
Multi-Faceted Role of Cancer-Associated Adipocytes in Colorectal Cancer


Abstract
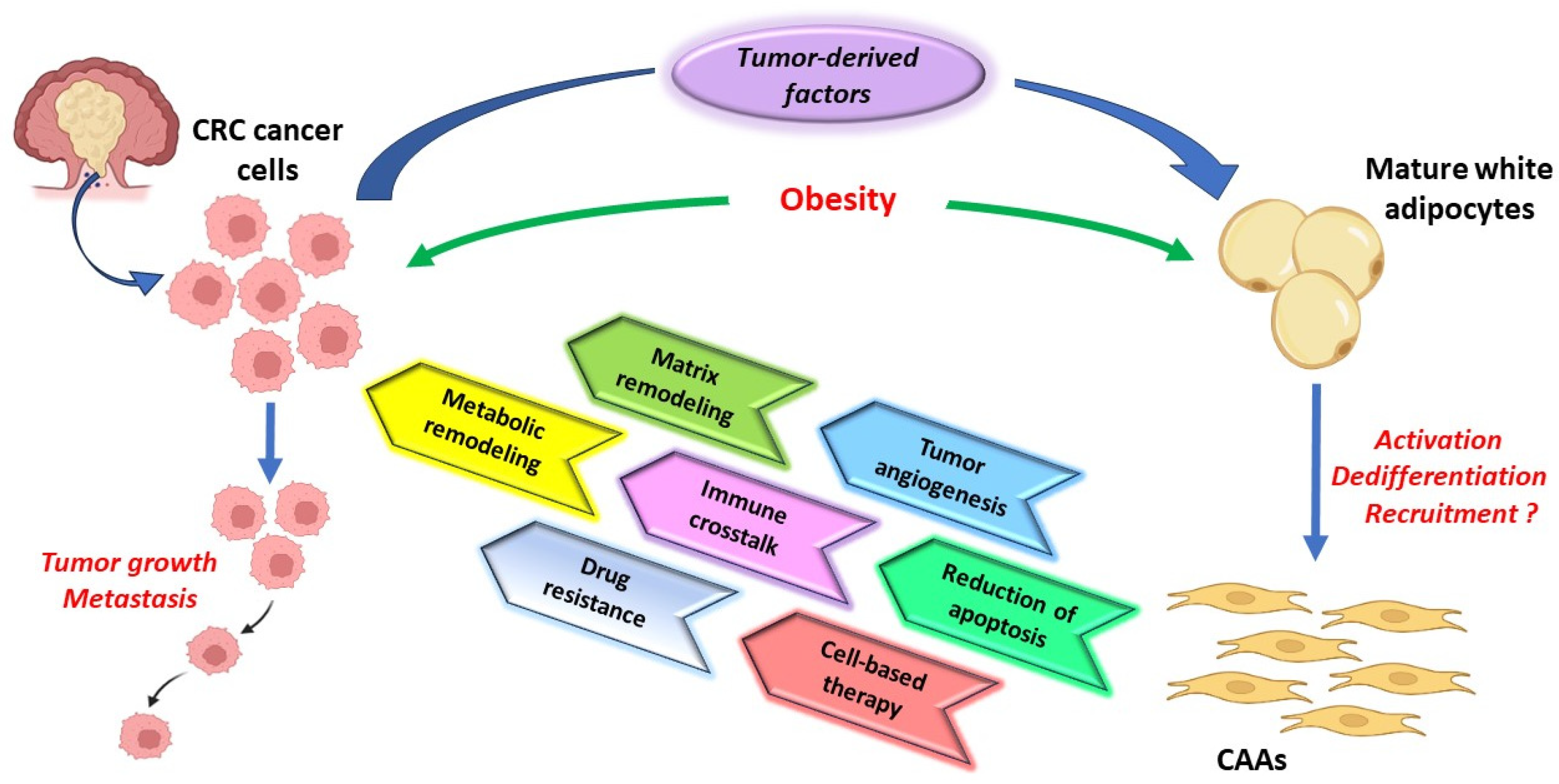
:1. Introduction
2. The Crosslink between Obesity and Colorectal Cancer
3. Cancer-Associated Adipocytes (CAAs) Characteristics
4. Potential Mechanisms of CAAs Differentiation in CRC
5. The CAAs Role in CRC Progression
5.1. CAAs Adipokines in CRC
5.2. CAAs Metabolites in CRC
5.3. CAAs Immunomodulatory Role in CRC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathways/Key Gene/Molecules/Receptors | Function | References | ||
|---|---|---|---|---|
| Adipokines | Leptin | c-Jun, Akt, and JAK/STAT3 axis IL-6, MMPs, and TGF-β ↑ | carcinogenesis metastasis | [5,14] |
| Adiponectin | AMPK axis | inhibition of tumor cell proliferation, angiogenesis, and metastasis | [5,64,65] | |
| Apelin | tumor growth and migration pathways | lymph node and distant metastasis | [75] | |
| Gherlin | gherlin gene ↓ | increase in TME inflammation | [58] | |
| Resistin | Toll-like receptor 4 ↑ IL-6 and TNF-α ↑ | promotion of tumor angiogenesis and metastasis | [1,76] | |
| Visfatin | ERK1/2, p38 MAPK, PI3K/mTOR, JNK, and JAK/STAT axis visfatin/SDF-1/Akt axis | support of tumor cell proliferation and metastasis inhibition of 5-FU therapeutic effect | [79,80] | |
| Insulin | PI3K/Akt pathway | increase in the resistance to 5-FU and cycloheximide cytotoxicity inhibition of tumor cell proliferation and metastasis | [81,84] | |
| IFGs | Src (tyrosine-protein kinase) | carcinogenesis proliferation of tumor cells | [85,86] | |
| VEGF | VEGF gene | tumor angiogenesis and metastasis | [96] | |
| IL-6 | STAT3 axis | support of tumor growth and metastasis | [5,88,89] | |
| TNF-α | NF-kB, Wnt/β-catenin, ERK1/2 axis | carcinogenesis and tumor cell migration | [90,91,92] | |
| PAI-1 | PAI1–tPA axis | increase in TGF-β expression in colitis CRC carcinogenesis and metastasis (?) | [94] | |
| CCL2 | CCL2 gene | increase in TME macrophages infiltration and induction of tumor progression | [14] | |
| MMP9 and MMP11 | MMP genes | induction of tumoral extracellular matrix remodeling and metastasis | [1,4,5,28] | |
| Metabolites | Lactate Pyruvate ATP | fatty acids oxidation | tumor growth and metastasis | [5,28] |
| FFAs | FFAs receptors (FABPs and CD36) | increase in ROS synthesis and fatty acids oxidation promotion of tumor growth and metastasis | [101,102,103] | |
| Glutamine | CXCL2-VEGFA axis | support of angiogenesis, tumor growth and metastasis | [108,109] | |
| Immune cells activity modulation | IL-6 leptin | JAK/STAT3 axis | promotion of MDSCs infiltration reduction in NK cells cytotoxicity modulation of the innate and adaptive immune response | [48,113] |
| FFAs | FFA uptake | induction of neutrophils recruitment | [5,111,112] | |
| FFAs Lactate Glycerin | metabolic remodeling VEGF | reduction in T cells antitumoral activity and dendritic cells activation tumor angiogenesis and metastasis | [5,28,113] | |
| PD-L1 | glycolysis and FAO | reduction in CD4+ and CD8+ T cells activity induction of M2-like macrophages polarization promotion of tumor growth and metastasis | [5,28] | |
| MAIT-derived IL-17 | IL-23/IL-17 axis | promotion of MDSCs recruitment increase in VEGF and Bcl-2 in TME and Bcl-x expression in tumor cells carcinogenesis and tumor growth | [110,118] |
5.4. CAAs and Potential Therapeutic Strategies in CRC: New Trends and Future Perspectives
| Potential Drugs | Target | Mechanism | Study Model | References |
|---|---|---|---|---|
| Adipocytes and CAAs-derived visfatin | 5-FU chemoresistance ↑ | SDF-1/CXCR4 pathway | DLD-1 and SW620 CRC cells and CRC patient tissue samples | [80] |
| FASN inhibitors (TVB-3664) | FASN activity ↓ | AMPK, Akt, and Erk1/2 pathway | CRC patient-derived xenograft model | [120] |
| CD36 (fatty acid transport proteins) inhibitors | CD36 blockage |
| Cd36 fl/fl mice inoculated with MC38 CRC cells mice CD36 shRNA and patients’ tumor tissue samples C57BL/6J mice inoculated with HCT116 CRC cells and CRC patient tissue samples | [124,125,128,129] |
| PPAR-ɣ antagonist (pioglitazone, cladosporol B) | PPAR | COX-2 and cyclin D1 expression ↓ | CB-17/lcr-scid/Jclw mice inoculated with HT-29 and SW480 CRC cells | [131,132] |
| MCT1 inhibitors (AZD3965) (?) | MCT1 | modulate the lactate export from tumor cells | phase I tumor clinical trial | [133] |
| CAAs-derived leptin inhibitors (Doxil) | Ob-R | tumor growth inhibition | BALB/c mice bearing C26 murine carcinoma | [134] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chaplin, A.; Rodriguez, R.M.; Segura-Sampedro, J.J.; Ochogavía-Seguí, A.; Romaguera, D.; Barceló-Coblijn, G. Insights behind the relationship between colorectal cancer and obesity: Is visceral adipose tissue the missing link? Int. J. Mol. Sci. 2022, 23, 13128. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [PubMed]
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; He, S. Multi-faceted role of cancer-associated adipocytes in the tumor microenvironment (Review). Mol. Med. Rep. 2021, 24, 866. [Google Scholar]
- Lo Iacono, M.L.; Modica, C.; Porcelli, G.; Brancato, O.R.; Muratore, G.; Bianca, P.; Gaggianesi, M.; Turdo, A.; Veschi, V.; Todaro, M.; et al. Targeting of the peritumoral adipose tissue microenvironment as an innovative antitumor therapeutic strategy. Biomolecules 2022, 12, 702. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar]
- Bejarano, L.; Jordāo, M.J.C.; Joyce, J.A. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar]
- Ten Hoorn, S.; de Back, T.R.; Sommeijer, D.W.; Vermeulen, L. Clinical value of consensus molecular subtypes in colorectal cancer: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2022, 114, 503–516. [Google Scholar]
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, P.A.G.; Stevens, G.A.; Ezzati, P.M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R.; et al. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36–46. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- Rybinska, I.; Mangano, N.; Tagliabue, E.; Triulzi, T. Cancer-associated adipocytes in breast cancer: Causes and consequences. Int. J. Mol. Sci. 2021, 22, 3775. [Google Scholar] [CrossRef] [PubMed]
- Bouche, C.; Quail, D. Fueling the tumor microenvironment with cancer-associated adipocytes. Cancer Res. 2023, 83, 1170–1172. [Google Scholar] [CrossRef]
- Dong, Y.; Zhou, J.; Zhu, Y.; Luo, L.; He, T.; Hu, H.; Liu, H.; Zhang, Y.; Luo, D.; Xu, S.; et al. Abdominal obesity and colorectal cancer risk: Systematic review and meta-analysis of prospective studies. Biosci. Rep. 2017, 37, BSR20170945. [Google Scholar] [CrossRef]
- Ye, P.; Xi, Y.; Huang, Z.; Xu, P. Linking obesity with colorectal cancer: Epidemiology and mechanistic insights. Cancers 2020, 12, 1408. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, J.; Yang, J.; Jin, Z.; Shi, W.; Qin, Y.; Yu, F.; He, J. Association between adult weight gain and colorectal cancer: A dose-response meta-analysis of observational studies. Int. J. Cancer 2015, 136, 2880–2889. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, S.; Aleksandrova, K.; Abar, L.; Vieria, A.R.; Vingeliene, S.; Polemiti, E.; Stevens, C.A.T.; Greenwood, D.C.; Chan, D.S.M.; Aune, D.; et al. Adult weight gain and colorectal adenomas-a systematic review and meta-analysis. Ann. Oncol. 2017, 28, 1217–1229. [Google Scholar] [CrossRef] [PubMed]
- Bull, C.J.; Bell, J.A.; Murphy, N.; Sanderson, E.; Smith, G.D.; Timpson, N.J.; Banbury, B.L.; Albanes, D.; Berndt, S.I.; Bézieau, S.; et al. Adiposity, metabolites, and colorectal cancer risk: Mendelian randomization study. BMC Med. 2020, 18, 396. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.M.; Brown, K.A.; Iyengar, N.M. Targeting obesity-related dysfunction in hormonally driven cancers. Br. J. Cancer 2021, 125, 495–509. [Google Scholar]
- Bhaskaran, K.; Dos-Santos-Silva, I.; Leon, D.A.; Douglas, I.J.; Smeeth, L. Association of BMI with overall and cause-specific mortality: A population-based cohort study of 3.6 million adults in the UK. Lancet Diabetes Endocrinol. 2018, 6, 944–953. [Google Scholar] [CrossRef]
- Afshar, S.; Kelly, S.B.; Seymour, K.; Lara, J.; Woodcock, S.; Mathers, J.C. The effects of bariatric surgery on colorectal cancer risk: Systematic review and meta-analysis. Obes. Surg. 2014, 24, 1793–1799. [Google Scholar] [CrossRef]
- Grigoras, A.; Amalinei, C.; Balan, R.A.; Giusca, S.E.; Caruntu, I.D. Perivascular adipose tissue in cardiovascular diseases-an update. Anatol. J. Cardiol. 2019, 22, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Wu, Y.; Fried, S.K. Adipose tissue heterogeneity: Implication of depot differences in adipose tissue for obesity complications. Mol. Asp. Med. 2013, 34, 1–11. [Google Scholar]
- Das, E.; Moon, J.H.; Lee, J.H.; Thakkar, N.; Pausova, Z.; Sung, H.K. Adipose tissue and modulation of hypertension. Curr. Hypertens. Rep. 2018, 20, 96. [Google Scholar]
- Carruthers, N.; Strieder-Barboza, C.; Caruso, J.; Flesher, C.; Baker, N.; Kerk, S.; Ky, A.; Ehlers, A.; Varban, O.; Costas, A.; et al. The human type 2 diabetes-specific visceral adipose tissue proteome and transcriptome in obesity. Sci. Rep. 2021, 11, 17394. [Google Scholar] [CrossRef]
- Riondino, S.; Roselli, M.; Palmirotta, R.; Della-Morte, D.; Ferroni, P.; Guadagni, F. Obesity and colorectal cancer: Role of adipokines in tumor initiation and progression. World J. Gastroenterol. 2014, 20, 5177–5190. [Google Scholar] [CrossRef] [PubMed]
- Notarnicola, M.; Miccolis, A.; Tutino, V.; Lorusso, D.; Caruso, M.G. Low levels of lipogenic enzymes in peritumoral adipose tissueof colorectal cancer patients. Lipids 2012, 47, 59–63. [Google Scholar] [CrossRef]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, J.; Sun, S.; Yuan, J.; Sun, S. Cancer-associated adipocytes as immunomodulators in cancer. Biomark. Res. 2021, 9, 2. [Google Scholar]
- Tang, Y.; Zhang, W.; Sheng, T.; He, X.; Xiong, X. Overview of the molecular mechanisms contributing to the formation of cancer-associated adipocytes (Review). Mol. Med. Rep. 2021, 24, 768. [Google Scholar] [CrossRef]
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Duong, M.N.; Geneste, A.; Fallone, F.; Li, X.; Dumontet, C.; Muller, C. The fat and the bad: Mature adipocytes, key actors in tumor progression and resistance. Oncotarget 2017, 8, 57622–57641. [Google Scholar] [CrossRef] [PubMed]
- Grigoraş, A.; Amalinei, C.; Balan, R.A.; Giuşcă, S.E.; Avădănei, E.R.; Lozneanu, L.; Căruntu, I.D. Adipocytes spectrum—From homeostasia to obesity and its associated pathology. Ann. Anat. 2018, 219, 102–120. [Google Scholar] [CrossRef]
- López-Canoa, J.N.; Couselo-Seijas, M.; González-Ferrero, T.; Almengló, C.; Álvarez, E.; González-Maestro, A.; González-Melchor, L.; Martínez-Sande, J.L.; García-Seara, J.; Fernández-López, J.; et al. The role of fatty acid-binding protein 4 in the characterization of atrial fibrillation and the prediction of outcomes after catheter ablation. Int. J. Mol. Sci. 2022, 23, 11107. [Google Scholar] [CrossRef]
- Cheng, H.S.; Yip, Y.S.; Lim, E.K.Y.; Wahli, W.; Tan, N.S. PPARs and Tumor Microenvironment: The emerging roles of the metabolic master regulators in tumor stromal–epithelial crosstalk and carcinogenesis. Cancers 2021, 13, 2153. [Google Scholar] [CrossRef]
- Tabuso, M.; Homer-Vanniasinkam, S.; Adya, R.; Arasaradnam, R. Role of tissue microenvironment resident adipocytes in colon cancer. World J. Gastroenterol. 2017, 23, 5829–5835. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A. The role of tumor microenvironment cells in colorectal cancer (CRC) cachexia. Int. J. Mol. Sci. 2021, 22, 1565. [Google Scholar] [CrossRef]
- Wada, S.; Yasunaga, Y.; Oka, K.; Dan, N.; Tanaka, E.; Morita, K.; Masuda, E.; Yanagawa, K.; Matsumoto, H.; Yoshioka, S.; et al. Submucosal fat accumulation in human colorectal tissue and its association with abdominal obesity and insulin resistance. United Eur. Gastroenterol. J. 2018, 6, 1065–1073. [Google Scholar] [CrossRef]
- Stephens, J.M.; Pekala, P.H. Transcriptional repression of the C/EBP-alpha and GLUT4 genes in 3T3-L1 adipocytes by tumor necrosis factor-alpha. Regulations is coordinate and independent of protein synthesis. J. Biol. Chem. 1992, 267, 13580–13584. [Google Scholar] [CrossRef]
- Song, T.; Kuang, S. Adipocyte dedifferentiation in health and diseases. Clin. Sci. 2019, 133, 2107–2119. [Google Scholar] [CrossRef]
- Gustafson, B.; Smith, U. Activation of canonical wingless-type MMTV integration site family (Wnt) signaling in mature adipocytes increases beta-catenin levels and leads to cell dedifferentiation and insulin resistance. J. Biol. Chem. 2010, 285, 14031–14041. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Denson, J.L.; Brüning, J.C. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015, 36, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Placencio, V.; DeClerck, Y. Plasminogen activator inhibitor-1 in cancer: Rationale and insight for future therapeutic testing. Cancer Res. 2015, 75, 2969–2974. [Google Scholar] [CrossRef]
- Sillen, M.; Declerck, P. A Narrative review on plasminogen activator inhibitor-1 and its (patho)physiological role: To target or not to target? Int. J. Mol. Sci. 2021, 22, 2721. [Google Scholar] [CrossRef]
- Matsumoto, T.; Kano, K.; Kondo, D.; Fukuda, N.; Iribe, Y.; Tanaka, N.; Matsubara, Y.; Sakuma, T.; Satomi, A.; Otaki, M.; et al. Mature adipocyte-derived dedifferentiated fat cells exhibit multilineage potential. J. Cell Physiol. 2008, 215, 210–222. [Google Scholar] [CrossRef]
- Lin, Q.; Gao, Z.; Alarcon, R.M.; Ye, J.; Yun, Z. A role of miR-27 in the regulation of adipogenesis. FEBS J. 2009, 276, 2348–2358. [Google Scholar] [CrossRef]
- Yu, J.; Lv, Y.; Wang, F.; Kong, X.; Di, W.; Liu, J.; Sheng, Y.; Lv, S.; Ding, G. MiR-27b-3p inhibition enhances browning of epididymal fat in high-fat diet induced obese mice. Front. Endocrinol. 2019, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, J.; Li, Z.; Sun, S.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Zhang, Y.; Sun, S.; et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J. Exp. Clin. Cancer Res. 2019, 38, 223. [Google Scholar] [CrossRef]
- Sanhueza, S.; Simón, L.; Cifuentes, M.; Quest, A. The adipocyte–macrophage relationship in cancer: A potential target for antioxidant therapy. Antioxidants 2023, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tseng, C.; Zhang, Y.; Sirin, O.; Corn, P.G.; Li-Ning-Tapia, E.M.; Troncoso, P.; Davis, J.; Pettaway, C.; Ward, J.; et al. CXCL1 mediates obesity-associated adipose stromal cell trafficking and function in the tumour microenvironment. Nat. Commun. 2016, 7, 11674. [Google Scholar] [CrossRef]
- Vaitkus, J.A.; Celi, F.S. The role of adipose tissue in cancer-associated cachexia. Exp. Biol. Med. (Maywood) 2017, 242, 473–481. [Google Scholar] [CrossRef]
- Han, J.; Meng, Q.; Shen, L.; Wu, G. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis. 2018, 17, 14. [Google Scholar] [CrossRef]
- Nimri, L.; Peri, I.; Yehuda-Shnaidman, E.; Schwartz, B. Adipocytes isolated from visceral and subcutaneous depots of donors differing in BMI crosstalk with colon cancer cells and modulate their invasive phenotype. Transl. Oncol. 2019, 12, 1404–1415. [Google Scholar] [CrossRef] [PubMed]
- Welte, G.; Alt, E.; Devarajan, E.; Krishnappa, S.; Jotzu, C.; Song, Y.H. Interleukin-8 derived from local tissue-resident stromal cells promotes tumor cell invasion. Mol. Carcinog. 2012, 51, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Yang, Y.; Liu, K.; Zhao, J.; Chen, X.; Liu, H.; Bai, R.; Li, X.; Jiang, Y.; Zhang, X. Combination curcumin and (−)- epigallocatechin-3-gallate inhibits colorectal carcinoma microenvironment-induced angiogenesis by JAK/STAT3/IL-8 pathway. Oncogenesis 2017, 6, e384. [Google Scholar] [CrossRef]
- Ayele, T.M.; Muche, Z.T.; Teklemariam, A.B.; Kassie, A.B.; Abebe, E.C. Role of JAK2/STAT3 signaling pathway in the tumorigenesis, chemotherapy resistance, and treatment of solid tumors: A systemic review. J. Inflamm. Res. 2022, 15, 1349–1364. [Google Scholar] [CrossRef]
- Zhong, J.; Krawczyk, S.A.; Chaerkady, R.; Huang, H.; Goel, R.; Bader, J.S.; Wong, G.W.; Corkey, B.E.; Pandey, A. Temporal profiling of the secretome during adipogenesis in humans. J. Proteome Res. 2010, 9, 5228–5238. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A. Role of the ghrelin system in colorectal cancer. Int. J. Mol. Sci. 2022, 23, 5380. [Google Scholar] [CrossRef]
- Engin, A. Diet-induced obesity and the mechanism of leptin resistance. Adv. Exp. Med. Biol. 2017, 960, 381–397. [Google Scholar]
- Koda, M.; Sulkowska, M.; Kanczuga-Koda, L.; Surmacz, E.; Sulkowski, S. Overexpression of the obesity hormone leptin in human colorectal cancer. J. Clin. Pathol. 2007, 60, 902–906. [Google Scholar] [CrossRef]
- Endo, H.; Hosono, K.; Uchiyama, T.; Sakai, E.; Sugiyama, M.; Takahashi, H.; Nakajima, N.; Wada, K.; Takeda, K.; Nakagama, H. Leptin acts as a growth factor for colorectal tumours at stages subsequent to tumour initiation in murine colon carcinogenesis. Gut 2011, 60, 1363–1371. [Google Scholar] [PubMed]
- Aleksandrova, K.; Boeing, H.; Jenab, M.; Bueno-de-Mesquita, H.B.; Jansen, E.; van Duijnhoven, F.J.; Rinaldi, S.; Fedirko, V.; Romieu, I.; Riboli, E.; et al. Leptin and soluble leptin receptor in risk of colorectal cancer in the european prospective investigation into cancer and nutrition cohort. Cancer Res. 2012, 72, 5328–5337. [Google Scholar]
- Jaffe, T.; Schwartz, B. Leptin promotes motility and invasiveness in human colon cancer cells by activating multiple signal-transduction pathways. Int. J. Cancer 2008, 123, 2543–2556. [Google Scholar]
- La Cava, A. Adiponectin: A relevant player in obesity-related colorectal cancer? Gut 2013, 62, 483–484. [Google Scholar] [CrossRef]
- Di Zazzo, E.; Polito, R.; Bartollino, S.; Nigro, E.; Porcile, C.; Bianco, A.; Daniele, A.; Moncharmont, B. Adiponectin as link factor between adipose tissue and cancer. Int. J. Mol. Sci. 2019, 20, 839. [Google Scholar]
- Saxena, A.; Baliga, M.S.; Ponemone, V.; Kaur, K.; Larsen, B.; Fletcher, E.; Greene, J.; Fayad, R. Mucus and adiponectin deficiency: Role in chronic inflammation-induced colon cancer. Int. J. Colorectal. Dis. 2013, 28, 1267–1279. [Google Scholar] [PubMed]
- Wei, E.K.; Giovannucci, E.; Fuchs, C.S.; Willett, W.C.; Mantzoros, C.S. Low plasma adiponectin levels and risk of colorectal cancer in men: A prospective study. J. Natl. Cancer Inst. 2005, 97, 1688–1694. [Google Scholar]
- Saetang, J.; Boonpipattanapong, T.; Palanusont, A.; Maneechay, W.; Sangkhathat, S. Alteration of leptin and adiponectin in multistep colorectal tumorigenesis. Asian Pac. J. Cancer Prev. 2016, 17, 2119–2123. [Google Scholar] [CrossRef]
- Polito, R.; Nigro, E.; Fei, L.; De Magistris, L.; Monaco, M.L.; D’Amico, R.; Naviglio, S.; Signoriello, G.; Daniele, A. Adiponectin is inversely associated with tumour grade in colorectal cancer patients. AntiCancer Res. 2020, 40, 3751–3757. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Schettino, P.; Polito, R.; Scudiero, O.; Monaco, M.L.; De Palma, G.D.; Daniele, A. Adiponectin and colon cancer: Evidence for inhibitory effects on viability and migration of human colorectal cell lines. Mol. Cell Biochem 2018, 448, 125–135. [Google Scholar]
- Tae, C.H.; Kim, S.E.; Jung, S.A.; Joo, Y.H.; Shim, K.N.; Jung, H.K.; Kim, T.H.; Cho, M.S.; Kim, K.H.; Kim, J.S. Involvement of adiponectin in early stage of colorectal carcinogenesis. BMC Cancer 2014, 14, 811. [Google Scholar]
- Choe, E.K.; Yi, J.W.; Chai, Y.J.; Park, K.J. Upregulation of the adipokine genes ADIPOR1 and SPP1 is related to poor survival outcomes in colorectal cancer. J. Surg. Oncol. 2018, 117, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Gialamas, S.P.; Petridou, E.T.; Tseleni-Balafouta, S.; Spyridopoulos, T.N.; Matsoukis, I.L.; Kondi-Pafiti, A.; Zografos, G.; Mantzoros, C.S. Serum adiponectin levels and tissue expression of adiponectin receptors are associated with risk, stage, and grade of colorectal cancer. Metabolism 2011, 60, 1530–1538. [Google Scholar]
- Wysocka, M.B.; Pietraszek-Gremplewicz, K.; Nowak, D. The role of apelin in cardiovascular diseases, obesity and cancer. Front. Physiol. 2018, 9, 15. [Google Scholar]
- Podgórska, M.; Diakowska, D.; Pietraszek-Gremplewicz, K.; Nienartowicz, M.; Nowak, D. Evaluation of apelin and apelin receptor level in the primary tumor and serum of colorectal cancer patients. J. Clin. Med. 2019, 8, 1513. [Google Scholar] [CrossRef]
- Pham, T.T.; Nimptsch, K.; Aleksandrova, K.; Jenab, M.; Reichmann, R.; Wu, K.; Tjønneland, A.; Kyrø, C.; Schulze, M.; Kaaks, R.; et al. Pre-diagnostic circulating resistin concentrations are not associated with colorectal cancer risk in the european prospective investigation into cancer and nutrition study. Cancers 2022, 14, 5499. [Google Scholar] [PubMed]
- Yang, G.; Fan, W.; Luo, B.; Xu, Z.; Wang, P.; Tang, S.; Xu, P.; Yu, M. Circulating resistin levels and risk of colorectal cancer: A meta-analysis. BioMed. Res. Int. 2016, 2016, 7367485. [Google Scholar]
- Chang, M.L.; Yang, Z.; Yang, S.S. Roles of adipokines in digestive diseases: Markers of inflammation, metabolic alteration and disease progression. Int. J. Mol. Sci. 2020, 21, 8308. [Google Scholar] [PubMed]
- Umar, M.I.; Hassan, W.; Murtaza, G.; Buabeid, M.; Arafa, E.; Irfan, H.M.; Asmawi, M.Z.; Huang, X. The adipokine component in the molecular regulation of cancer cell survival, proliferation and metastasis. Pathol. Oncol. Res. 2021, 27, 1609828. [Google Scholar]
- Zhao, Q.; Long, Y.; Cheng, W.; Huang, Y.; Li, J.; Li, Y.; Li, X.; Guo, X.; Li, Y.; Li, G.; et al. Visfatin inhibits colon cancer cell apoptosis and decreases chemosensitivity to 5-FU by promoting the SDF-1/CXCR4/Akt axis. Int. J. Oncol. 2022, 60, 75. [Google Scholar]
- Chen, J.; Katsifis, A.; Hu, C.; Huang, X.F. Insulin decreases therapeutic efficacy in colon cancer cell line HT29 via the activation of the PI3K/Akt pathway. Curr. Drug Discov. Technol. 2011, 8, 119–125. [Google Scholar] [CrossRef]
- Vigneri, P.G.; Tirro, E.; Pennisi, M.S.; Massimino, M.; Stella, S.; Romano, C.; Manzella, L. The insulin/IGF system in colorectal cancer development and resistance to therapy. Front. Oncol. 2015, 5, 230. [Google Scholar]
- Garcia-Echeverria, C.; Sellers, W.R. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene 2008, 27, 5511–5526. [Google Scholar] [CrossRef]
- Zhu, S.; Bjorge, J.D.; Fujita, D.J. PTP1B contributes to the oncogenic properties of colon cancer cells through Src activation. Cancer Res. 2007, 67, 10129–10137. [Google Scholar] [CrossRef] [PubMed]
- Sekharam, M.; Nasir, A.; Kaiser, H.E.; Coppola, D. Insulin-like growth factor 1 receptor activates c-SRC and modifies transformation and motility of colon cancer in vitro. Anticancer Res. 2003, 23, 1517–1524. [Google Scholar] [PubMed]
- Abbruzzese, C.; Diodoro, M.G.; Sperduti, I.; Mileo, A.M.; Pattaro, G.; De Salvo, L.; Cosimelli, M.; Perrotti, N.; Paggi, M.G. Detection of phosphorylated insulin receptor in colorectal adenoma and adenocarcinoma: Implications for prognosis and clinical outcome. J. Cell Physiol. 2015, 230, 562–567. [Google Scholar] [PubMed]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Rasic, I.; Rebic, V.; Rasic, A.; Aksamija, G.; Radovic, S. The association of simultaneous increase in interleukin-6, C reactive protein, and matrix metalloproteinase-9 serum levels with increasing stages of colorectal cancer. J. Oncol. 2018, 2018, 2830503. [Google Scholar] [CrossRef]
- Himbert, C.; Ose, J.; Lin, T.; Warby, C.A.; Gigic, B.; Steindorf, K.; Schrotz-King, P.; Abbenhardt-Martin, C.; Zielske, L.; Boehm, J.; et al. Inflammation- and angiogenesis-related biomarkers are correlated with cancer-related fatigue in colorectal cancer patients: Results from the ColoCare Study. Eur. J. Cancer Care 2019, 28, e13055. [Google Scholar] [CrossRef]
- Wei, X.; Li, X.; Kong, F.; Ma, L.; Sui, Y.; Chen, D.; Xu, F. TNF-alpha activates Wnt signaling pathway to promote the invasion of human colon cancer stem cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2018, 34, 982–988. [Google Scholar] [PubMed]
- Kaltschmidt, C.; Banz-Jansen, C.; Benhidjeb, T.; Beshay, M.; Forster, C.; Greiner, J.; Hamelmann, E.; Jorch, N.; Mertzlufft, F.; Pfitzenmaier, J.; et al. A Role for NF-kappaB in organ specific cancer and cancer stem cells. Cancers 2019, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, Z. TNF-alpha promotes colon cancer cell migration and invasion by upregulating TROP-2. Oncol. Lett. 2018, 15, 3820–3827. [Google Scholar] [PubMed]
- Kim, E.R.; Yang, M.H.; Lim, Y.J.; Lee, J.H.; Chang, D.K.; Kim, Y.H.; Son, H.J.; Kim, J.J.; Rhee, J.C.; Kim, J.Y. Association between plasma levels of plasminogen activator inhibitor-1 and colorectal neoplasms. Gut Liver 2013, 7, 519–523. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Chen, F.; Lai, C.W.; Chiang, I.L.; Perrigoue, J.; Stojmirovic, A.; Li, K.; Muegge, B.D.; Jain, U.; VanDussen, K.L.; et al. PAI-1 augments mucosal damage in colitis. Sci. Transl. Med. 2019, 11, eaat0852. [Google Scholar] [CrossRef]
- McClellan, J.L.; Davis, J.M.; Steiner, J.L.; Enos, R.T.; Jung, S.H.; Carson, J.A.; Pena, M.M.; Carnevale, K.A.; Berger, F.G.; Murphy, E.A. Linking tumor-associated macrophages, inflammation, and intestinal tumorigenesis: Role of MCP-1. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1087–G1095. [Google Scholar] [CrossRef]
- Abdulla, M.H.; Shaik, A.S.; Vaali-Mohammed, M.A.; Al Khayal, K.A.; Traiki, T.B.; Zubaidi, A.M.; Al-Johani, T.; Shakoor, Z.; Al-Obeed, O.A. Expression of VEGF, EGF and HGF in early- and late-stage colorectal cancer. Mol. Clin. Oncol. 2021, 15, 251. [Google Scholar] [CrossRef]
- Bendardaf, R.; Buhmeida, A.; Hilska, M.; Laato, M.; Syrjänen, S.; Syrjänen, K.; Collan, Y.; Pyrhönen, S. VEGF-1 expression in colorectal cancer is associated with disease localization, stage, and long-term disease-specific survival. Anticancer Res. 2008, 28, 3865–3870. [Google Scholar] [PubMed]
- Martins, S.F.; Garcia, E.A.; Luz, M.A.; Pardal, F.; Rodrigues, M.; Filho, A.L. Clinicopathological correlation and prognostic significance of VEGF-A, VEGF-C, VEGFR-2 and VEGFR-3 expression in colorectal cancer. Cancer Genom. Proteom. 2013, 10, 55–67. [Google Scholar]
- Wang, J.; Chen, G.L.; Cao, S.; Zhao, M.C.; Liu, Y.Q.; Chen, X.X.; Qian, C. Adipogenic niches for melanoma cell colonization and growth in bone marrow. Lab. Investig. 2017, 97, 737–745. [Google Scholar] [CrossRef]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar]
- Wen, Y.A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Evers, B.M.; Gao, T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef]
- Capece, D.; Franzoso, G. Rewired lipid metabolism as an actionable vulnerability of aggressive colorectal carcinoma. Mol. Cell. Oncol. 2022, 9, 2024051. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, W.; Zhang, Y.; Zhu, T.; Hua, Y.; Li, H.; Zhang, Q.; Xia, M. FABP4 promotes invasion and metastasis of colon cancer by regulating fatty acid transport. Cancer Cell Int. 2020, 20, 512. [Google Scholar] [CrossRef]
- Zaytseva, Y. Lipid metabolism as a targetable metabolic vulnerability in colorectal cancer. Cancers 2021, 13, 301. [Google Scholar] [CrossRef]
- Feng, M.; Xiong, G.; Cao, Z.; Yang, G.; Zheng, S.; Qiu, J.; You, L.; Zheng, L.; Zhang, T.; Zhao, Y. LAT2 regulates glutamine-dependent mTOR activation to promote glycolysis and chemoresistance in pancreatic cancer. J. Exp. Clin. Cancer Res. 2018, 37, 274. [Google Scholar] [CrossRef]
- Turowski, G.A.; Rashid, Z.; Hong, F.; Madri, J.A.; Basson, M.D. Glutamine modulates phenotype and stimulates proliferation in human colon cancer cell lines. Cancer Res. 1994, 54, 5974–5980. [Google Scholar]
- Natsume, M.; Shimura, T.; Iwasaki, H.; Okuda, Y.; Hayashi, K.; Takahashi, S.; Kataoka, H. Omental adipocytes promote peritoneal metastasis of gastric cancer through the CXCL2-VEGFA axis. Br. J. Cancer 2020, 123, 459–470. [Google Scholar]
- Zhang, X.; Li, Q.; Du, A.; Li, Y.; Shi, Q.; Chen, Y.; Zhao, Y.; Wang, B.; Pan, F. Adipocytic glutamine synthetase upregulation via altered histone methylation promotes 5FU chemoresistance in peritoneal carcinomatosis of colorectal cancer. Front. Oncol. 2021, 11, 748730. [Google Scholar] [CrossRef]
- Del Cornò, M.; Conti, L.; Gessani, S. Innate lymphocytes in adipose tissue homeostasis and their alterations in obesity and colorectal cancer. Front. Immunol. 2018, 9, 2556. [Google Scholar]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar]
- Harmon, C.; Robinson, M.W.; Hand, F.; Almuaili, D.; Mentor, K.; Houlihan, D.D.; Hoti, E.; Lynch, L.; Geoghegan, J.; O’Farrelly, C. Lactate-mediated acidification of tumor microenvironment induces apoptosis of liver-resident NK cells in colorectal liver metastasis. Cancer Immunol. Res. 2019, 7, 335–346. [Google Scholar]
- Haeryfar, S.M.M.; Shaler, C.R.; Rudak, P.T. Mucosa-associated invariant T cells in malignancies: A faithful friend or formidable foe? Cancer Immunol. Immunother. 2018, 67, 1885–1896. [Google Scholar] [CrossRef]
- Carolan, E.; Tobin, L.M.; Mangan, B.A.; Corrigan, M.; Gaoatswe, G.; Byrne, G.; Geoghegan, J.; Cody, D.; O’Connell, J.; Winter, D.; et al. Altered distribution and increased IL-17 production by mucosal-associated invariant T cells in adult and childhood obesity. J. Immunol. 2015, 194, 5775–5780. [Google Scholar] [CrossRef]
- Ling, L.; Lin, Y.; Zheng, W.; Hong, S.; Tang, X.; Zhao, P.; Li, M.; Ni, J.; Li, C.; Wang, L.; et al. Circulating and tumor-infiltrating mucosal associated invariant T (MAIT) cells in colorectal cancer patients. Sci. Rep. 2016, 6, 20358. [Google Scholar] [CrossRef]
- Won, E.J.; Ju, J.K.; Cho, Y.N.; Jin, H.M.; Park, K.J.; Kim, T.J.; Kwon, Y.S.; Kee, H.J.; Kim, J.C.; Kee, S.J.; et al. Clinical relevance of circulating mucosal-associated invariant T cell levels and their anti-cancer activity in patients with mucosal-associated cancer. Oncotarget 2016, 7, 76274–76290. [Google Scholar] [CrossRef]
- Razi, S.; Noveiry, B.B.; Keshavarz-Fathi, M.; Rezaei, N. IL-17 and colorectal cancer: From carcinogenesis to treatment. Cytokine 2019, 116, 7–12. [Google Scholar]
- Glatz, J.F.C.; Luiken, J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Rychahou, P.G.; Le, A.T.; Scott, T.L.; Flight, R.M.; Kim, J.T.; Harris, J.; Liu, J.; Wang, C.; Morris, A.; et al. Preclinical evaluation of novel fatty acid synthase inhibitors in primary colorectal cancer cells and a patient-derived xenograft model of colorectal cancer. Oncotarget 2018, 9, 24787–24800. [Google Scholar] [CrossRef]
- Hoxha, M.; Zappacosta, B. A review on the role of fatty acids in colorectal cancer progression. Front. Pharmacol. 2022, 13, 1032806. [Google Scholar] [CrossRef]
- Mullen, G.E.; Yet, L. Progress in the development of fatty acid synthase inhibitors as anticancer targets. Bioorg. Med. Chem. Lett. 2015, 25, 4363–4369. [Google Scholar] [CrossRef]
- Buckley, D.; Duke, G.; Heuer, T.S.; O’Farrell, M.; Wagman, A.S.; McCulloch, W.; Kemble, G. Fatty acid synthase—Modern tumor cell biology insights into a classical oncology target. Pharmacol. Ther. 2017, 177, 23–31. [Google Scholar] [CrossRef]
- Wang, H.; Franco, F.; Tsui, Y.C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernandez-Garcia, J.; Tsai, C.H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. (Lond.) 2018, 38, 27. [Google Scholar]
- Wang, J.; Li, Y. CD36 tango in cancer: Signaling pathways and functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 49. [Google Scholar] [CrossRef]
- Drury, J.; Rychahou, P.G.; He, D.; Jafari, N.; Wang, C.; Lee, E.Y.; Weiss, H.L.; Evers, B.M.; Zaytseva, Y.Y. Inhibition of fatty acid synthase upregulates expression of CD36 to sustain proliferation of colorectal cancer cells. Front. Oncol. 2020, 10, 1185. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, M.; Yang, K.; Chi, T.; Liao, Z.; Wei, P. PPAR-α modulators as current and potential cancer treatments. Front. Oncol. 2021, 11, 599995. [Google Scholar] [CrossRef]
- Takano, S.; Kubota, T.; Nishibori, H.; Hasegawa, H.; Ishii, Y.; Nitori, N.; Ochiai, H.; Okabayashi, K.; Kitagawa, Y.; Watanabe, M.; et al. Pioglitazone, a ligand for peroxisome proliferator-activated receptor-gamma acts as an inhibitor of colon cancer liver metastasis. Anticancer Res. 2008, 28, 3593–3599. [Google Scholar]
- Zurlo, D.; Ziccardi, P.; Votino, C.; Colangelo, T.; Cerchia, C.; Piaz, F.D.; Dallavalle, S.; Moricca, S.; Novellino, E.; Lavecchia, A.; et al. The antiproliferative and proapoptotic effects of cladosporols A and B are related to their different binding mode as PPARγ ligands. Biochem. Pharmacol. 2016, 108, 22–35. [Google Scholar] [CrossRef]
- Silva, A.; Antunes, B.; Batista, A.; Pinto-Ribeiro, F.; Baltazar, F.; Afonso, J. In vivo anticancer activity of AZD3965: A systematic review. Molecules 2022, 27, 181. [Google Scholar] [CrossRef]
- Darban, S.A.; Nikoofal, S.; Amiri, N.; Kiamanesh, N.; Mehrabian, A.; Zendehbad, B.; Gholizadeh, Z.; Jaafari, M.R. Targeting the leptin receptor: To evaluate therapeutic efficacy and anti-tumor effects of Doxil, in vitro and in vivo in mice bearing C26 colon carcinoma tumor. Colloids Surf. B Biointerfaces 2018, 164, 107–115. [Google Scholar] [CrossRef]
- Munteanu, R.; Onaciu, A.; Moldovan, C.; Zimta, A.A.; Gulei, D.; Paradiso, A.; Lazar, V.; Berindan-Neagoe, I. Adipocyte-based cell therapy in oncology: The role of cancer-associated adipocytes and their reinterpretation as delivery platforms. Pharmaceutics 2020, 12, 402. [Google Scholar] [CrossRef]
- Sharifi-Azad, M.; Fathi, M.; Cho, W.; Barzegari, A.; Dadashi, H.; Dadashpour, M.; Jahanban-Esfahlan, R. Recent advances in targeted drug delivery systems for resistant colorectal cancer. Cancer Cell Int. 2022, 22, 196. [Google Scholar] [CrossRef]
- Wang, K.; Shen, R.; Meng, T.; Hu, F.; Yuan, H. Nano-drug delivery systems based on different targeting mechanisms in the targeted therapy of colorectal cancer. Molecules 2022, 27, 2981. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigoraș, A.; Amalinei, C. Multi-Faceted Role of Cancer-Associated Adipocytes in Colorectal Cancer. Biomedicines 2023, 11, 2401. https://doi.org/10.3390/biomedicines11092401
Grigoraș A, Amalinei C. Multi-Faceted Role of Cancer-Associated Adipocytes in Colorectal Cancer. Biomedicines. 2023; 11(9):2401. https://doi.org/10.3390/biomedicines11092401
Chicago/Turabian StyleGrigoraș, Adriana, and Cornelia Amalinei. 2023. "Multi-Faceted Role of Cancer-Associated Adipocytes in Colorectal Cancer" Biomedicines 11, no. 9: 2401. https://doi.org/10.3390/biomedicines11092401