
Establishing Molecular Subgroups of CD8+ T Cell-Associated Genes in the Ovarian Cancer Tumour Microenvironment and Predicting the Immunotherapy Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Pre-Processing
2.2. Weighted Gene Correlation Network Analysis (WGCNA)
2.3. Immunotyping
2.4. Specimen Collection
2.5. Quantitative Reverse Transcription PCR (qRT-PCR) and Immunohistochemistry Analysis
2.6. Statistical Analysis
3. Results
3.1. CD8+ T Cell-Associated Genes
3.2. Immunophenotyping
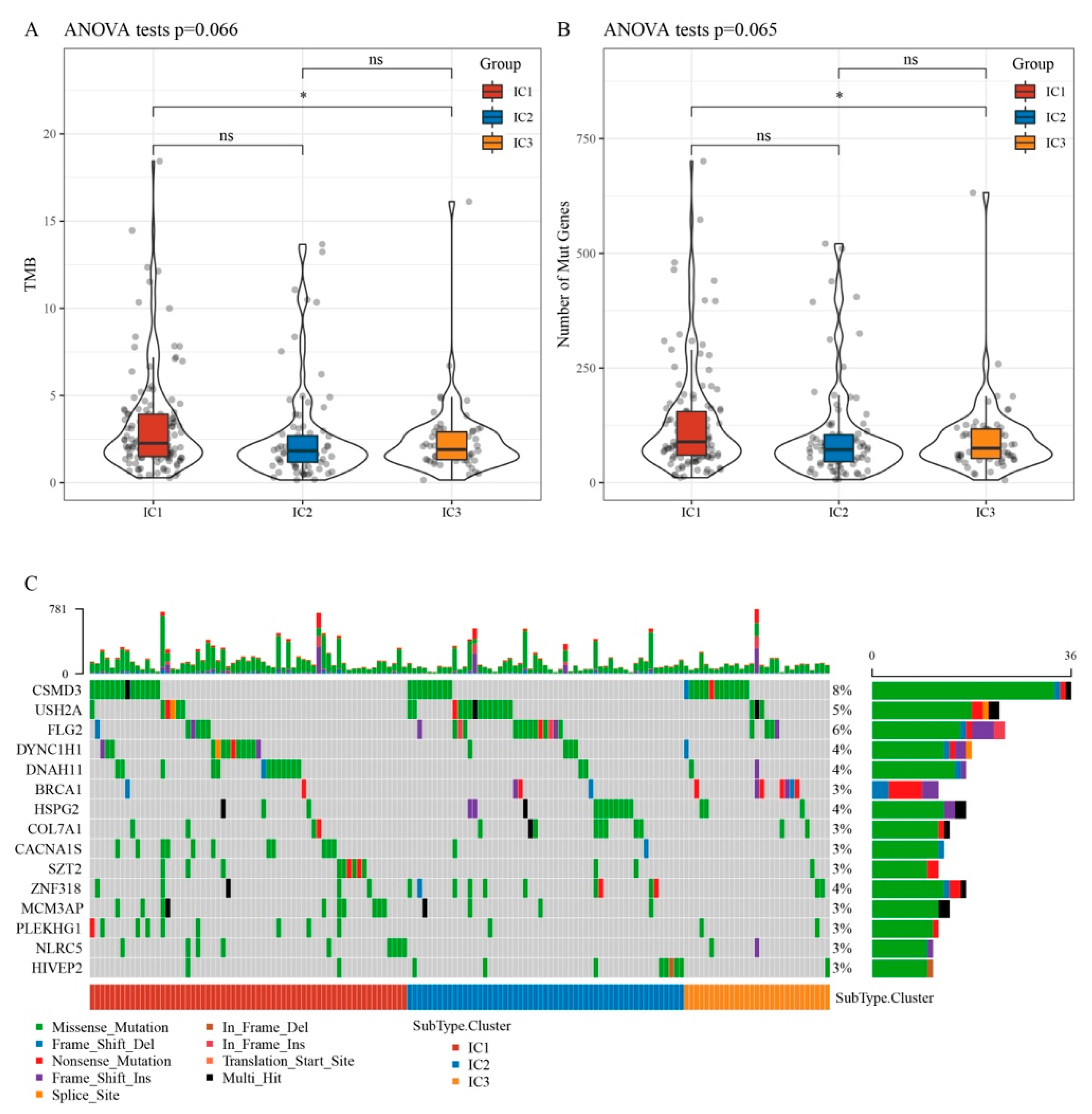
3.3. Relationship between Immunophenotyping and TMB and Gene Mutation
3.4. Immunophenotyping Chemokines and Immunological Checkpoint Genes
3.5. Immune Signatures and Pathways in Different ICs
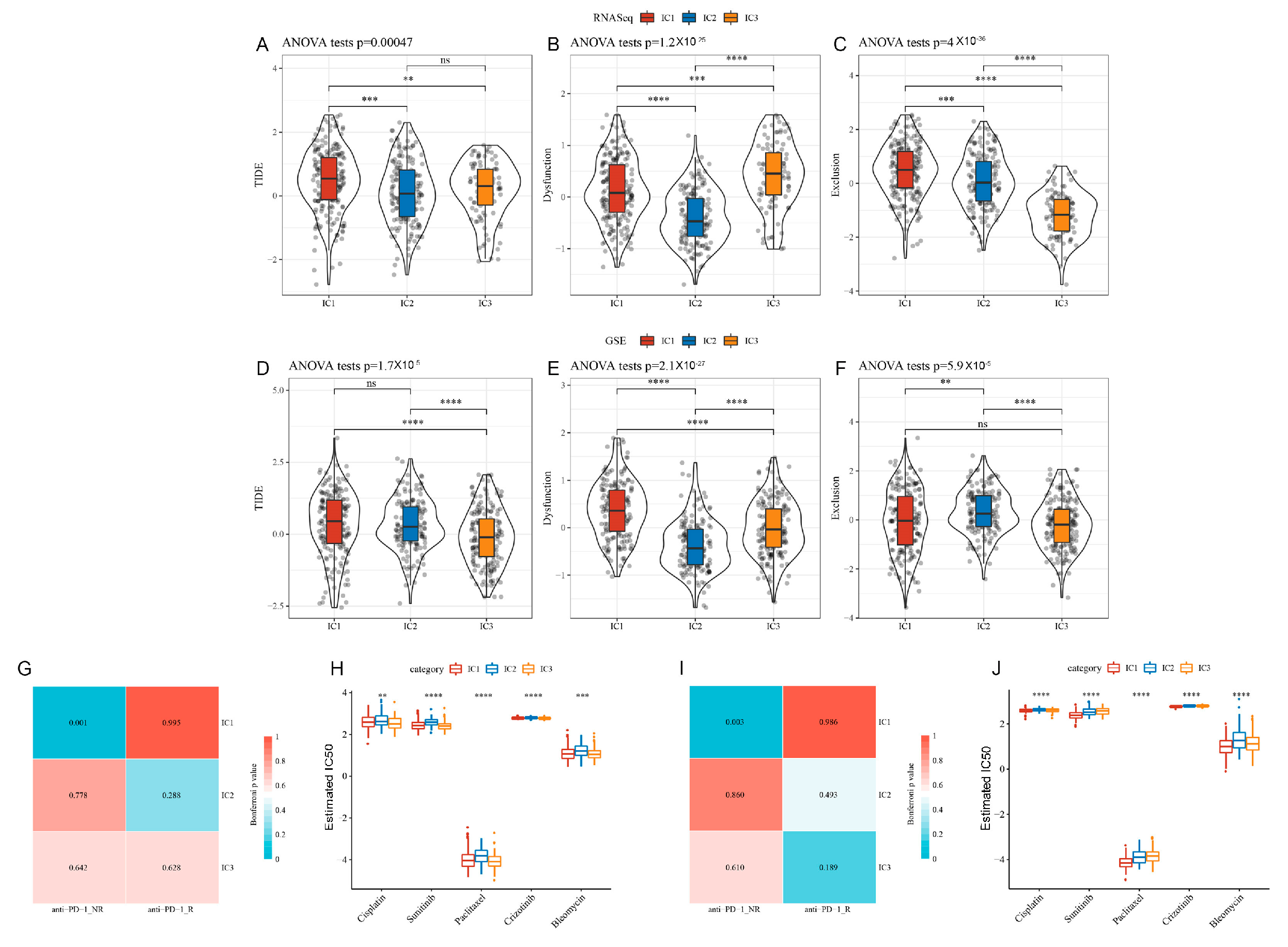
3.6. Tumour Immune Dysfunction and Exclusion (TIDE) among ICs
3.7. Differential Analysis of ICs in Immunotherapy/Chemotherapy
3.8. Prognostic Risk Model
3.9. Prognostic Risk Model Validation
3.10. Relationships between RiskScore, Clinical Characteristics, and Molecular Subtypes
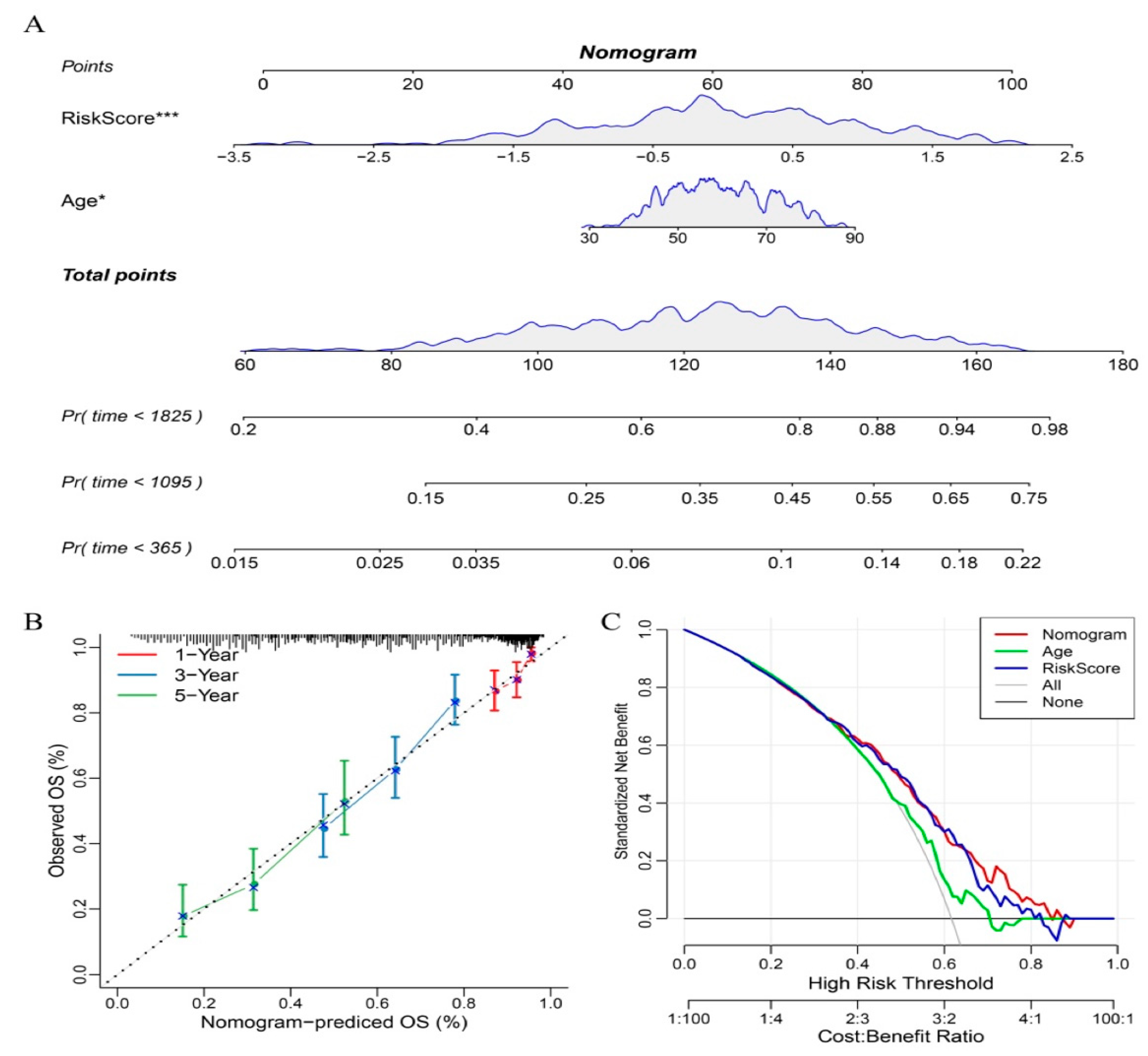
3.11. Nomograms and Forest Plots
3.12. Predicting Immunotherapy Efficacy with the Constructed Risk Model
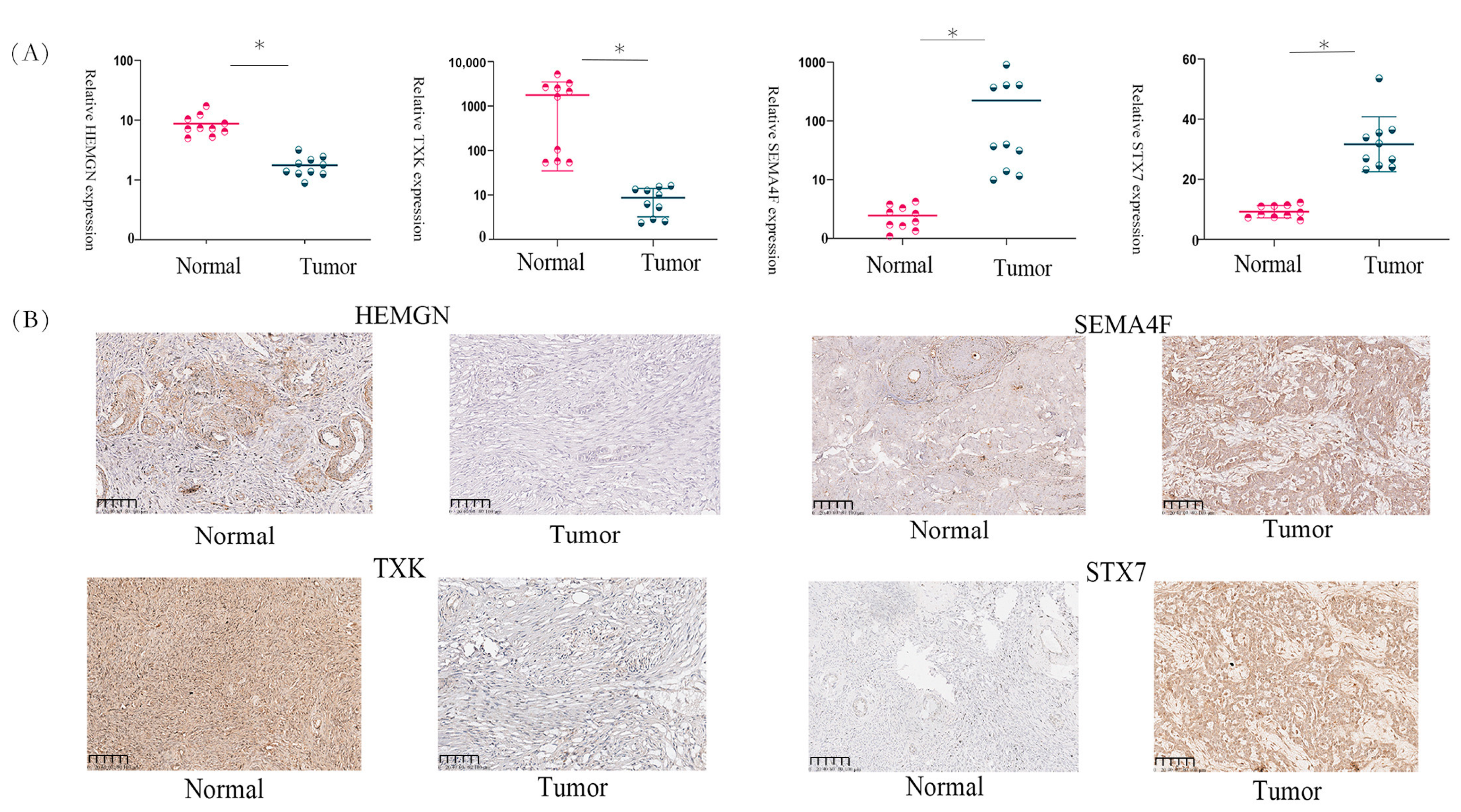
3.13. Gene Expression in OC Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Doherty, J.A.; Peres, L.C.; Wang, C.; Way, G.P.; Greene, C.S.; Schildkraut, J.M. Challenges and opportunities in studying the epidemiology of ovarian cancer subtypes. Curr. Epidemiol. Rep. 2017, 4, 211–220. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous recombination deficiency: Exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef]
- Mantia-Smaldone, G.M.; Corr, B.; Chu, C.S. Immunotherapy in ovarian cancer. Hum. Vaccin. Immunother. 2012, 8, 1179–1191. [Google Scholar] [CrossRef]
- Kandalaft, L.E.; Powell, D.J., Jr.; Singh, N.; Coukos, G. Immunotherapy for ovarian cancer: What’s next? J. Clin. Oncol. 2011, 29, 925–933. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordāo, M.J.C.; Joyce, J.A. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Dey, P.; Kimmelman, A.C.; DePinho, R.A. Metabolic codependencies in the tumor microenvironment. Cancer Discov. 2021, 11, 1067–1081. [Google Scholar] [CrossRef]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial cd8+ tumor-infiltrating lymphocytes and a high cd8+/regulatory t cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for pd-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- Park, J.; Lee, J.Y.; Kim, S. How to use immune checkpoint inhibitor in ovarian cancer? J. Gynecol. Oncol. 2019, 30, e105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral t cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating ny-eso-1-specific cd8+ t cells are negatively regulated by lag-3 and pd-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef]
- Danilova, L.; Ho, W.J.; Zhu, Q.; Vithayathil, T.; De Jesus-Acosta, A.; Azad, N.S.; Laheru, D.A.; Fertig, E.J.; Anders, R.; Jaffee, E.M.; et al. Programmed cell death ligand-1 (pd-l1) and cd8 expression profiling identify an immunologic subtype of pancreatic ductal adenocarcinomas with favorable survival. Cancer Immunol. Res. 2019, 7, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Xu, J.; Wang, M.; Xu, G.; Zhang, N.; Wang, G.; Zhao, Y. Lncrna hcp5 promotes follicular thyroid carcinoma progression via mirnas sponge. Cell Death Dis. 2018, 9, 372. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon receptor signaling pathways regulating pd-l1 and pd-l2 expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Myers, A.C.; Chen, L.; Pardoll, D.M.; Truong-Tran, Q.A.; Lane, A.P.; McDyer, J.F.; Fortuno, L.; Schleimer, R.P. Constitutive and inducible expression of b7 family of ligands by human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2005, 33, 280–289. [Google Scholar] [CrossRef]
- Takikawa, O.; Tagawa, Y.; Iwakura, Y.; Yoshida, R.; Truscott, R.J. Interferon-gamma-dependent/independent expression of indoleamine 2,3-dioxygenase. Studies with interferon-gamma-knockout mice. Adv. Exp. Med. Biol. 1999, 467, 553–557. [Google Scholar]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef]
- Masiero, M.; Simões, F.C.; Han, H.D.; Snell, C.; Peterkin, T.; Bridges, E.; Mangala, L.S.; Wu, S.Y.; Pradeep, S.; Li, D.; et al. A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor eltd1 as a key regulator of angiogenesis. Cancer Cell 2013, 24, 229–241. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef] [PubMed]
- Musella, A.; Vertechy, L.; Romito, A.; Marchetti, C.; Giannini, A.; Sciuga, V.; Bracchi, C.; Tomao, F.; Di Donato, V.; De Felice, F.; et al. Bevacizumab in ovarian cancer: State of the art and unanswered questions. Chemotherapy 2017, 62, 111–120. [Google Scholar] [CrossRef]
- Liu, J.F.; Herold, C.; Gray, K.P.; Penson, R.T.; Horowitz, N.; Konstantinopoulos, P.A.; Castro, C.M.; Hill, S.J.; Curtis, J.; Luo, W.; et al. Assessment of combined nivolumab and bevacizumab in relapsed ovarian cancer: A phase 2 clinical trial. JAMA Oncol. 2019, 5, 1731–1738. [Google Scholar] [CrossRef]
- Fialová, A.; Partlová, S.; Sojka, L.; Hromádková, H.; Brtnický, T.; Fučíková, J.; Kocián, P.; Rob, L.; Bartůňková, J.; Spíšek, R. Dynamics of t-cell infiltration during the course of ovarian cancer: The gradual shift from a th17 effector cell response to a predominant infiltration by regulatory t-cells. Int. J. Cancer 2013, 132, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Gasparri, M.L.; Attar, R.; Palaia, I.; Perniola, G.; Marchetti, C.; Di Donato, V.; Farooqi, A.A.; Papadia, A.; Panici, P.B. Tumor infiltrating lymphocytes in ovarian cancer. Asian Pac. J. Cancer Prev. 2015, 16, 3635–3638. [Google Scholar] [CrossRef]
- Mihara, S.; Suzuki, N. Role of txk, a member of the tec family of tyrosine kinases, in immune-inflammatory diseases. Int. Rev. Immunol. 2007, 26, 333–348. [Google Scholar] [CrossRef]
- Wang, H.; Frelin, L.; Pevsner, J. Human syntaxin 7: A pep12p/vps6p homologue implicated in vesicle trafficking to lysosomes. Gene 1997, 199, 39–48. [Google Scholar] [CrossRef]
- Zhang, D.D.; Zhang, J.G.; Wang, Y.Z.; Liu, Y.; Liu, G.L.; Li, X.Y. Per-arnt-sim kinase (pask): An emerging regulator of mammalian glucose and lipid metabolism. Nutrients 2015, 7, 7437–7450. [Google Scholar] [CrossRef] [PubMed]
- Leng, K.; Xu, Y.; Kang, P.; Qin, W.; Cai, H.; Wang, H.; Ji, D.; Jiang, X.; Li, J.; Li, Z.; et al. Akirin2 is modulated by mir-490-3p and facilitates angiogenesis in cholangiocarcinoma through the il-6/stat3/vegfa signaling pathway. Cell Death Dis. 2019, 10, 262. [Google Scholar] [CrossRef] [PubMed]
- Fekete, J.T.; Ősz, Á.; Pete, I.; Nagy, G.R.; Vereczkey, I.; Győrffy, B. Predictive biomarkers of platinum and taxane resistance using the transcriptomic data of 1816 ovarian cancer patients. Gynecol. Oncol. 2020, 156, 654–661. [Google Scholar] [CrossRef]
- Ding, Y.; He, D.; Florentin, D.; Frolov, A.; Hilsenbeck, S.; Ittmann, M.; Kadmon, D.; Miles, B.; Rowley, D.; Ayala, G. Semaphorin 4f as a critical regulator of neuroepithelial interactions and a biomarker of aggressive prostate cancer. Clin. Cancer Res. 2013, 19, 6101–6111. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, O.; Sugier, P.E.; Guibon, J.; Boland-Augé, A.; Lonjou, C.; Bacq-Daian, D.; Olaso, R.; Rubino, C.; Souchard, V.; Rachedi, F.; et al. Gene network and biological pathways associated with susceptibility to differentiated thyroid carcinoma. Sci. Rep. 2021, 11, 8932. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lv, S.X. The effect of jak2 knockout on inhibition of liver tumor growth by inducing apoptosis, autophagy and anti-proliferation via stats and pi3k/akt signaling pathways. Biomed. Pharmacother. 2016, 84, 1202–1212. [Google Scholar] [CrossRef]
- Zahra, A.; Hall, M.; Chatterjee, J.; Sisu, C.; Karteris, E. In silico study to predict the structural and functional consequences of snps on biomarkers of ovarian cancer (oc) and bpa exposure-associated oc. Int. J. Mol. Sci. 2022, 23, 1725. [Google Scholar] [CrossRef]
- Hart, K.M.; Usherwood, E.J.; Berwin, B.L. Cx3cr1 delineates temporally and functionally distinct subsets of myeloid-derived suppressor cells in a mouse model of ovarian cancer. Immunol. Cell Biol. 2014, 92, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, S.; Futamura, M.; Kamino, H.; Nakamura, Y.; Kitamura, N.; Miyamoto, Y.; Miyamoto, T.; Shinogi, D.; Goda, O.; Arakawa, H. Identification of neep21, encoding neuron-enriched endosomal protein of 21 kda, as a transcriptional target of tumor suppressor p53. Int. J. Oncol. 2010, 37, 1133–1141. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Liang, L.; Li, J.; Zeng, J.; Yao, H.; Wu, L. Establishing Molecular Subgroups of CD8+ T Cell-Associated Genes in the Ovarian Cancer Tumour Microenvironment and Predicting the Immunotherapy Response. Biomedicines 2023, 11, 2399. https://doi.org/10.3390/biomedicines11092399
Zhu Y, Liang L, Li J, Zeng J, Yao H, Wu L. Establishing Molecular Subgroups of CD8+ T Cell-Associated Genes in the Ovarian Cancer Tumour Microenvironment and Predicting the Immunotherapy Response. Biomedicines. 2023; 11(9):2399. https://doi.org/10.3390/biomedicines11092399
Chicago/Turabian StyleZhu, Yunshu, Leilei Liang, Jian Li, Jia Zeng, Hongwen Yao, and Lingying Wu. 2023. "Establishing Molecular Subgroups of CD8+ T Cell-Associated Genes in the Ovarian Cancer Tumour Microenvironment and Predicting the Immunotherapy Response" Biomedicines 11, no. 9: 2399. https://doi.org/10.3390/biomedicines11092399