

Single-Cell RNA Sequencing and Microarray Analysis Reveal the Role of Lipid-Metabolism-Related Genes and Cellular Immune Infiltration in Pre-Eclampsia and Identify Novel Biomarkers for Pre-Eclampsia

Abstract
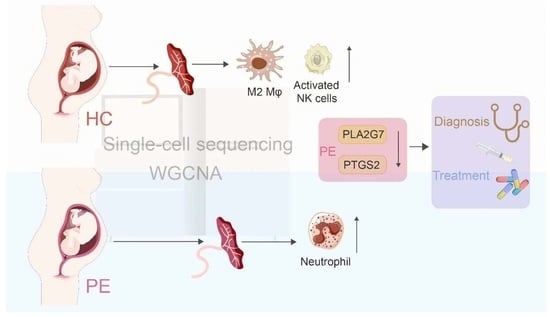
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Patients and Sample Collection
2.2. Data Recruitment and Processing
2.3. Identification of DEGs and Construction of Weighted Gene Co-Expression Networks
2.4. KEGG and GO Enrichment Analysis
2.5. ROC Curve Analysis and Expression Analysis
2.6. Correlation Analysis between Core Genes and Infiltrated Immune Cells
2.7. Prediction of Networks Mutually Regulated by miRNAs and TFs
2.8. Laboratory Measurements
- PLA2G7: TCAATGACAACTCCTGCAAACTG (sense primer);
- PLA2G7: TCCTCCTCTTGTTTCAGGGTTCT (antisense primer);
- PTGS2: GGGTTGCTGGTGGTAGGAATG (sense primer);
- PTGS2: CATAAAGCGTTTGCGGTACTCAT (antisense primer);
- GAPDH: GGAAGCTTGTCATCAATGGAAATC (sense primer);
- GAPDH: TGATGACCCTTTTGGCTCCC (antisense primer).
2.9. Statistics
3. Results
3.1. scRNA-Seq Data Preprocessing and PCA
3.2. Cell-Type Annotation and Single-Cell Differentiation Trajectory Analysis
3.3. Immune Characterization Analysis
3.4. Identification of DEGs and Construction of Co-Expression Networks
3.5. Identification of LMRG-PE and Functional Enrichment Analysis
3.6. Expression Analysis of Core Genes at the Single-Cell Level and Bulk RNA-seq Level
3.7. Associations of Core Genes with Immune Cells
3.8. GSEA and GSVA of Two Hub Genes
3.9. Drug–Gene Networks and Prediction of Key miRNAs and TFs
3.10. Validation of the mRNA Expression Levels of Hub Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ACOG. Gestational Hypertension and Preeclampsia: ACOG Practice Bulletin Number 202. Obstet. Gynecol. 2019, 133, 1. [Google Scholar] [CrossRef]
- Ives, C.W.; Sinkey, R.; Rajapreyar, I.; Tita, A.T.N.; Oparil, S. Preeclampsia-Pathophysiology and Clinical Presentations: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 1690–1702. [Google Scholar] [CrossRef] [PubMed]
- Gammill, H.S.; Chettier, R.; Brewer, A.; Roberts, J.M.; Shree, R.; Tsigas, E.; Ward, K. Cardiomyopathy and Preeclampsia. Circulation 2018, 138, 2359–2366. [Google Scholar] [CrossRef]
- Laissue, P.; Vaiman, D. Exploring the Molecular Aetiology of Preeclampsia by Massive Parallel Sequencing of DNA. Curr. Hypertens. Rep. 2020, 22, 31. [Google Scholar] [CrossRef]
- Rana, S.; Burke, S.D.; Karumanchi, S.A. Imbalances in circulating angiogenic factors in the pathophysiology of preeclampsia and related disorders. Am. J. Obstet. Gynecol. 2022, 226, S1019–S1034. [Google Scholar] [CrossRef]
- Khaire, A.A.; Thakar, S.R.; Wagh, G.N.; Joshi, S.R. Placental lipid metabolism in preeclampsia. J. Hypertens. 2021, 39, 127–134. [Google Scholar] [CrossRef]
- Canfield, J.; Arlier, S.; Mong, E.F.; Lockhart, J.; VanWye, J.; Guzeloglu-Kayisli, O.; Schatz, F.; Magness, R.R.; Lockwood, C.J.; Tsibris, J.C.M.; et al. Decreased LIN28B in preeclampsia impairs human trophoblast differentiation and migration. FASEB J. 2019, 33, 2759–2769. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, L.; van der Meiden-van Roest, A.J.; Savelkoul, C.; Vogelvang, T.E.; Lely, A.T.; Franx, A.; van Rijn, B.B. Recurrence of pre-eclampsia and the risk of future hypertension and cardiovascular disease: A systematic review and meta-analysis. BJOG Int. J. Obstet. Gynaecol. 2018, 125, 1642–1654. [Google Scholar] [CrossRef]
- Sanapo, L.; Bublitz, M.H.; Bourjeily, G. Sleep Disordered Breathing, a Novel, Modifiable Risk Factor for Hypertensive Disorders of Pregnancy. Curr. Hypertens. Rep. 2020, 22, 28. [Google Scholar] [CrossRef] [PubMed]
- Theilen, L.H.; Meeks, H.; Fraser, A.; Esplin, M.S.; Smith, K.R.; Varner, M.W. Long-term mortality risk and life expectancy following recurrent hypertensive disease of pregnancy. Am. J. Obstet. Gynecol. 2018, 219, 107.e1–107.e6. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, E.; Rolnik, D.L.; Zhou, W.; Estrada-Gutierrez, G.; Koga, K.; Francisco, R.P.V.; Whitehead, C.; Hyett, J.; da Silva Costa, F.; Nicolaides, K.; et al. Pre-eclampsia. Nat. Rev. Dis. Primers 2023, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Melzer, K.; Schutz, Y.; Boulvain, M.; Kayser, B. Physical activity and pregnancy: Cardiovascular adaptations, recommendations and pregnancy outcomes. Sports Med. 2010, 40, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Duhig, K.E.; Myers, J.; Seed, P.T.; Sparkes, J.; Lowe, J.; Hunter, R.M.; Shennan, A.H.; Chappell, L.C. Placental growth factor testing to assess women with suspected pre-eclampsia: A multicentre, pragmatic, stepped-wedge cluster-randomised controlled trial. Lancet 2019, 393, 1807–1818. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-eclampsia: Pathophysiology and clinical implications. Br. Med. J. 2019, 366, l2381. [Google Scholar] [CrossRef] [PubMed]
- ACOG. ACOG Committee Opinion No. 743-Low-Dose Aspirin Use During Pregnancy. Obstet. Gynecol. 2018, 132, e44–e52. [Google Scholar] [CrossRef] [PubMed]
- Duley, L.; Henderson-Smart, D.J.; Meher, S.; King, J.F. Antiplatelet agents for preventing pre-eclampsia and its complications. Cochrane Database Syst. Rev. 2007, 10, CD004659. [Google Scholar] [CrossRef] [PubMed]
- Rolnik, D.L.; Wright, D.; Poon, L.C.; O’Gorman, N.; Syngelaki, A.; de Paco Matallana, C.; Akolekar, R.; Cicero, S.; Janga, D.; Singh, M.; et al. Aspirin versus Placebo in Pregnancies at High Risk for Preterm Preeclampsia. N. Engl. J. Med. 2017, 377, 613–622. [Google Scholar] [CrossRef]
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennström, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Predictive Value of the sFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N. Engl. J. Med. 2016, 374, 13–22. [Google Scholar] [CrossRef]
- Richter, A.E.; Scherjon, S.A.; Dikkers, R.; Bos, A.F.; Kooi, E.M.W. Antenatal Magnesium Sulfate and Preeclampsia Differentially Affect Neonatal Cerebral Oxygenation. Neonatology 2020, 117, 331–340. [Google Scholar] [CrossRef]
- ACOG. Gestational Hypertension and Preeclampsia: ACOG Practice Bulletin, Number 222. Obstet. Gynecol. 2020, 135, e237–e260. [Google Scholar] [CrossRef]
- Luo, S.; Cao, N.; Tang, Y.; Gu, W. Identification of key microRNAs and genes in preeclampsia by bioinformatics analysis. PLoS ONE 2017, 12, e0178549. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Li, C.; Liu, C.X. Immune cell infiltration landscape and immune marker molecular typing in preeclampsia. Bioengineered 2021, 12, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Moon, J.Y.; Lim, B.Y.; Kim, S.M.; Park, C.W.; Kim, B.J.; Jun, J.K.; Norwitz, E.R.; Choi, M.H.; Park, J.S. Increased biosynthesis and accumulation of cholesterol in maternal plasma, but not amniotic fluid in pre-eclampsia. Sci. Rep. 2019, 9, 1550. [Google Scholar] [CrossRef] [PubMed]
- Girardi, G. Complement activation, a threat to pregnancy. Semin. Immunopathol. 2018, 40, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zheng, Q.; Jin, L. Dynamic Function and Composition Changes of Immune Cells During Normal and Pathological Pregnancy at the Maternal-Fetal Interface. Front. Immunol. 2019, 10, 2317. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Yang, Z.; Han, Y.; Yu, H. Correlation of long-chain fatty acid oxidation with oxidative stress and inflammation in pre-eclampsia-like mouse models. Placenta 2015, 36, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Jain, A.; Baumann, M.; Korner, M.; Surbek, D.; Butikofer, P.; Albrecht, C. Increased placental phospholipid levels in pre-eclamptic pregnancies. Int. J. Mol. Sci. 2013, 14, 3487–3499. [Google Scholar] [CrossRef]
- Nelson, D.B.; Ziadie, M.S.; McIntire, D.D.; Rogers, B.B.; Leveno, K.J. Placental pathology suggesting that preeclampsia is more than one disease. Am. J. Obstet. Gynecol. 2014, 210, 66.e1–66.e7. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, Y.; Zhang, K.; Huang, Y.; Yan, Y.; Wang, F.; Wu, J.; Wang, X.; Xu, Z.; Chen, Y.; et al. Cadmium-induced immune abnormality is a key pathogenic event in human and rat models of preeclampsia. Environ. Pollut. 2016, 218, 770–782. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, Y.; Zhang, K.; Yan, Y.; Wu, J.; Wang, F.; Zhao, Y.; Xu, H.; Jiang, W.; Yu, D.; et al. Progesterone attenuates hypertension and autoantibody levels to the angiotensin II type 1 receptor in response to elevated cadmium during pregnancy. Placenta 2018, 62, 16–24. [Google Scholar] [CrossRef]
- Liu, H.; Cheng, F.; Xu, Q.; Huang, W.; Wang, S.; Sun, R.; Ye, D.; Zhang, D. Lipoxin A4 suppresses angiotensin II type 1 receptor autoantibody in preeclampsia via modulating caspase-1. Cell Death Dis. 2020, 11, 78. [Google Scholar] [CrossRef]
- Stadler, J.T.; Scharnagl, H.; Wadsack, C.; Marsche, G. Preeclampsia Affects Lipid Metabolism and HDL Function in Mothers and Their Offspring. Antioxidants 2023, 12, 795. [Google Scholar] [CrossRef]
- Bu, C.; Wang, Z.; Ren, Y.; Chen, D.; Jiang, S.W. Syncytin-1 nonfusogenic activities modulate inflammation and contribute to preeclampsia pathogenesis. Cell. Mol. Life Sci. 2022, 79, 290. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.; Lei, D.; Chen, T.; Liu, Y.; Fan, C. The Enzyme 15-Hydroxyprostaglandin Dehydrogenase Inhibits a Shift to the Mesenchymal Pattern of Trophoblasts and Decidual Stromal Cells Accompanied by Prostaglandin Transporter in Preeclampsia. Int. J. Mol. Sci. 2023, 24, 5111. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wen, T.; Li, Q.; Chen, Z.; Peng, X.; Wei, C.; Wei, Y.; Peng, J.; Zhang, W. Single-Cell Sequencing Revealed Pivotal Genes Related to Prognosis of Myocardial Infarction Patients. Comput. Math. Methods Med. 2022, 2022, 6534126. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shui, K.; Zhang, Y.; Lv, Y.; Deng, W.; Ullah, S.; Zhang, L.; Xue, Y. CGDB: A database of circadian genes in eukaryotes. Nucleic Acids Res. 2017, 45, D397–D403. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef]
- Fan, Y.; Xia, J. miRNet-Functional Analysis and Visual Exploration of miRNA-Target Interactions in a Network Context. Methods Mol. Biol. 2018, 1819, 215–233. [Google Scholar] [CrossRef]
- Barrett, H.L.; Kubala, M.H.; Scholz Romero, K.; Denny, K.J.; Woodruff, T.M.; McIntyre, H.D.; Callaway, L.K.; Dekker Nitert, M. Placental lipase expression in pregnancies complicated by preeclampsia: A case-control study. Reprod. Biol. Endocrinol. 2015, 13, 100. [Google Scholar] [CrossRef] [PubMed]
- Hentschke, M.R.; Poli-de-Figueiredo, C.E.; da Costa, B.E.; Kurlak, L.O.; Williams, P.J.; Mistry, H.D. Is the atherosclerotic phenotype of preeclamptic placentas due to altered lipoprotein concentrations and placental lipoprotein receptors? Role of a small-for-gestational-age phenotype. J. Lipid Res. 2013, 54, 2658–2664. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Chen, M.; Bai, H.; He, G.L.; Liu, Q.Q.; Guan, L.B.; Liu, X.H.; Fan, P. Association of the G994T and R92H genotypes of platelet-activating factor acetylhydrolase with risk of preeclampsia in Chinese women. Pregnancy Hypertens. 2020, 20, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jia, X.; Huang, Y.; Wang, J.; Lu, C.; Yuan, X.; Xu, J.; Zhu, H. Vitamin D stimulates miR-26b-5p to inhibit placental COX-2 expression in preeclampsia. Sci. Rep. 2021, 11, 11168. [Google Scholar] [CrossRef] [PubMed]
- Voutetakis, A.; Pervanidou, P.; Kanaka-Gantenbein, C. Aspirin for the Prevention of Preeclampsia and Potential Consequences for Fetal Brain Development. JAMA Pediatr. 2019, 173, 619–620. [Google Scholar] [CrossRef]
- Szczuko, M.; Kikut, J.; Komorniak, N.; Bilicki, J.; Celewicz, Z.; Ziętek, M. The Role of Arachidonic and Linoleic Acid Derivatives in Pathological Pregnancies and the Human Reproduction Process. Int. J. Mol. Sci. 2020, 21, 9628. [Google Scholar] [CrossRef] [PubMed]
- Steegers, E.A.P.; von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Pre-eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef]
- Yi, Y.; Cheng, J.C.; Klausen, C.; Leung, P.C.K. TGF-beta1 inhibits human trophoblast cell invasion by upregulating cyclooxygenase-2. Placenta 2018, 68, 44–51. [Google Scholar] [CrossRef]
- Deng, M.; Yin, Y.; Zhang, Q.; Zhou, X.; Hou, G. Identification of Inflammation-Related Biomarker Lp-PLA2 for Patients with COPD by Comprehensive Analysis. Front. Immunol. 2021, 12, 670971. [Google Scholar] [CrossRef]
- Tu, B.; Fang, R.; Zhu, Z.; Chen, G.; Peng, C.; Ning, R. Comprehensive analysis of arachidonic acid metabolism-related genes in diagnosis and synovial immune in osteoarthritis: Based on bulk and single-cell RNA sequencing data. Inflamm. Res. 2023, 72, 955–970. [Google Scholar] [CrossRef]
- Feng, X.; Meng, X.; Guo, S.; Li, K.; Wang, L.; Ai, J. Identification of key genes and immune cell infiltration in recurrent implantation failure: A study based on integrated analysis of multiple microarray studies. Am. J. Reprod. Immunol. 2022, 88, e13607. [Google Scholar] [CrossRef]
- Han, Y.; Lee, S.; Lee, J.H.; Yoo, H.J. Potential Mechanisms of Improved Activity of Natural Killer Cells Induced by the Consumption of F-MRP for 8 weeks. Mol. Nutr. Food Res. 2021, 65, e2100337. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Xu, B.; Fan, C. Single-Cell RNA Sequencing and Microarray Analysis Reveal the Role of Lipid-Metabolism-Related Genes and Cellular Immune Infiltration in Pre-Eclampsia and Identify Novel Biomarkers for Pre-Eclampsia. Biomedicines 2023, 11, 2328. https://doi.org/10.3390/biomedicines11082328
Liu Y, Xu B, Fan C. Single-Cell RNA Sequencing and Microarray Analysis Reveal the Role of Lipid-Metabolism-Related Genes and Cellular Immune Infiltration in Pre-Eclampsia and Identify Novel Biomarkers for Pre-Eclampsia. Biomedicines. 2023; 11(8):2328. https://doi.org/10.3390/biomedicines11082328
Chicago/Turabian StyleLiu, Yujie, Borui Xu, and Cuifang Fan. 2023. "Single-Cell RNA Sequencing and Microarray Analysis Reveal the Role of Lipid-Metabolism-Related Genes and Cellular Immune Infiltration in Pre-Eclampsia and Identify Novel Biomarkers for Pre-Eclampsia" Biomedicines 11, no. 8: 2328. https://doi.org/10.3390/biomedicines11082328