
Effect of Kinins on the Hepatic Oxidative Stress in Mice Treated with a Methionine-Choline Deficient Diet
 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. In Situ Liver Perfusion (Experimental Design)
2.3. Biochemical Analysis
2.3.1. Glucose
2.3.2. Cholesterol and Triglycerides
2.3.3. Aminotransferases
2.4. Histological Analysis
2.5. Cell Death Pathway Analysis by Cathepsin B and Caspase-3 Activity
2.6. Oxidative Stress Analysis
2.6.1. Catalase Activity
2.6.2. Glutathione S-Transferase Activity
2.6.3. Superoxide Dismutase Activity
2.6.4. Levels of Malondialdehyde
2.6.5. Levels of Carbonyl Protein
2.7. Statistical Analysis
3. Results
3.1. Experimental Model
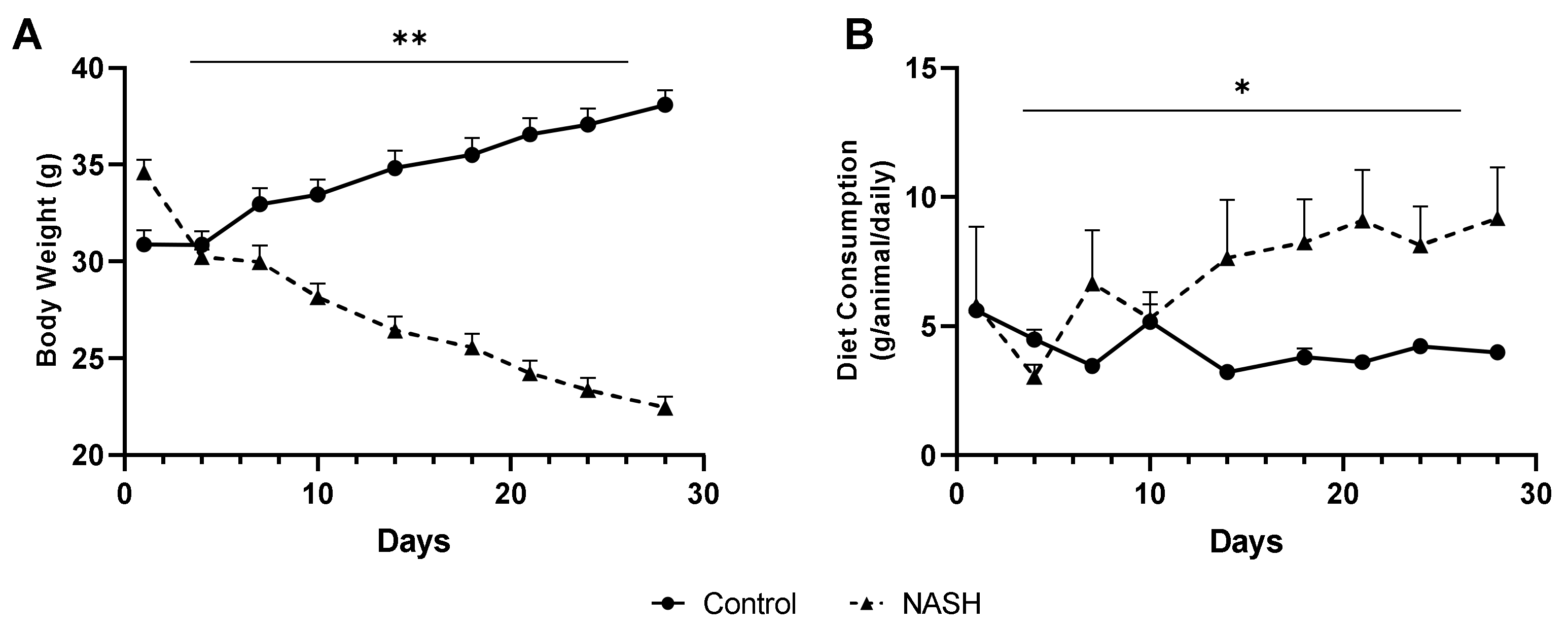
3.1.1. Quantification of Body Weight and Diet Consumption
3.1.2. Descriptive Parameters
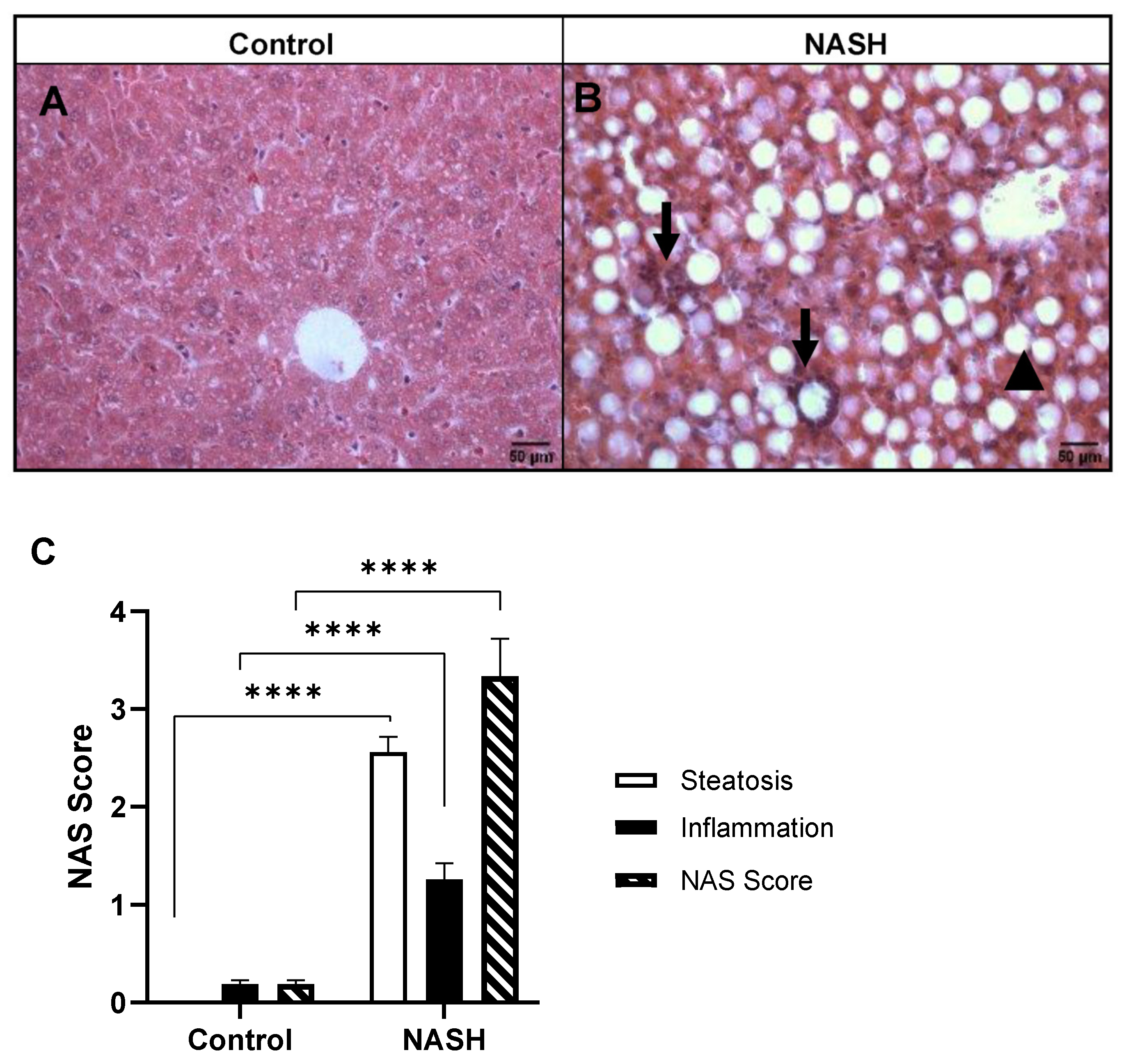
3.2. Histopathological Analysis
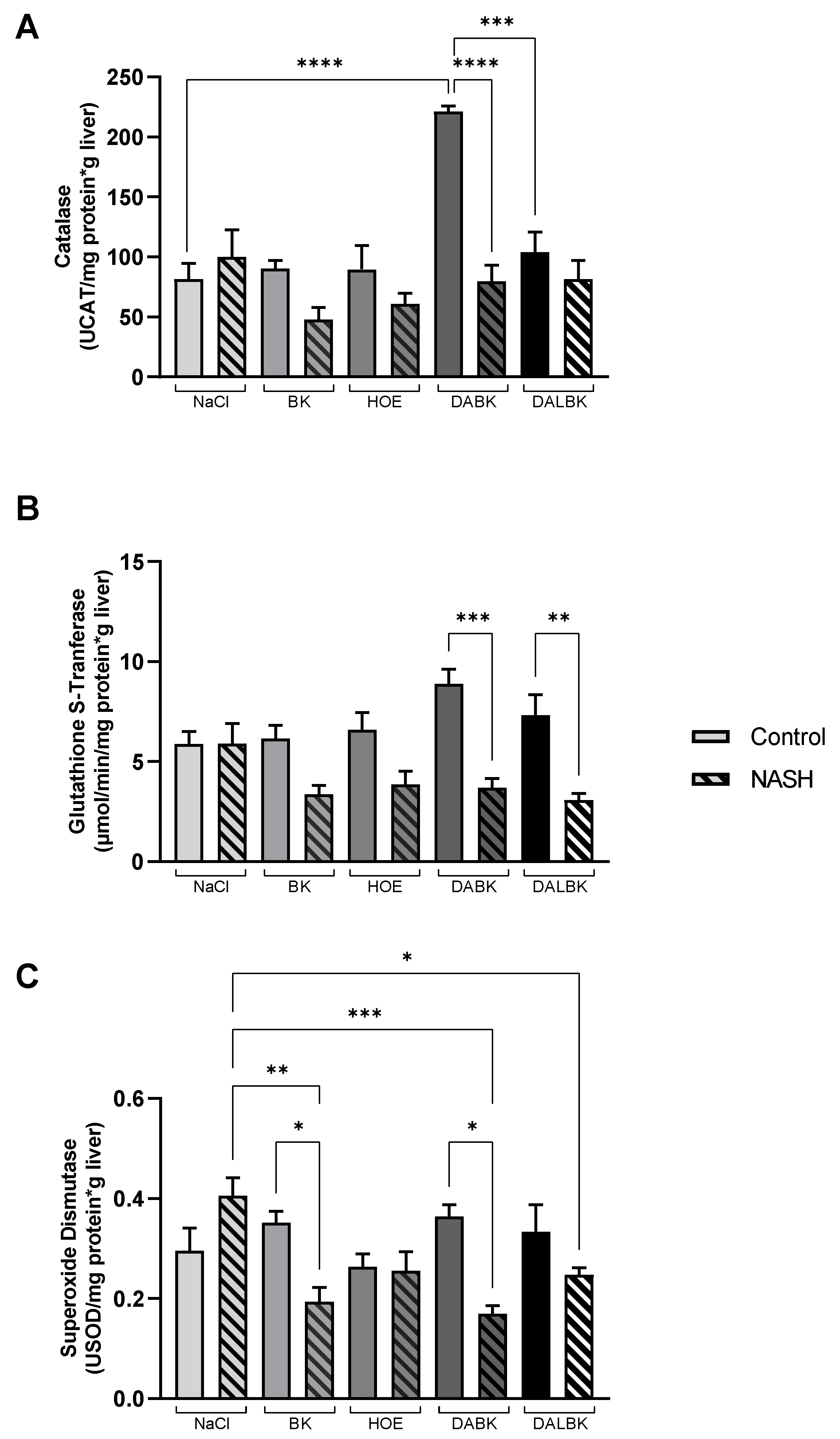
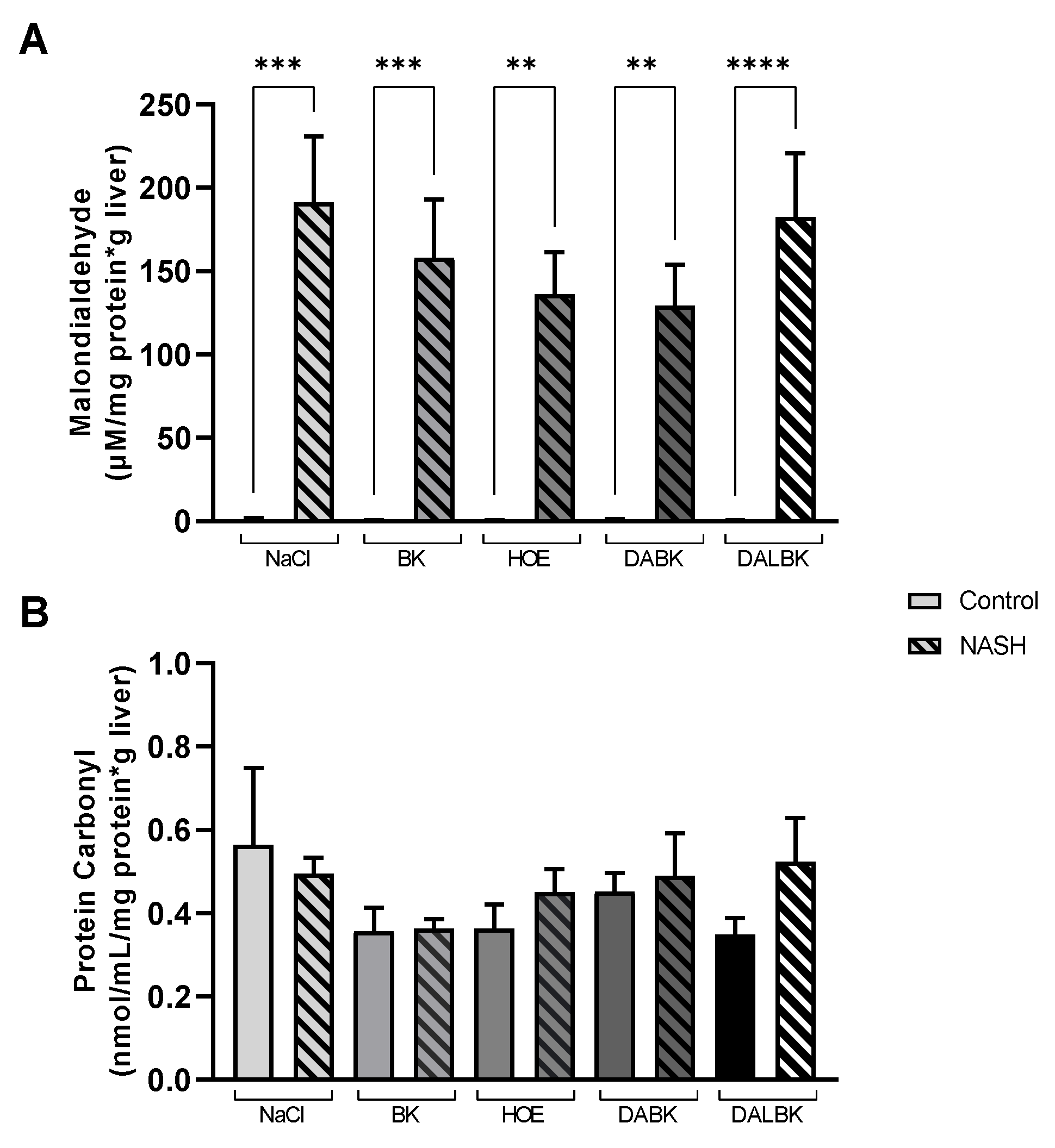
3.3. Oxidative Stress Analysis
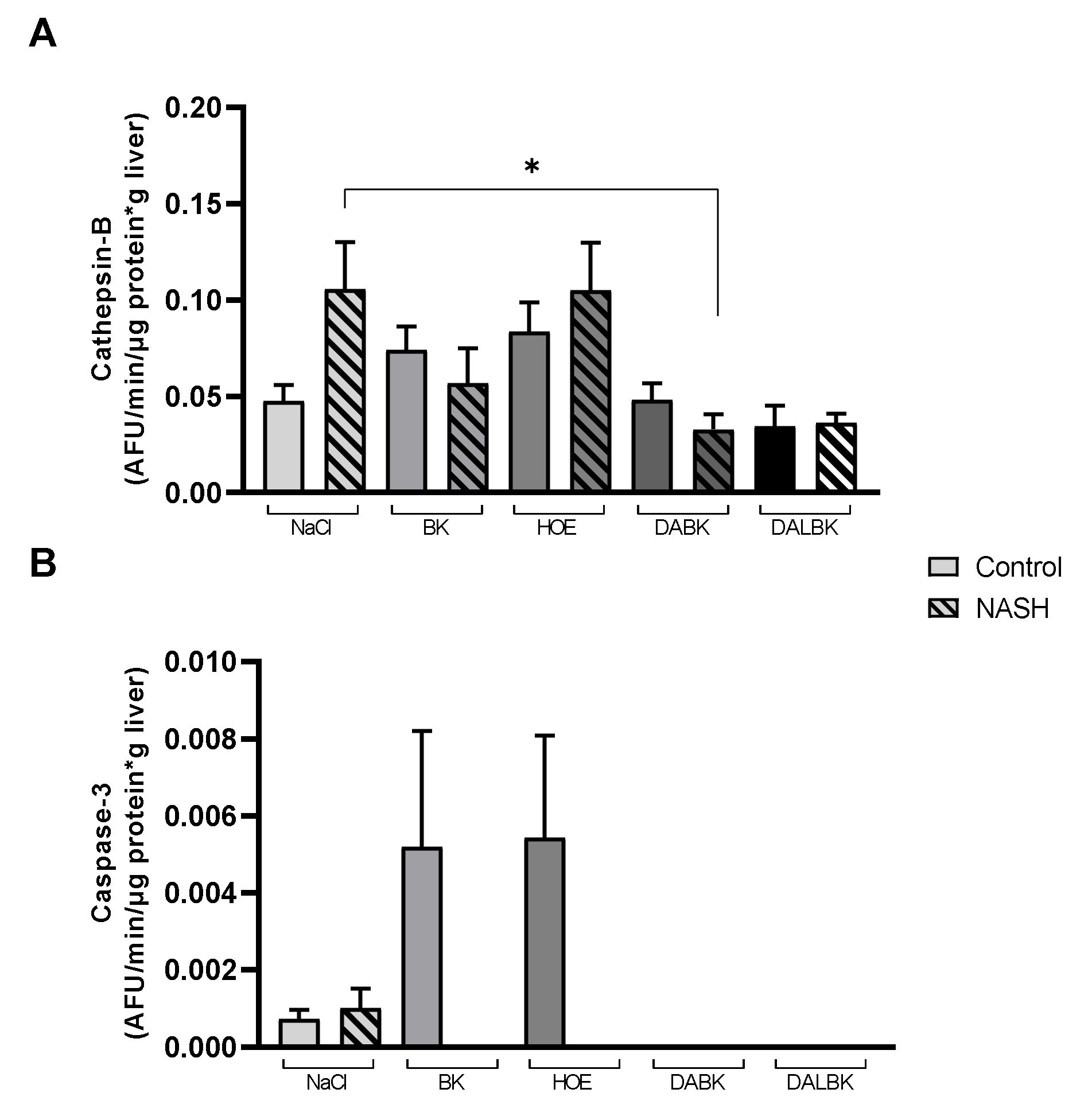
3.4. Cell Death Pathway Enzymes
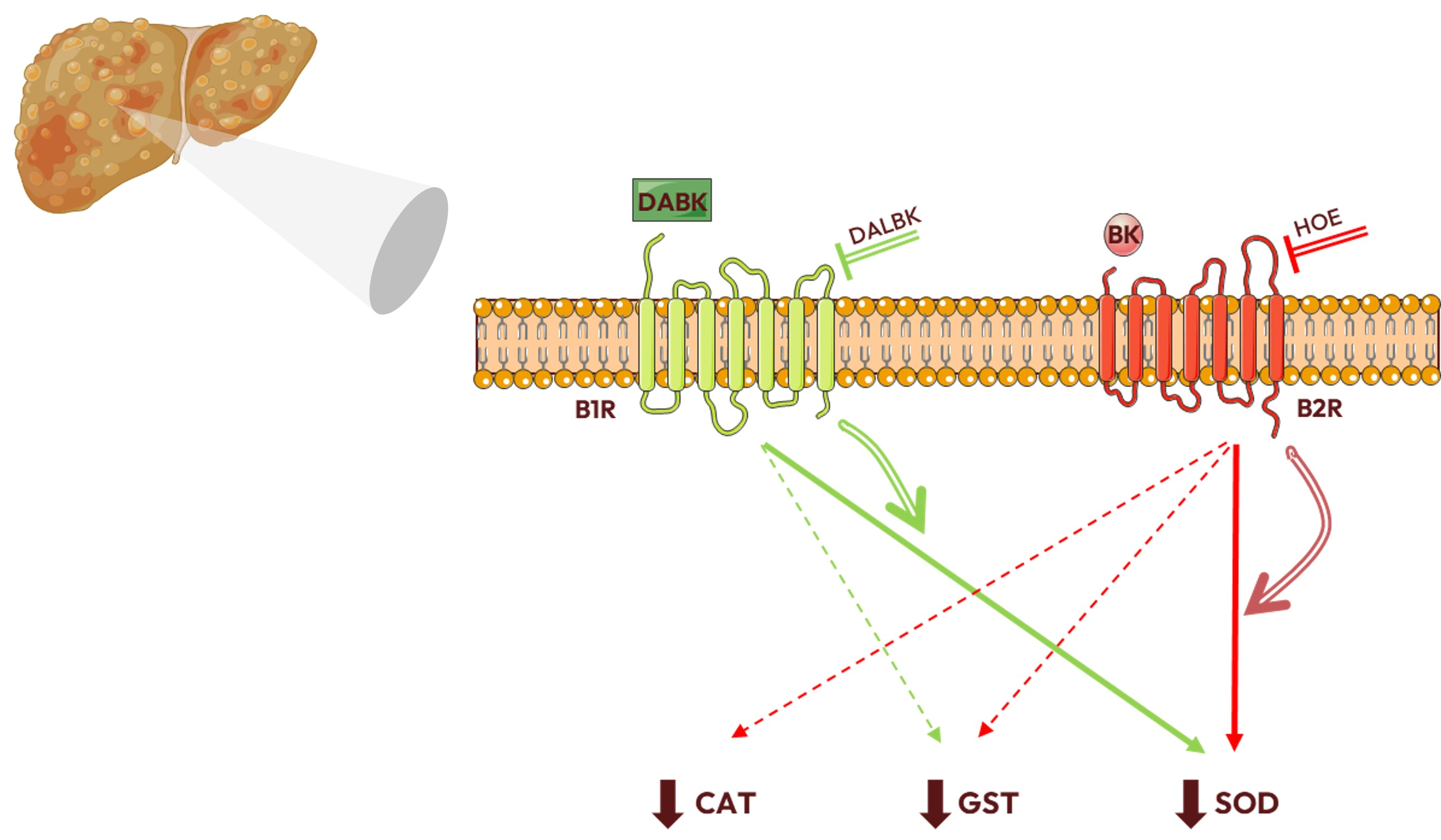
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; Kleiner, D.E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism 2016, 65, 1080–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Ore, A.; Akinloye, O.A. Oxidative Stress and Antioxidant Biomarkers in Clinical and Experimental Models of Non-Alcoholic Fatty Liver Disease. Medicina 2019, 55, 26. [Google Scholar] [CrossRef]
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 160–174. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [Green Version]
- Tessari, P.; Coracina, A.; Cosma, A.; Tiengo, A. Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 291–302. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Tariq, Z.; Green, C.J.; Hodson, L. Are oxidative stress mechanisms the common denominator in the progression from hepatic steatosis towards non-alcoholic steatohepatitis (NASH)? Liver Int. 2014, 34, e180–e190. [Google Scholar] [CrossRef] [PubMed]
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front Med. 2021, 8, 595371. [Google Scholar] [CrossRef] [PubMed]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Benzie, I.F. Lipid peroxidation: A review of causes, consequences, measurement and dietary influences. Int. J. Food Sci. Nutr. 1996, 47, 233–261. [Google Scholar] [CrossRef]
- Parlati, L.; Régnier, M.; Guillou, H.; Postic, C. New targets for NAFLD. JHEP Rep. 2021, 3, 100346. [Google Scholar] [CrossRef]
- Varela-Rey, M.; Embade, N.; Ariz, U.; Lu, S.C.; Mato, J.M.; Martínez-Chantar, M.L. Non-alcoholic steatohepatitis and animal models: Understanding the human disease. Int. J. Biochem. Cell Biol. 2009, 41, 969–976. [Google Scholar] [CrossRef]
- Zhu, H.; Ge, K.; Lu, J.; Jia, C. Downregulation of GNAI3 Promotes the Pathogenesis of Methionine/Choline-Deficient Diet-Induced Nonalcoholic Fatty Liver Disease. Gut Liver 2020, 14, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- Kaplan, A.P.; Joseph, K.; Silverberg, M. Pathways for bradykinin formation and inflammatory disease. J. Allergy Clin. Immunol. 2002, 109, 195–209. [Google Scholar] [CrossRef]
- Hall, J.M. Bradykinin receptors: Pharmacological properties and biological roles. Pharmacol. Ther. 1992, 56, 131–190. [Google Scholar] [PubMed]
- Kuhr, F.; Lowry, J.; Zhang, Y.; Brovkovych, V.; Skidgel, R.A. Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides 2010, 44, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marceau, F.; Lussier, A.; Regoli, D.; Giroud, J.P. Pharmacology of kinins: Their relevance to tissue injury and inflammation. Gen. Pharmacol. 1983, 14, 209–229. [Google Scholar] [CrossRef]
- Cyr, M.; Lepage, Y.; Blais, C., Jr.; Gervais, N.; Cugno, M.; Rouleau, J.L.; Adam, A. Bradykinin and des-Arg(9)-bradykinin metabolic pathways and kinetics of activation of human plasma. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H275–H283. [Google Scholar] [CrossRef]
- Prado, J.L.; Limãos, E.A.; Roblero, J.; Freitas, J.O.; Prado, E.S.; Paiva, A.C. Recovery and conversion of kinins in exsanguinated rat preparations. Naunyn. Schmiedebergs Arch. Pharmacol. 1975, 290, 191–205. [Google Scholar] [CrossRef]
- Kouyoumdjian, M.; Nagaoka, M.R.; Borges, D.R. Kallikrein-kinin system in hepatic experimental models. Peptides 2005, 26, 1301–1307. [Google Scholar] [CrossRef]
- Paio, M.A.; Kouyoumdjian, M.; Borges, D.R.; Nagaoka, M.R. Differential bradykinin B1 and B2 receptor regulation in cell death induced by hepatic ischaemia/reperfusion injury. Clin. Sci. 2014, 127, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Kayashima, Y.; Smithies, O.; Kakoki, M. The kallikrein-kinin system and oxidative stress. Curr. Opin. Nephrol. Hypertens 2012, 21, 92–96. [Google Scholar] [CrossRef] [Green Version]
- CIOMS International Guiding Principles for Biomedical Research Involving Animals. Altern. Lab Anim. 1985, 12, ii.
- Loureiro-Silva, M.R.; Molina, H.M.; Borges, D.R. Portal hypertensive response to bradykinin in inflamed or cirrhotic rat livers is mediated by B2-type receptors. J. Gastroenterol. Hepatol. 2001, 16, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; van den Hoek, A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef] [Green Version]
- Mostafa, M.; Abdelkader, A.; Evans, J.J.; Hagen, C.E.; Hartley, C.P. Fatty Liver Disease: A Practical Approach. Arch. Pathol. Lab Med. 2020, 144, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Cotrin, S.S.; Puzer, L.; de Souza Judice, W.A.; Juliano, L.; Carmona, A.K.; Juliano, M.A. Positional-scanning combinatorial libraries of fluorescence resonance energy transfer peptides to define substrate specificity of carboxydipeptidases: Assays with human cathepsin B. Anal. Biochem. 2004, 335, 244–252. [Google Scholar] [CrossRef]
- Higuchi, H.; Bronk, S.F.; Taniai, M.; Canbay, A.; Gores, G.J. Cholestasis increases tumor necrosis factor-related apoptotis-inducing ligand (TRAIL)-R2/DR5 expression and sensitizes the liver to TRAIL-mediated cytotoxicity. J. Pharmacol. Exp. Ther. 2002, 303, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.; Wang, B.T.; Wu, Z.C.; Yu, W.T.; Lin, P.J.; Tsai, W.L.; Shiesh, S.C. Glycine ameliorates liver injury and vitamin D deficiency induced by bile duct ligation. Clin. Chim. Acta 2013, 420, 150–154. [Google Scholar] [CrossRef]
- Góth, L. A simple method for determination of serum catalase activity and revision of reference range. Clin. Chim. Acta 1991, 196, 143–151. [Google Scholar] [CrossRef]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef]
- Sertorio, M.N.; César, H.; de Souza, E.A.; Mennitti, L.V.; Santamarina, A.B.; De Souza Mesquita, L.M.; Jucá, A.; Casagrande, B.P.; Estadella, D.; Aguiar, O., Jr.; et al. Parental High-Fat High-Sugar Diet Intake Programming Inflammatory and Oxidative Parameters of Reproductive Health in Male Offspring. Front. Cell Dev. Biol. 2022, 10, 867127. [Google Scholar] [CrossRef]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Methods Enzymol. 1978, 52, 302–310. [Google Scholar]
- Levine, R.L.; Williams, J.A.; Stadtman, E.R.; Shacter, E. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 1994, 233, 346–357. [Google Scholar] [PubMed]
- Machado, M.V.; Cortez-Pinto, H. Cell death and nonalcoholic steatohepatitis: Where is ballooning relevant? Expert Rev. Gastroenterol. Hepatol. 2011, 5, 213–222. [Google Scholar] [CrossRef]
- Akazawa, Y.; Nakao, K. To die or not to die: Death signaling in nonalcoholic fatty liver disease. J. Gastroenterol. 2018, 53, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, S.; Rajak, S.; Upadhyay, A.; Tewari, A.; Anthony Sinha, R. Current treatment paradigms and emerging therapies for NAFLD/NASH. Front. Biosci. 2021, 26, 206–237. [Google Scholar] [CrossRef]
- Chen, X.; Liu, C.; Ruan, L. G-Protein-Coupled Receptors 120 Agonist III Improves Hepatic Inflammation and ER Stress in Steatohepatitis. Dig Dis. Sci. 2021, 66, 1090–1096. [Google Scholar] [CrossRef]
- Kong, L.; An, X.; Hu, L.; Zhang, S.; Liu, L.; Zhao, S.; Wang, R.; Nan, Y. Resveratrol ameliorates nutritional steatohepatitis through the mmu-miR-599/PXR pathway. Int. J. Mol. Med. 2022, 49, 47. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, J.; Huang, Y.; Li, H.; Yan, S.; Lin, J.; Chen, Y.; Wu, L.; Liu, B.; Wang, G.; et al. Polydatin attenuates diet-induced nonalcoholic steatohepatitis and fibrosis in mice. Int. J. Biol. Sci. 2018, 14, 1411–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Han, J.; Man, K.; Li, X.; Du, J.; Chu, E.S.; Go, M.Y.; Sung, J.J.; Yu, J. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. J. Hepatol. 2016, 64, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Dela Peña, A.; Leclercq, I.; Field, J.; George, J.; Jones, B.; Farrell, G. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology 2005, 129, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Green, R.M. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 2004, 40, 47–51. [Google Scholar] [CrossRef]
- Marcolin, E.; Forgiarini, L.F.; Tieppo, J.; Dias, A.S.; Freitas, L.A.; Marroni, N.P. Methionine- and choline-deficient diet induces hepatic changes characteristic of non-alcoholic steatohepatitis. Arq. Gastroenterol. 2011, 48, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; Almeida Pereira, T.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef] [Green Version]
- Rizki, G.; Arnaboldi, L.; Gabrielli, B.; Yan, J.; Lee, G.S.; Ng, R.K.; Turner, S.M.; Badger, T.M.; Pitas, R.E.; Maher, J.J. Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. J. Lipid Res. 2006, 47, 2280–2290. [Google Scholar] [CrossRef] [Green Version]
- Van Herck, M.A.; Vonghia, L.; Francque, S.M. Animal Models of Nonalcoholic Fatty Liver Disease—A Starter’s Guide. Nutrients 2017, 9, 1072. [Google Scholar] [CrossRef] [Green Version]
- Caballero, F.; Fernández, A.; Matías, N.; Martínez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernández-Checa, J.C.; García-Ruiz, C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef] [Green Version]
- Mato, J.M.; Martínez-Chantar, M.L.; Lu, S.C. Methionine metabolism and liver disease. Annu. Rev. Nutr. 2008, 28, 273–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzorno, J. Glutathione! Integr. Med. 2014, 13, 8–12. [Google Scholar]
- Vance, D.E. Role of phosphatidylcholine biosynthesis in the regulation of lipoprotein homeostasis. Curr. Opin. Lipidol. 2008, 19, 229–234. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859 Pt B, 1558–1572. [Google Scholar] [CrossRef]
- Alshawsh, M.A.; Alsalahi, A.; Alshehade, S.A.; Saghir, S.A.M.; Ahmeda, A.F.; Al Zarzour, R.H.; Mahmoud, A.M. A Comparison of the Gene Expression Profiles of Non-Alcoholic Fatty Liver Disease between Animal Models of a High-Fat Diet and Methionine-Choline-Deficient Diet. Molecules 2022, 27, 858. [Google Scholar] [CrossRef]
- Montandon, S.A.; Somm, E.; Loizides-Mangold, U.; de Vito, C.; Dibner, C.; Jornayvaz, F.R. Multi-technique comparison of atherogenic and MCD NASH models highlights changes in sphingolipid metabolism. Sci. Rep. 2019, 9, 16810. [Google Scholar] [CrossRef] [Green Version]
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2241–2257. [Google Scholar] [CrossRef]
- Sueyoshi, T.; Enjyoji, K.; Shimada, T.; Kato, H.; Iwanaga, S.; Bando, Y.; Kominami, E.; Katunuma, N. A new function of kininogens as thiol-proteinase inhibitors: Inhibition of papain and cathepsins B, H and L by bovine, rat and human plasma kininogens. FEBS Lett. 1985, 182, 193–195. [Google Scholar] [CrossRef] [Green Version]
- Assy, N.; Nassar, F.; Nasser, G.; Grosovski, M. Olive oil consumption and non-alcoholic fatty liver disease. World J. Gastroenterol. 2009, 15, 1809–1815. [Google Scholar] [CrossRef]
- Mizunoe, Y.; Kobayashi, M.; Tagawa, R.; Nakagawa, Y.; Shimano, H.; Higami, Y. Association between Lysosomal Dysfunction and Obesity-Related Pathology: A Key Knowledge to Prevent Metabolic Syndrome. Int. J. Mol. Sci. 2019, 20, 3688. [Google Scholar] [CrossRef] [Green Version]
- Dias, J.P.; Talbot, S.; Sénécal, J.; Carayon, P.; Couture, R. Kinin B1 receptor enhances the oxidative stress in a rat model of insulin resistance: Outcome in hypertension, allodynia and metabolic complications. PLoS ONE 2010, 5, e12622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, J.P.; Couture, R. Suppression of vascular inflammation by kinin B1 receptor antagonism in a rat model of insulin resistance. J. Cardiovasc. Pharmacol. 2012, 60, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Mikrut, K.; Paluszak, J.; Kozlik, J.; Sosnowski, P.; Krauss, H.; Grześkowiak, E. The effect of bradykinin on the oxidative state of rats with acute hyperglycaemia. Diabetes Res. Clin. Pract. 2001, 51, 79–85. [Google Scholar] [CrossRef]
- Dong, R.; Xu, X.; Li, G.; Feng, W.; Zhao, G.; Zhao, J.; Wang, D.W.; Tu, L. Bradykinin inhibits oxidative stress-induced cardiomyocytes senescence via regulating redox state. PLoS ONE 2013, 8, e77034. [Google Scholar] [CrossRef] [PubMed]
- Borges, D.R.; Limãos, E.A.; Prado, J.L.; Camargo, A.C. Catabolism of vasoactive polypeptides by perfused rat liver. Naunyn. Schmiedeberg Arch. Pharmacol. 1976, 295, 33–40. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | NASH | p | |
|---|---|---|---|
| Final Body Weight (g) | 37.7 ± 0.8 | 22.0 ± 0.5 | **** |
| Liver Weight (g) | 1.81 ± 0.04 | 1.28 ± 0.07 | **** |
| Liver Index (%) | 4.8 ± 0.1 | 5.8 ± 0.3 | ** |
| Glycemia (mg/dL) | 206 ± 10 | 103 ± 5 | **** |
| Cholesterol (mg/dL) | 120 ± 4 | 89 ± 5 | **** |
| Triglycerides (mg/dL) | 122 ± 4 | 123 ± 8 | |
| ALT (U/L) | 153 ± 20 | 414 ± 48 | **** |
| AST (U/L) | 303 ± 28 | 433 ± 44 | * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomaz, M.S.; Sertorio, M.N.; Gazarini, M.L.; Ribeiro, D.A.; Pisani, L.P.; Nagaoka, M.R. Effect of Kinins on the Hepatic Oxidative Stress in Mice Treated with a Methionine-Choline Deficient Diet. Biomedicines 2023, 11, 2199. https://doi.org/10.3390/biomedicines11082199
Thomaz MS, Sertorio MN, Gazarini ML, Ribeiro DA, Pisani LP, Nagaoka MR. Effect of Kinins on the Hepatic Oxidative Stress in Mice Treated with a Methionine-Choline Deficient Diet. Biomedicines. 2023; 11(8):2199. https://doi.org/10.3390/biomedicines11082199
Chicago/Turabian StyleThomaz, Mariana Silva, Marcela Nascimento Sertorio, Marcos Leoni Gazarini, Daniel Araki Ribeiro, Luciana Pellegrini Pisani, and Marcia Regina Nagaoka. 2023. "Effect of Kinins on the Hepatic Oxidative Stress in Mice Treated with a Methionine-Choline Deficient Diet" Biomedicines 11, no. 8: 2199. https://doi.org/10.3390/biomedicines11082199