 15 m | Focal seizures with impaired awareness (hypomotor events) followed by generalized tonic seizures | Multifocal epileptiform abnormalities, which were maximum bifrontal and paucity of generalized discharges; generalized paroxysmal fast activity (GPFA) or burst attenuation (BA) pattern were conspicuously absent | Thickened, dysmorphic CC with abnormal CSP | Sodium, valproate, Phenobarbitone, Levetiracetam, Clobazam, Perampanel, and Zonisamide |
| 30 | Yang et al., 2022 (patient 1) [25] | NA | NA | NA | NA | NA |
| 31 | Yang et al., 2022 (patient 2) [25] | NA | NA | NA | NA | NA |
| 32 | Yang et al., 2022 (patient 3) [25] | NA | NA | NA | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muthaffar, O.Y.; Jan, M.M.S.; Alyazidi, A.S.; Alotibi, T.K.; Alsulami, E.A. Insight into Genetic Mutations of SZT2: Is It a Syndrome? Biomedicines 2023, 11, 2402. https://doi.org/10.3390/biomedicines11092402
Muthaffar OY, Jan MMS, Alyazidi AS, Alotibi TK, Alsulami EA. Insight into Genetic Mutations of SZT2: Is It a Syndrome? Biomedicines. 2023; 11(9):2402. https://doi.org/10.3390/biomedicines11092402
Chicago/Turabian StyleMuthaffar, Osama Y., Mohammed M. S. Jan, Anas S. Alyazidi, Taif K. Alotibi, and Eman A. Alsulami. 2023. "Insight into Genetic Mutations of SZT2: Is It a Syndrome?" Biomedicines 11, no. 9: 2402. https://doi.org/10.3390/biomedicines11092402