
When Size Really Matters: The Eccentricities of Dystrophin Transcription and the Hazards of Quantifying mRNA from Very Long Genes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
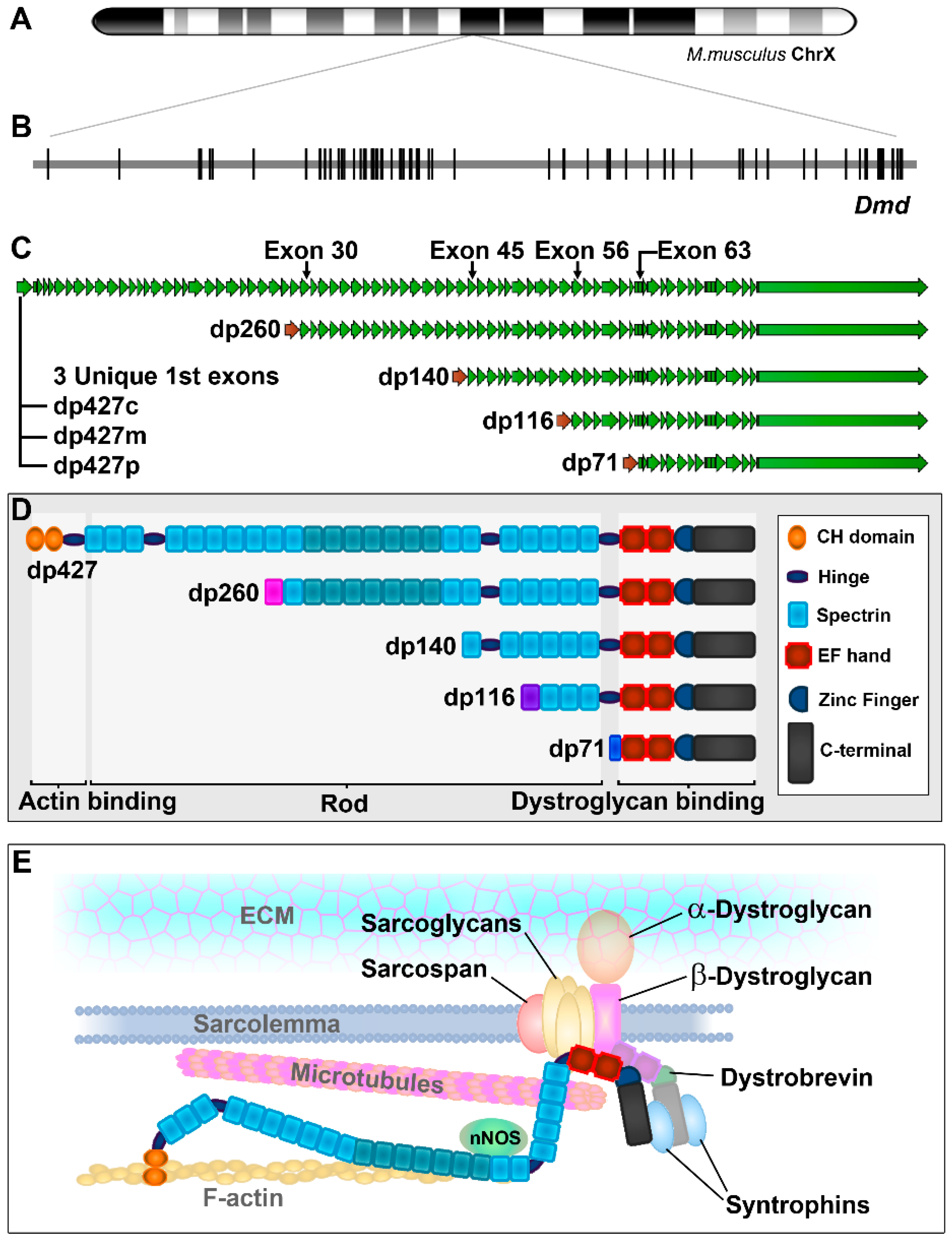
1.1. The Dystrophin Gene
1.2. Dystrophin and Muscular Dystrophy
1.3. Quantifying Dystrophin Protein
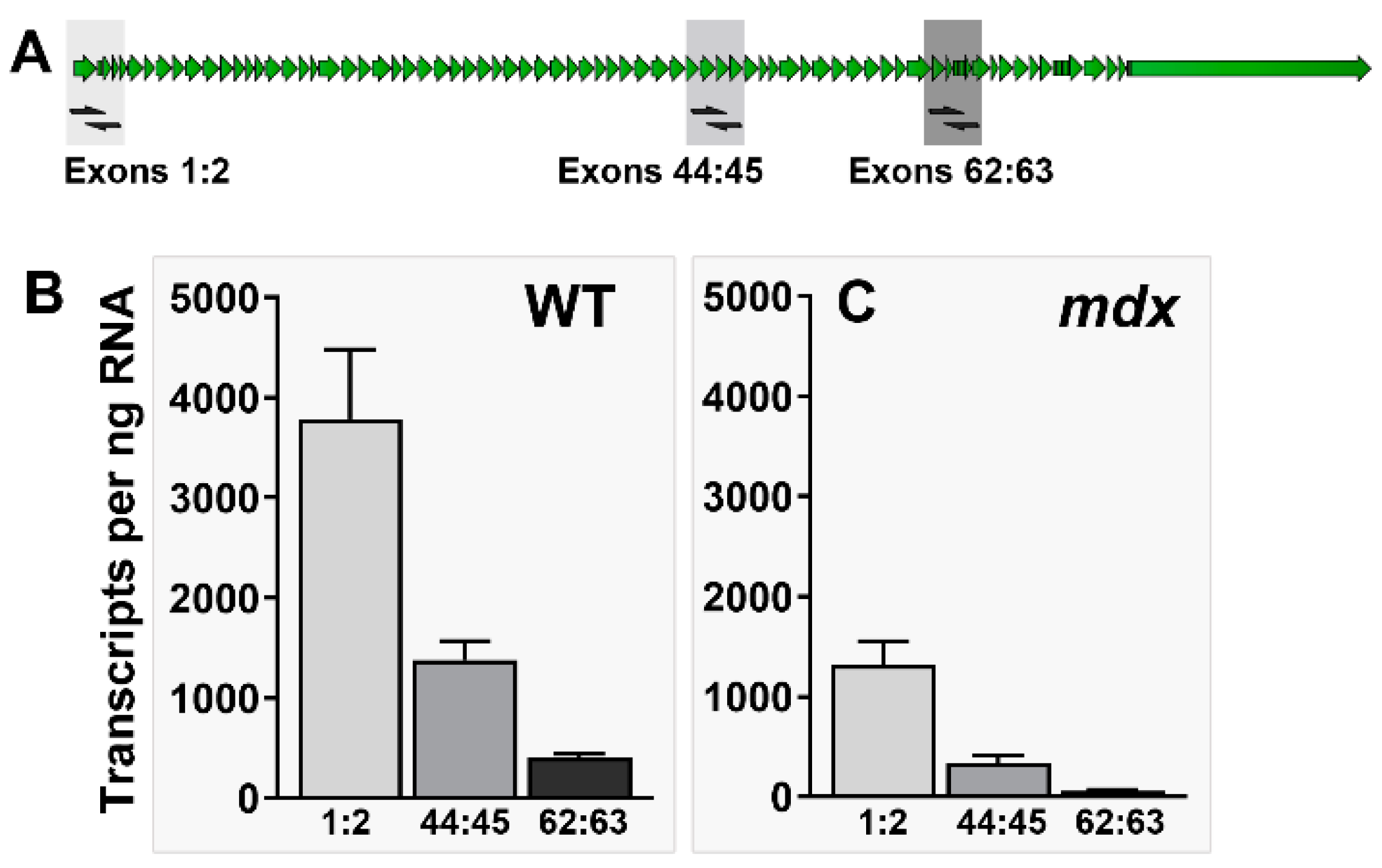
1.4. Quantifying Dystrophin mRNA and Transcript Imbalance
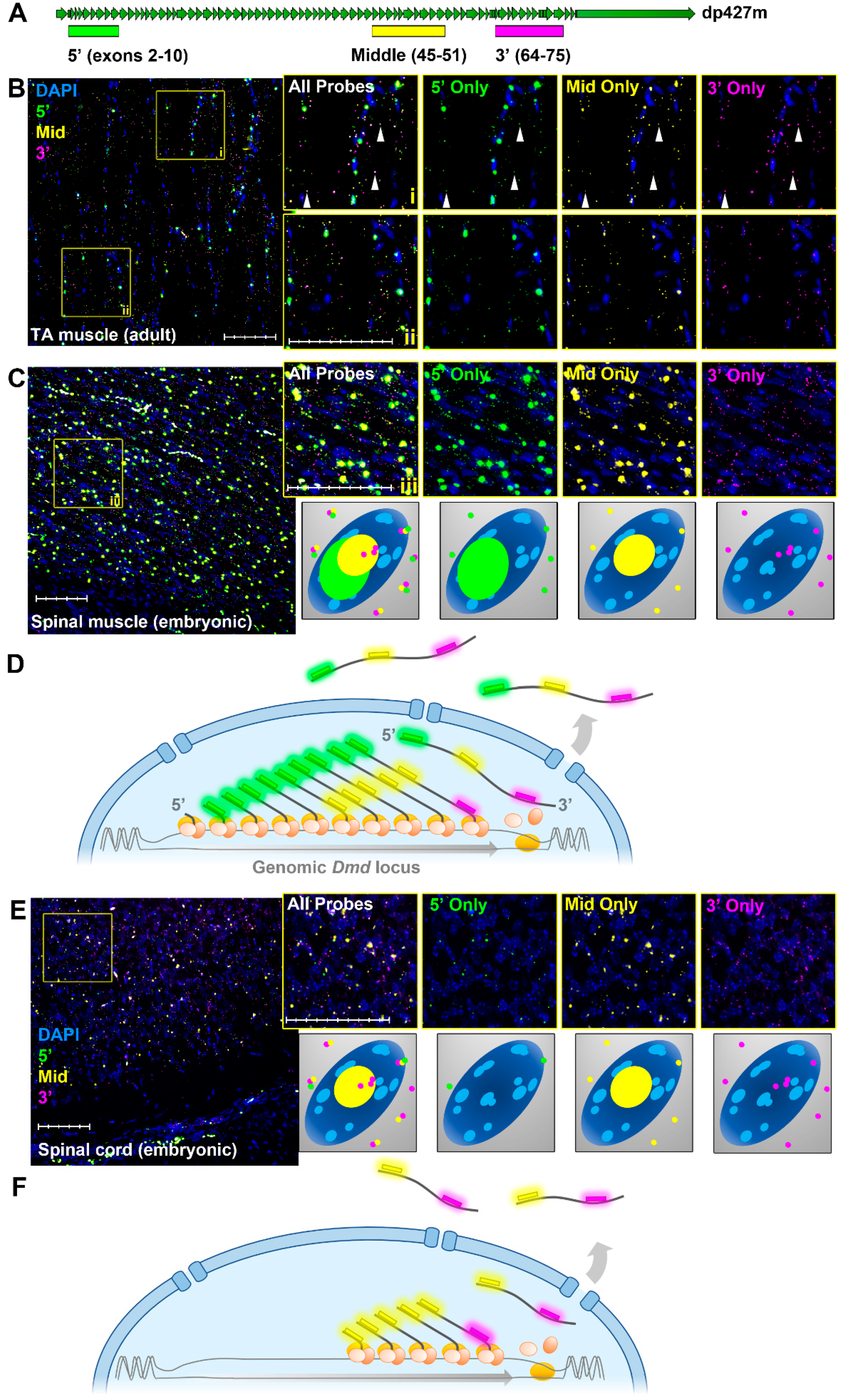
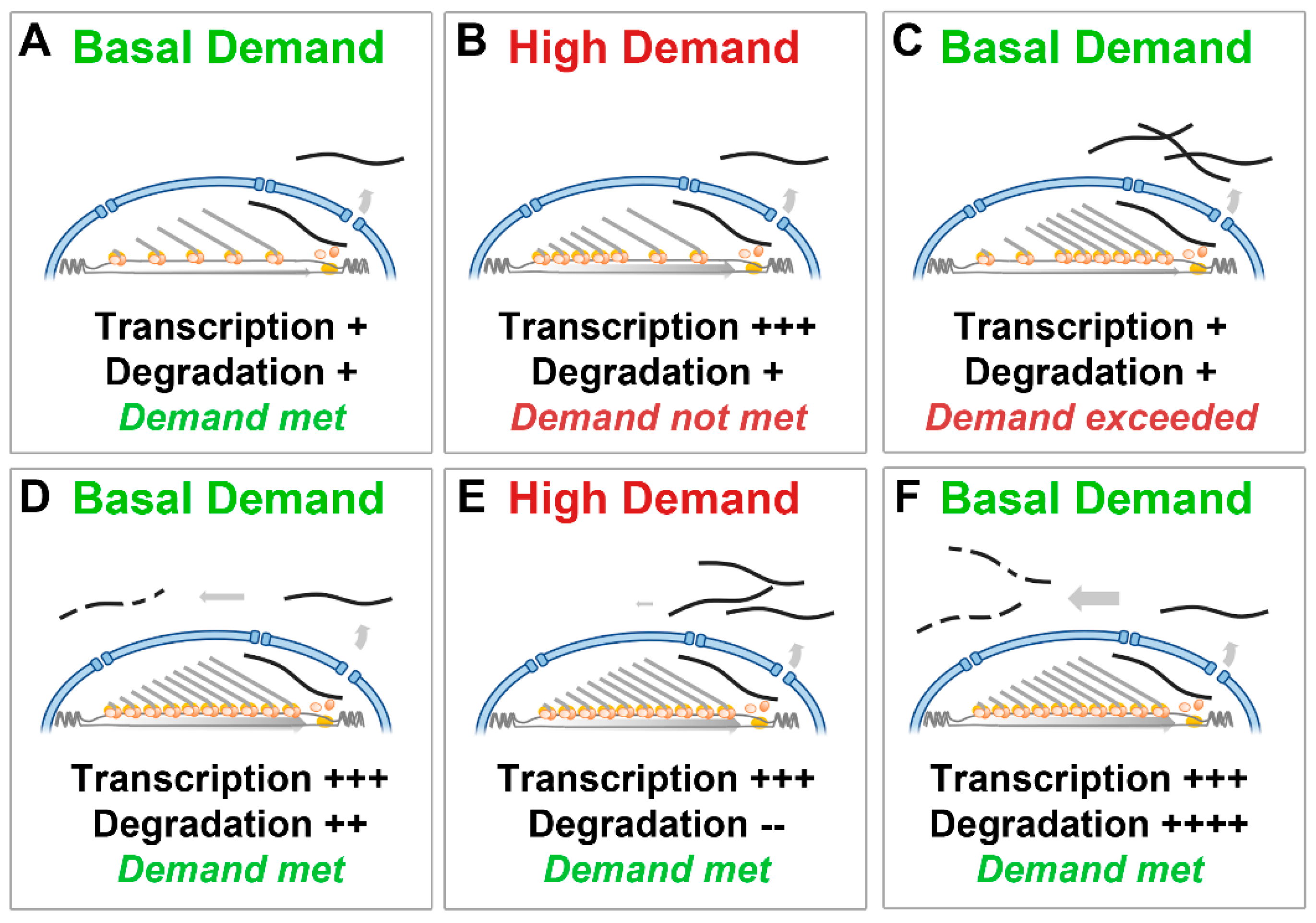
1.5. Dystrophin Transcriptional Model
1.6. Quantitative Measurement of Unconventional Dystrophin Expression
1.6.1. cDNA Synthesis
1.6.2. Comparing Healthy and Dystrophic Transcripts
1.6.3. Measuring Transcriptional Changes over Time
1.6.4. Quantifying Exon Skipping
1.6.5. Dystrophin in the Transcriptomic Era
2. Methods
2.1. Sample Collection and Preparation
2.2. Multiplex FISH: Sample Preparation
2.3. RNAscope Multiplex Assay
2.4. Imaging
2.5. RNAseq Analysis
3. Discussion
3.1. Dystrophin Transcriptional Model
3.2. Dystrophin Expression and RNAseq
3.3. Dystrophin Transcription: Caveats and Alternative Hypotheses
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tennyson, C.N.; Klamut, H.J.; Worton, R.G. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat. Genet. 1995, 9, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Gazzoli, I.; Pulyakhina, I.; Verwey, N.E.; Ariyurek, Y.; Laros, J.F.; ’t Hoen, P.A.; Aartsma-Rus, A. Non-sequential and multi-step splicing of the dystrophin transcript. RNA Biol. 2016, 13, 290–305. [Google Scholar] [CrossRef] [Green Version]
- Warner, L.E.; DelloRusso, C.; Crawford, R.W.; Rybakova, I.N.; Patel, J.R.; Ervasti, J.M.; Chamberlain, J.S. Expression of Dp260 in muscle tethers the actin cytoskeleton to the dystrophin-glycoprotein complex and partially prevents dystrophy. Hum. Mol. Genet. 2002, 11, 1095–1105. [Google Scholar] [CrossRef]
- Molza, A.E.; Mangat, K.; Le Rumeur, E.; Hubert, J.F.; Menhart, N.; Delalande, O. Structural Basis of Neuronal Nitric-oxide Synthase Interaction with Dystrophin Repeats 16 and 17. J. Biol. Chem. 2015, 290, 29531–29541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Thomas, G.D.; Yue, Y.; Yang, H.T.; Li, D.; Long, C.; Judge, L.; Bostick, B.; Chamberlain, J.S.; Terjung, R.L.; et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Investig. 2009, 119, 624–635. [Google Scholar] [CrossRef] [Green Version]
- Belanto, J.J.; Mader, T.L.; Eckhoff, M.D.; Strandjord, D.M.; Banks, G.B.; Gardner, M.K.; Lowe, D.A.; Ervasti, J.M. Microtubule binding distinguishes dystrophin from utrophin. Proc. Natl. Acad. Sci. USA 2014, 111, 5723. [Google Scholar] [CrossRef]
- Prins, K.W.; Humston, J.L.; Mehta, A.; Tate, V.; Ralston, E.; Ervasti, J.M. Dystrophin is a microtubule-associated protein. J. Cell Biol. 2009, 186, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef] [Green Version]
- Hildyard, J.C.W.; Crawford, A.H.; Rawson, F.; Riddell, D.O.; Harron, R.C.M.; Piercy, R.J. Single-transcript multiplex in situ hybridisation reveals unique patterns of dystrophin isoform expression in the developing mammalian embryo. Wellcome Open Res. 2020, 5, 76. [Google Scholar] [CrossRef] [Green Version]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Boyce, F.M.; Beggs, A.H.; Feener, C.; Kunkel, L.M. Dystrophin is transcribed in brain from a distant upstream promoter. Proc. Natl. Acad. Sci. USA 1991, 88, 1276–1280. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, D.C.; Monaco, A.P.; Derry, J.M.; Walker, A.P.; Barnard, E.A.; Barnard, P.J. Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum. Mol. Genet. 1992, 1, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Klamut, H.J.; Gangopadhyay, S.B.; Worton, R.G.; Ray, P.N. Molecular and functional analysis of the muscle-specific promoter region of the Duchenne muscular dystrophy gene. Mol. Cell. Biol. 1990, 10, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nudel, U.; Zuk, D.; Einat, P.; Zeelon, E.; Levy, Z.; Neuman, S.; Yaffe, D. Duchenne muscular dystrophy gene product is not identical in muscle and brain. Nature 1989, 337, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Lidov, H.G.; Selig, S.; Kunkel, L.M. Dp140: A novel 140 kDa CNS transcript from the dystrophin locus. Hum. Mol. Genet. 1995, 4, 329–335. [Google Scholar] [CrossRef]
- Crawford, A.H.; Hildyard, J.C.W.; Rushing, S.A.M.; Wells, D.J.; Diez-Leon, M.; Piercy, R.J. Validation of DE50-MD dogs as a model for the brain phenotype of Duchenne muscular dystrophy. Dis. Models Mech. 2022, 15, dmm049291. [Google Scholar] [CrossRef]
- D’Souza, V.N.; Nguyen, T.M.; Morris, G.E.; Karges, W.; Pillers, D.A.; Ray, P.N. A novel dystrophin isoform is required for normal retinal electrophysiology. Hum. Mol. Genet. 1995, 4, 837–842. [Google Scholar] [CrossRef]
- Byers, T.J.; Lidov, H.G.; Kunkel, L.M. An alternative dystrophin transcript specific to peripheral nerve. Nat. Genet. 1993, 4, 77–81. [Google Scholar] [CrossRef]
- Bar, S.; Barnea, E.; Levy, Z.; Neuman, S.; Yaffe, D.; Nudel, U. A novel product of the Duchenne muscular dystrophy gene which greatly differs from the known isoforms in its structure and tissue distribution. Biochem. J. 1990, 272, 557–560. [Google Scholar] [CrossRef]
- Durbeej, M.; Jung, D.; Hjalt, T.; Campbell, K.P.; Ekblom, P. Transient expression of Dp140, a product of the Duchenne muscular dystrophy locus, during kidney tubulogenesis. Dev. Biol. 1997, 181, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Doorenweerd, N.; Mahfouz, A.; van Putten, M.; Kaliyaperumal, R.; T’ Hoen, P.A.C.; Hendriksen, J.G.M.; Aartsma-Rus, A.M.; Verschuuren, J.J.G.M.; Niks, E.H.; Reinders, M.J.T.; et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci. Rep. 2017, 7, 12575. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Tan, S.; Hermanowski, J.; Böhm, S.; Pacheco, S.; McCauley, J.M.; Greener, M.J.; Hinits, Y.; Hughes, S.M.; Sharpe, P.T.; et al. The dystrotelin, dystrophin and dystrobrevin superfamily: New paralogues and old isoforms. BMC Genom. 2007, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Segurel, L.; Wyman, M.J.; Przeworski, M. Determinants of mutation rate variation in the human germline. Annu. Rev. Genom. Hum. Genet. 2014, 15, 47–70. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar] [CrossRef] [Green Version]
- White, S.J.; den Dunnen, J.T. Copy number variation in the genome; the human DMD gene as an example. Cytogenet. Genome Res. 2006, 115, 240–246. [Google Scholar] [CrossRef]
- Ankala, A.; Kohn, J.N.; Hegde, A.; Meka, A.; Ephrem, C.L.; Askree, S.H.; Bhide, S.; Hegde, M.R. Aberrant firing of replication origins potentially explains intragenic nonrecurrent rearrangements within genes, including the human DMD gene. Genome Res. 2012, 22, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef]
- Bushby, K.M.; Gardner-Medwin, D. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. I. Natural history. J. Neurol. 1993, 240, 98–104. [Google Scholar] [CrossRef]
- Comi, G.P.; Prelle, A.; Bresolin, N.; Moggio, M.; Bardoni, A.; Gallanti, A.; Vita, G.; Toscano, A.; Ferro, M.T.; Bordoni, A.; et al. Clinical variability in Becker muscular dystrophy. Genetic, biochemical and immunohistochemical correlates. Brain 1994, 117 Pt 1, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Morandi, L.; Mora, M.; Bernasconi, P.; Mantegazza, R.; Gebbia, M.; Balestrini, M.R.; Cornelio, F. Very small dystrophin molecule in a family with a mild form of Becker dystrophy. Neuromuscul. Disord. NMD 1993, 3, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Farrokhi, V.; Walsh, J.; Palandra, J.; Brodfuehrer, J.; Caiazzo, T.; Owens, J.; Binks, M.; Neelakantan, S.; Yong, F.; Dua, P.; et al. Dystrophin and mini-dystrophin quantification by mass spectrometry in skeletal muscle for gene therapy development in Duchenne muscular dystrophy. Gene Ther. 2022, 29, 608–615. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Morgan, J.; Lonkar, P.; Neubert, H.; Owens, J.; Binks, M.; Montolio, M.; Phadke, R.; Datson, N.; Van Deutekom, J.; et al. Report of a TREAT-NMD/World Duchenne Organisation Meeting on Dystrophin Quantification Methodology. J. Neuromuscul. Dis. 2019, 6, 147–159. [Google Scholar] [CrossRef]
- Godfrey, C.; Muses, S.; McClorey, G.; Wells, K.E.; Coursindel, T.; Terry, R.L.; Betts, C.; Hammond, S.; O’Donovan, L.; Hildyard, J.; et al. How much dystrophin is enough: The physiological consequences of different levels of dystrophin in the mdx mouse. Hum. Mol. Genet. 2015, 24, 4225–4237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, A.; Stantzou, A.; Petrova, O.N.; Hildyard, J.; Tensorer, T.; Matouk, M.; Petkova, M.V.; Richard, I.; Manoliu, T.; Goyenvalle, A.; et al. Dystrophin myonuclear domain restoration governs treatment efficacy in dystrophic muscle. Proc. Natl. Acad. Sci. USA 2023, 120, e2206324120. [Google Scholar] [CrossRef] [PubMed]
- Dickson, G.; Pizzey, J.A.; Elsom, V.E.; Love, D.; Davies, K.E.; Walsh, F.S. Distinct dystrophin mRNA species are expressed in embryonic and adult mouse skeletal muscle. FEBS Lett. 1988, 242, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Burghes, A.H.; Logan, C.; Hu, X.; Belfall, B.; Worton, R.G.; Ray, P.N. A cDNA clone from the Duchenne/Becker muscular dystrophy gene. Nature 1987, 328, 434–437. [Google Scholar] [CrossRef]
- Scott, M.O.; Sylvester, J.E.; Heiman-Patterson, T.; Shi, Y.J.; Fieles, W.; Stedman, H.; Burghes, A.; Ray, P.; Worton, R.; Fischbeck, K.H. Duchenne muscular dystrophy gene expression in normal and diseased human muscle. Science 1988, 239, 1418–1420. [Google Scholar] [CrossRef]
- Muntoni, F.; Strong, P.N. Transcription of the dystrophin gene in Duchenne muscular dystrophy muscle. FEBS Lett. 1989, 252, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, J.S.; Farwell, N.J.; Chamberlain, J.R.; Cox, G.A.; Caskey, C.T. PCR analysis of dystrophin gene mutation and expression. J. Cell. Biochem. 1991, 46, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Abbs, S.; Bobrow, M. Analysis of quantitative PCR for the diagnosis of deletion and duplication carriers in the dystrophin gene. J. Med. Genet. 1992, 29, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Rodriguez, R.; Hiller, M.; Jimenez-Gracia, L.; van der Pal, Z.; Balog, J.; Adamzek, K.; Aartsma-Rus, A.; Spitali, P. Premature termination codons in the DMD gene cause reduced local mRNA synthesis. Proc. Natl. Acad. Sci. USA 2020, 117, 16456–16464. [Google Scholar] [CrossRef] [PubMed]
- Maquat, L.E.; Tarn, W.Y.; Isken, O. The pioneer round of translation: Features and functions. Cell 2010, 142, 368–374. [Google Scholar] [CrossRef] [Green Version]
- Hildyard, J.C.W.; Rawson, F.; Wells, D.J.; Piercy, R.J. Multiplex in situ hybridization within a single transcript: RNAscope reveals dystrophin mRNA dynamics. PLoS ONE 2020, 15, e0239467. [Google Scholar] [CrossRef]
- Spitali, P.; van den Bergen, J.C.; Verhaart, I.E.; Wokke, B.; Janson, A.A.; van den Eijnde, R.; den Dunnen, J.T.; Laros, J.F.; Verschuuren, J.J.; Hoen, P.A.T.; et al. DMD transcript imbalance determines dystrophin levels. FASEB J. 2013, 27, 4909–4916. [Google Scholar] [CrossRef] [Green Version]
- Tennyson, C.N.; Shi, Q.; Worton, R.G. Stability of the human dystrophin transcript in muscle. Nucleic Acids Res. 1996, 24, 3059–3064. [Google Scholar] [CrossRef] [Green Version]
- Waldrop, M.A.; Moore, S.A.; Mathews, K.D.; Darbro, B.W.; Medne, L.; Finkel, R.; Connolly, A.M.; Crawford, T.O.; Drachman, D.; Wein, N.; et al. Intron mutations and early transcription termination in Duchenne and Becker muscular dystrophy. Hum. Mutat. 2022, 43, 511–528. [Google Scholar] [CrossRef]
- Muniz, L.; Nicolas, E.; Trouche, D. RNA polymerase II speed: A key player in controlling and adapting transcriptome composition. EMBO J. 2021, 40, e105740. [Google Scholar] [CrossRef]
- Singh, J.; Padgett, R.A. Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol. 2009, 16, 1128–1133. [Google Scholar] [CrossRef] [Green Version]
- Hildyard, J.C.W.; Riddell, D.O.; Harron, R.C.M.; Rawson, F.; Foster, E.M.A.; Massey, C.; Taylor-Brown, F.; Wells, D.J.; Piercy, R.J. The skeletal muscle phenotype of the DE50-MD dog model of Duchenne muscular dystrophy. Wellcome Open Res. 2022, 7, 238. [Google Scholar] [CrossRef] [PubMed]
- Hildyard, J.C.W.; Wells, D.J.; Piercy, R.J. Identification of qPCR reference genes suitable for normalising gene expression in the developing mouse embryo. Wellcome Open Res. 2021, 6, 197. [Google Scholar] [CrossRef] [PubMed]
- Donandt, T.; Todorow, V.; Hintze, S.; Graupner, A.; Schoser, B.; Walter, M.C.; Meinke, P. Nuclear Small Dystrophin Isoforms during Muscle Differentiation. Life 2023, 13, 1367. [Google Scholar] [CrossRef] [PubMed]
- Terry, E.E.; Zhang, X.; Hoffmann, C.; Hughes, L.D.; Lewis, S.A.; Li, J.; Wallace, M.J.; Riley, L.A.; Douglas, C.M.; Gutierrez-Monreal, M.A.; et al. Transcriptional profiling reveals extraordinary diversity among skeletal muscle tissues. eLife 2018, 7, e34613. [Google Scholar] [CrossRef]
- Chemello, F.; Wang, Z.; Li, H.; McAnally, J.R.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Degenerative and regenerative pathways underlying Duchenne muscular dystrophy revealed by single-nucleus RNA sequencing. Proc. Natl. Acad. Sci. USA 2020, 117, 29691–29701. [Google Scholar] [CrossRef]
- Schmitt, B.M.; Rudolph, K.L.; Karagianni, P.; Fonseca, N.A.; White, R.J.; Talianidis, I.; Odom, D.T.; Marioni, J.C.; Kutter, C. High-resolution mapping of transcriptional dynamics across tissue development reveals a stable mRNA-tRNA interface. Genome Res. 2014, 24, 1797–1807. [Google Scholar] [CrossRef] [Green Version]
- The Galaxy Community; Afgan, E.; Nekrutenko, A.; Blankenberg, D.; Goecks, J.; Schatz, M.C.; Ostrovsky, A.E.; Mahmoud, A.; Lonie, A.J.; Syme, A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Barthelemy, F.; Defour, A.; Levy, N.; Krahn, M.; Bartoli, M. Muscle Cells Fix Breaches by Orchestrating a Membrane Repair Ballet. J. Neuromuscul. Dis. 2018, 5, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Carmeille, R.; Bouvet, F.; Tan, S.; Croissant, C.; Gounou, C.; Mamchaoui, K.; Mouly, V.; Brisson, A.R.; Bouter, A. Membrane repair of human skeletal muscle cells requires Annexin-A5. Biochim. Biophys. Acta 2016, 1863, 2267–2279. [Google Scholar] [CrossRef] [PubMed]
- Hershey, J.W.B.; Sonenberg, N.; Mathews, M.B. Principles of Translational Control. Cold Spring Harb. Perspect. Biol. 2019, 11, a032607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, C.A.; Howard, M.T. Conserved regions of the DMD 3’ UTR regulate translation and mRNA abundance in cultured myotubes. Neuromuscul. Disord. NMD 2014, 24, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.R.; Goff, S.P. Upf1 senses 3’UTR length to potentiate mRNA decay. Cell 2010, 143, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Greener, M.J.; Sewry, C.A.; Muntoni, F.; Roberts, R.G. The 3’-untranslated region of the dystrophin gene—Conservation and consequences of loss. Eur. J. Hum. Genet. 2002, 10, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Roy, B.; Jacobson, A. The intimate relationships of mRNA decay and translation. Trends Genet. 2013, 29, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Verhaart, I.E.C.; van Vliet-van den Dool, L.; Sipkens, J.A.; de Kimpe, S.J.; Kolfschoten, I.G.M.; van Deutekom, J.C.T.; Liefaard, L.; Ridings, J.E.; Hood, S.R.; Aartsma-Rus, A. The Dynamics of Compound, Transcript, and Protein Effects After Treatment With 2OMePS Antisense Oligonucleotides in mdx Mice. Mol. Therapy. Nucleic Acids 2014, 3, e148. [Google Scholar] [CrossRef]
- Novak, J.S.; Spathis, R.; Dang, U.J.; Fiorillo, A.A.; Hindupur, R.; Tully, C.B.; Mazala, D.A.G.; Canessa, E.; Brown, K.J.; Partridge, T.A.; et al. Interrogation of Dystrophin and Dystroglycan Complex Protein Turnover After Exon Skipping Therapy. J. Neuromuscul. Dis. 2021, 8, S383–S402. [Google Scholar] [CrossRef]
- Wilton, S.D.; Dye, D.E.; Blechynden, L.M.; Laing, N.G. Revertant fibres: A possible genetic therapy for Duchenne muscular dystrophy? Neuromuscul. Disord. 1997, 7, 329–335. [Google Scholar] [CrossRef]
- Wilton, S.D.; Dye, D.E.; Laing, N.G. Dystrophin gene transcripts skipping the mdx mutation. Muscle Nerve 1997, 20, 728–734. [Google Scholar] [CrossRef]
- Bouge, A.L.; Murauer, E.; Beyne, E.; Miro, J.; Varilh, J.; Taulan, M.; Koenig, M.; Claustres, M.; Tuffery-Giraud, S. Targeted RNA-Seq profiling of splicing pattern in the DMD gene: Exons are mostly constitutively spliced in human skeletal muscle. Sci. Rep. 2017, 7, 39094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lederfein, D.; Levy, Z.; Augier, N.; Mornet, D.; Morris, G.; Fuchs, O.; Yaffe, D.; Nudel, U. A 71-kilodalton protein is a major product of the Duchenne muscular dystrophy gene in brain and other nonmuscle tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 5346–5350. [Google Scholar] [CrossRef] [PubMed]
- Rau, F.; Laine, J.; Ramanoudjame, L.; Ferry, A.; Arandel, L.; Delalande, O.; Jollet, A.; Dingli, F.; Lee, K.Y.; Peccate, C.; et al. Abnormal splicing switch of DMD’s penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat. Commun. 2015, 6, 7205. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, M.; Anthony, K. Dystrophin Dp71 and the Neuropathophysiology of Duchenne Muscular Dystrophy. Mol. Neurobiol. 2019, 57, 1748–1767. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.D.; Brenner, S.E.; Dudoit, S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010, 38, e131. [Google Scholar] [CrossRef] [Green Version]
- Mason, P.B.; Struhl, K. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol. Cell 2005, 17, 831–840. [Google Scholar] [CrossRef]
- Carey, L.B. RNA polymerase errors cause splicing defects and can be regulated by differential expression of RNA polymerase subunits. eLife 2015, 4, e09945. [Google Scholar] [CrossRef]
- Gout, J.F.; Thomas, W.K.; Smith, Z.; Okamoto, K.; Lynch, M. Large-scale detection of in vivo transcription errors. Proc. Natl. Acad. Sci. USA 2013, 110, 18584–18589. [Google Scholar] [CrossRef]
- Lynch, M. Evolution of the mutation rate. Trends Genet. 2010, 26, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, E.; Jaryszak, D.L.; Valliere, C.R. Response of satellite cells to focal skeletal muscle injury. Muscle Nerve 1985, 8, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, A.; Kaczmarek, M.; Cialowicz, M.; Clemente, F.M.; Wolanski, P.; Badicu, G.; Murawska-Cialowicz, E. The Role of Satellite Cells in Skeletal Muscle Regeneration-The Effect of Exercise and Age. Biology 2021, 10, 1056. [Google Scholar] [CrossRef]
- de Leon, M.B.; Montanez, C.; Gomez, P.; Morales-Lazaro, S.L.; Tapia-Ramirez, V.; Valadez-Graham, V.; Recillas-Targa, F.; Yaffe, D.; Nudel, U.; Cisneros, B. Dystrophin Dp71 expression is down-regulated during myogenesis: Role of Sp1 and Sp3 on the Dp71 promoter activity. J. Biol. Chem. 2005, 280, 5290–5299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farea, M.; Rani, A.Q.M.; Maeta, K.; Nishio, H.; Matsuo, M. Dystrophin Dp71ab is monoclonally expressed in human satellite cells and enhances proliferation of myoblast cells. Sci. Rep. 2020, 10, 17123. [Google Scholar] [CrossRef] [PubMed]
- Scherer, S. A Short Guide to the Human Genome; Cold Spring Harbor Laboratory Press: Huntington, NY, USA, 2009. [Google Scholar]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hildyard, J.C.W.; Piercy, R.J. When Size Really Matters: The Eccentricities of Dystrophin Transcription and the Hazards of Quantifying mRNA from Very Long Genes. Biomedicines 2023, 11, 2082. https://doi.org/10.3390/biomedicines11072082
Hildyard JCW, Piercy RJ. When Size Really Matters: The Eccentricities of Dystrophin Transcription and the Hazards of Quantifying mRNA from Very Long Genes. Biomedicines. 2023; 11(7):2082. https://doi.org/10.3390/biomedicines11072082
Chicago/Turabian StyleHildyard, John C. W., and Richard J. Piercy. 2023. "When Size Really Matters: The Eccentricities of Dystrophin Transcription and the Hazards of Quantifying mRNA from Very Long Genes" Biomedicines 11, no. 7: 2082. https://doi.org/10.3390/biomedicines11072082