

The Unfolded Protein Response and Its Implications for Novel Therapeutic Strategies in Inflammatory Bowel Disease

Abstract
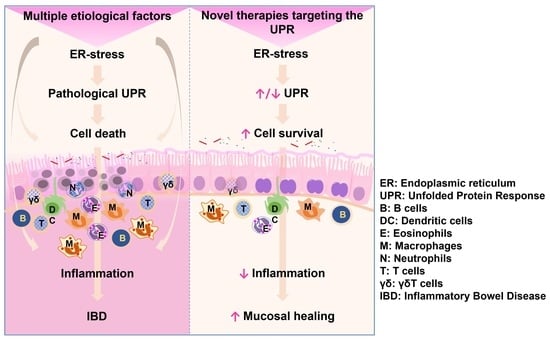
:
1. Endoplasmic Reticulum Stress (ER Stress)
2. The Unfolded Protein Response (UPR)
2.1. IRE1α/β
2.2. PERK
2.3. ATF6α
3. ER Stress and UPR in Inflammatory Bowel Disease (IBD)
3.1. Homeostasis of the Intestinal Epithelial Barrier
3.2. Pathogenesis of IBD
3.2.1. Apoptosis of IECs
3.2.2. Disruption of the Mucosal Barrier
3.2.3. Excessive Inflammation
3.2.4. Aberrant UPR Activation
4. Pharmacologic Intervention of the UPR as a Potential Therapeutic Strategy in IBD Treatment
4.1. Targeting the UPR Sensors
4.2. Non-Specific Targeting of the UPR
4.3. EPICERTIN, a KDELR Ligand Modulator of the UPR

5. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Voeltz, G.K.; Rolls, M.M.; Rapoport, T.A. Structural organization of the endoplasmic reticulum. EMBO Rep. 2002, 3, 944–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Maity, S. ER Stress-Sensor Proteins and ER-Mitochondrial Crosstalk-Signaling Beyond (ER) Stress Response. Biomolecules 2021, 11, 173. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M. Endoplasmic reticulum stress responses. Cell. Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef]
- Cao, S.S. Epithelial ER Stress in Crohn’s Disease and Ulcerative Colitis. Inflamm. Bowel Dis. 2016, 22, 984–993. [Google Scholar] [CrossRef]
- Wilfling, F.; Haas, J.T.; Walther, T.C.; Farese, R.V., Jr. Lipid droplet biogenesis. Curr. Opin. Cell Biol. 2014, 29, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Pineau, L.; Colas, J.; Dupont, S.; Beney, L.; Fleurat-Lessard, P.; Berjeaud, J.M.; Berges, T.; Ferreira, T. Lipid-induced ER stress: Synergistic effects of sterols and saturated fatty acids. Traffic 2009, 10, 673–690. [Google Scholar] [CrossRef]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Hou, N.S.; Gutschmidt, A.; Choi, D.Y.; Pather, K.; Shi, X.; Watts, J.L.; Hoppe, T.; Taubert, S. Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E2271–E2280. [Google Scholar] [CrossRef]
- Radanovic, T.; Ernst, R. The Unfolded Protein Response as a Guardian of the Secretory Pathway. Cells 2021, 10, 2965. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Jana, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernandez, E.; Sassano, M.L.; et al. Non-canonical function of IRE1alpha determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell. Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Oikonomou, C.; Hendershot, L.M. Disposing of misfolded ER proteins: A troubled substrate’s way out of the ER. Mol. Cell. Endocrinol. 2020, 500, 110630. [Google Scholar] [CrossRef]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell. Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Klausner, R.D.; Donaldson, J.G.; Lippincott-Schwartz, J. Brefeldin A: Insights into the control of membrane traffic and organelle structure. J. Cell Biol. 1992, 116, 1071–1080. [Google Scholar] [CrossRef]
- Citterio, C.; Vichi, A.; Pacheco-Rodriguez, G.; Aponte, A.M.; Moss, J.; Vaughan, M. Unfolded protein response and cell death after depletion of brefeldin A-inhibited guanine nucleotide-exchange protein GBF1. Proc. Natl. Acad. Sci. USA 2008, 105, 2877–2882. [Google Scholar] [CrossRef]
- Blum, A.; Khalifa, S.; Nordstrom, K.; Simon, M.; Schulz, M.H.; Schmitt, M.J. Transcriptomics of a KDELR1 knockout cell line reveals modulated cell adhesion properties. Sci. Rep. 2019, 9, 10611. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M.J.; Pelham, H.R. Ligand-induced redistribution of a human KDEL receptor from the Golgi complex to the endoplasmic reticulum. Cell 1992, 68, 353–364. [Google Scholar] [CrossRef]
- Jin, H.; Komita, M.; Aoe, T. The Role of BiP Retrieval by the KDEL Receptor in the Early Secretory Pathway and its Effect on Protein Quality Control and Neurodegeneration. Front. Mol. Neurosci. 2017, 10, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadana, R.; Dessauer, C.W. Physiological roles for G protein-regulated adenylyl cyclase isoforms: Insights from knockout and overexpression studies. Neurosignals 2009, 17, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Cela, I.; Dufrusine, B.; Rossi, C.; Luini, A.; De Laurenzi, V.; Federici, L.; Sallese, M. KDEL Receptors: Pathophysiological Functions, Therapeutic Options, and Biotechnological Opportunities. Biomedicines 2022, 10, 1234. [Google Scholar] [CrossRef] [PubMed]
- Giannotta, M.; Ruggiero, C.; Grossi, M.; Cancino, J.; Capitani, M.; Pulvirenti, T.; Consoli, G.M.; Geraci, C.; Fanelli, F.; Luini, A.; et al. The KDEL receptor couples to Galphaq/11 to activate Src kinases and regulate transport through the Golgi. EMBO J. 2012, 31, 2869–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggiero, C.; Grossi, M.; Fragassi, G.; Di Campli, A.; Di Ilio, C.; Luini, A.; Sallese, M. The KDEL receptor signalling cascade targets focal adhesion kinase on focal adhesions and invadopodia. Oncotarget 2018, 9, 10228–10246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancino, J.; Capalbo, A.; Di Campli, A.; Giannotta, M.; Rizzo, R.; Jung, J.E.; Di Martino, R.; Persico, M.; Heinklein, P.; Sallese, M.; et al. Control systems of membrane transport at the interface between the endoplasmic reticulum and the Golgi. Dev. Cell 2014, 30, 280–294. [Google Scholar] [CrossRef] [Green Version]
- Ruggiero, C.; Fragassi, G.; Grossi, M.; Picciani, B.; Di Martino, R.; Capitani, M.; Buccione, R.; Luini, A.; Sallese, M. A Golgi-based KDELR-dependent signalling pathway controls extracellular matrix degradation. Oncotarget 2015, 6, 3375–3393. [Google Scholar] [CrossRef] [Green Version]
- Trychta, K.A.; Back, S.; Henderson, M.J.; Harvey, B.K. KDEL Receptors Are Differentially Regulated to Maintain the ER Proteome under Calcium Deficiency. Cell Rep. 2018, 25, 1829–1840.e6. [Google Scholar] [CrossRef]
- Hamada, H.; Suzuki, M.; Yuasa, S.; Mimura, N.; Shinozuka, N.; Takada, Y.; Suzuki, M.; Nishino, T.; Nakaya, H.; Koseki, H.; et al. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol. Cell. Biol. 2004, 24, 8007–8017. [Google Scholar] [CrossRef] [Green Version]
- Mimura, N.; Hamada, H.; Kashio, M.; Jin, H.; Toyama, Y.; Kimura, K.; Iida, M.; Goto, S.; Saisho, H.; Toshimori, K.; et al. Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 2007, 14, 1475–1485. [Google Scholar] [CrossRef]
- Mesaeli, N.; Nakamura, K.; Zvaritch, E.; Dickie, P.; Dziak, E.; Krause, K.H.; Opas, M.; MacLennan, D.H.; Michalak, M. Calreticulin is essential for cardiac development. J. Cell Biol. 1999, 144, 857–868. [Google Scholar] [CrossRef]
- Luo, S.; Mao, C.; Lee, B.; Lee, A.S. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell. Biol. 2006, 26, 5688–5697. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Reimold, A.M.; Etkin, A.; Clauss, I.; Perkins, A.; Friend, D.S.; Zhang, J.; Horton, H.F.; Scott, A.; Orkin, S.H.; Byrne, M.C.; et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000, 14, 152–157. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [Green Version]
- Luo, K.; Cao, S.S. Endoplasmic reticulum stress in intestinal epithelial cell function and inflammatory bowel disease. Gastroenterol. Res. Pract. 2015, 2015, 328791. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Lee, A.H.; Chu, G.C.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005, 24, 4368–4380. [Google Scholar] [CrossRef] [Green Version]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, S.; Giovanelli, P.; Alvarado-Vazquez, P.A.; Alonso, S.; Song, M.; Sandoval, T.A.; Chae, C.S.; Tan, C.; Fonseca, M.M.; Gutierrez, S.; et al. IRE1alpha-XBP1 signaling in leukocytes controls prostaglandin biosynthesis and pain. Science 2019, 365, eaau6499. [Google Scholar] [CrossRef]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H.; et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 2006, 312, 572–576. [Google Scholar] [CrossRef] [Green Version]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol. Biol. Cell 2011, 22, 4390–4405. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [Green Version]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namba, T.; Tanaka, K.; Ito, Y.; Ishihara, T.; Hoshino, T.; Gotoh, T.; Endo, M.; Sato, K.; Mizushima, T. Positive role of CCAAT/enhancer-binding protein homologous protein, a transcription factor involved in the endoplasmic reticulum stress response in the development of colitis. Am. J. Pathol. 2009, 174, 1786–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.D.; Jousse, C.; Marciniak, S.J.; Zhang, Y.; Novoa, I.; Scheuner, D.; Kaufman, R.J.; Ron, D.; Harding, H.P. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004, 23, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.S.; Song, B.; Kaufman, R.J. PKR protects colonic epithelium against colitis through the unfolded protein response and prosurvival signaling. Inflamm. Bowel Dis. 2012, 18, 1735–1742. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.P.; Kumar, K.U.; Kaufman, R.J. Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J. Biol. Chem. 1998, 273, 2416–2423. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, S.; Kim, C.N.; Yeh, W.C.; Mak, T.W.; Bhalla, K.; Barber, G.N. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J. 1998, 17, 6888–6902. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell. Biol. 2005, 25, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Nadanaka, S.; Okada, T.; Yoshida, H.; Mori, K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell. Biol. 2007, 27, 1027–1043. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, L.; Selim, J.; Genty, D.; Baste, J.M.; Piton, N.; Boukhalfa, I.; Hamzaoui, M.; Pareige, P.; Richard, V. Electron microscopy approach for the visualization of the epithelial and endothelial glycocalyx. Morphologie 2017, 101, 55–63. [Google Scholar] [CrossRef]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef]
- Wan, Y.; Yang, L.; Jiang, S.; Qian, D.; Duan, J. Excessive Apoptosis in Ulcerative Colitis: Crosstalk Between Apoptosis, ROS, ER Stress, and Intestinal Homeostasis. Inflamm. Bowel Dis. 2022, 28, 639–648. [Google Scholar] [CrossRef]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Coleman, O.I.; Haller, D. ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front. Immunol. 2019, 10, 2825. [Google Scholar] [CrossRef]
- Okai, S.; Usui, F.; Yokota, S.; Hori, I.Y.; Hasegawa, M.; Nakamura, T.; Kurosawa, M.; Okada, S.; Yamamoto, K.; Nishiyama, E.; et al. High-affinity monoclonal IgA regulates gut microbiota and prevents colitis in mice. Nat. Microbiol. 2016, 1, 16103. [Google Scholar] [CrossRef]
- Cornick, S.; Tawiah, A.; Chadee, K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers 2015, 3, e982426. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti, A.; Wang, X.; Novoa, I.; Jungreis, R.; Schlessinger, K.; Cho, J.H.; West, A.B.; Ron, D. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J. Clin. Investig. 2001, 107, 585–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, H.S.; Tortori, C.J.; Castelo-Branco, M.T.; Carvalho, A.T.; Margallo, V.S.; Delgado, C.F.; Dines, I.; Elia, C.C. Apoptosis in the intestinal mucosa of patients with inflammatory bowel disease: Evidence of altered expression of FasL and perforin cytotoxic pathways. Int. J. Color. Dis. 2005, 20, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Ma, C.; Su, F.; Jiang, Y.; Lai, R.; Zhang, T.; Sun, K.; Fan, L.; Cai, Z.; Li, Z.; et al. Raf kinase inhibitor protein mediates intestinal epithelial cell apoptosis and promotes IBDs in humans and mice. Gut 2017, 66, 597–610. [Google Scholar] [CrossRef]
- Qiu, W.; Wu, B.; Wang, X.; Buchanan, M.E.; Regueiro, M.D.; Hartman, D.J.; Schoen, R.E.; Yu, J.; Zhang, L. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J. Clin. Investig. 2011, 121, 1722–1732. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto-Furusho, J.K.; Fonseca-Camarillo, G.; Furuzawa-Carballeda, J.; Sarmiento-Aguilar, A.; Barreto-Zuniga, R.; Martinez-Benitez, B.; Lara-Velazquez, M.A. Caspase recruitment domain (CARD) family (CARD9, CARD10, CARD11, CARD14 and CARD15) are increased during active inflammation in patients with inflammatory bowel disease. J. Inflamm. 2018, 15, 13. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Zhang, L.; Wang, Y.; He, X. Mechanisms of HuR in regulation of epithelial cell apoptosis in rat ulcerative colitis. Cell. Signal. 2021, 82, 109957. [Google Scholar] [CrossRef]
- McGuckin, M.A.; Eri, R.D.; Das, I.; Lourie, R.; Florin, T.H. Intestinal secretory cell ER stress and inflammation. Biochem. Soc. Trans. 2011, 39, 1081–1085. [Google Scholar] [CrossRef]
- Tsuru, A.; Fujimoto, N.; Takahashi, S.; Saito, M.; Nakamura, D.; Iwano, M.; Iwawaki, T.; Kadokura, H.; Ron, D.; Kohno, K. Negative feedback by IRE1beta optimizes mucin production in goblet cells. Proc. Natl. Acad. Sci. USA 2013, 110, 2864–2869. [Google Scholar] [CrossRef]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.; Price, G.R.; Tauro, S.B.; Taupin, D.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008, 5, e54. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cheng, J.; Shen, J.; Wang, S.; Guo, C.; Fan, X. Ghrelin Inhibits Intestinal Epithelial Cell Apoptosis Through the Unfolded Protein Response Pathway in Ulcerative Colitis. Front. Pharmacol. 2021, 12, 661853. [Google Scholar] [CrossRef]
- Fukata, M.; Chen, A.; Vamadevan, A.S.; Cohen, J.; Breglio, K.; Krishnareddy, S.; Hsu, D.; Xu, R.; Harpaz, N.; Dannenberg, A.J.; et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 2007, 133, 1869–1869.E14. [Google Scholar] [CrossRef] [Green Version]
- Fukata, M.; Shang, L.; Santaolalla, R.; Sotolongo, J.; Pastorini, C.; Espana, C.; Ungaro, R.; Harpaz, N.; Cooper, H.S.; Elson, G.; et al. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm. Bowel Dis. 2011, 17, 1464–1473. [Google Scholar] [CrossRef]
- Guo, J.; Liao, M.; Wang, J. TLR4 signaling in the development of colitis-associated cancer and its possible interplay with microRNA-155. Cell Commun. Signal. 2021, 19, 90. [Google Scholar] [CrossRef]
- Rees, W.D.; Stahl, M.; Jacobson, K.; Bressler, B.; Sly, L.M.; Vallance, B.A.; Steiner, T.S. Enteroids Derived From Inflammatory Bowel Disease Patients Display Dysregulated Endoplasmic Reticulum Stress Pathways, Leading to Differential Inflammatory Responses and Dendritic Cell Maturation. J. Crohn’s Colitis 2020, 14, 948–961. [Google Scholar] [CrossRef]
- Hasnain, S.Z.; Tauro, S.; Das, I.; Tong, H.; Chen, A.C.; Jeffery, P.L.; McDonald, V.; Florin, T.H.; McGuckin, M.A. IL-10 promotes production of intestinal mucus by suppressing protein misfolding and endoplasmic reticulum stress in goblet cells. Gastroenterology 2013, 144, 357–368.e9. [Google Scholar] [CrossRef]
- Birchenough, G.M.; Nystrom, E.E.; Johansson, M.E.; Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Vaishnava, S.; Behrendt, C.L.; Ismail, A.S.; Eckmann, L.; Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 2008, 105, 20858–20863. [Google Scholar] [CrossRef]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef] [Green Version]
- Andrews, C.; McLean, M.H.; Durum, S.K. Cytokine Tuning of Intestinal Epithelial Function. Front. Immunol. 2018, 9, 1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechuga, S.; Braga-Neto, M.B.; Naydenov, N.G.; Rieder, F.; Ivanov, A.I. Understanding disruption of the gut barrier during inflammation: Should we abandon traditional epithelial cell lines and switch to intestinal organoids? Front. Immunol. 2023, 14, 1108289. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, R.; Tsuchiya, K.; Nemoto, Y.; Akiyama, J.; Nakamura, T.; Kanai, T.; Watanabe, M. Requirement of Notch activation during regeneration of the intestinal epithelia. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G23–G35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velcich, A.; Yang, W.; Heyer, J.; Fragale, A.; Nicholas, C.; Viani, S.; Kucherlapati, R.; Lipkin, M.; Yang, K.; Augenlicht, L. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 2002, 295, 1726–1729. [Google Scholar] [CrossRef]
- An, G.; Wei, B.; Xia, B.; McDaniel, J.M.; Ju, T.; Cummings, R.D.; Braun, J.; Xia, L. Increased susceptibility to colitis and colorectal tumors in mice lacking core 3-derived O-glycans. J. Exp. Med. 2007, 204, 1417–1429. [Google Scholar] [CrossRef]
- Dawson, P.A.; Huxley, S.; Gardiner, B.; Tran, T.; McAuley, J.L.; Grimmond, S.; McGuckin, M.A.; Markovich, D. Reduced mucin sulfonation and impaired intestinal barrier function in the hyposulfataemic NaS1 null mouse. Gut 2009, 58, 910–919. [Google Scholar] [CrossRef]
- Park, S.W.; Zhen, G.; Verhaeghe, C.; Nakagami, Y.; Nguyenvu, L.T.; Barczak, A.J.; Killeen, N.; Erle, D.J. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc. Natl. Acad. Sci. USA 2009, 106, 6950–6955. [Google Scholar] [CrossRef]
- Wei, X.; Yang, Z.; Rey, F.E.; Ridaura, V.K.; Davidson, N.O.; Gordon, J.I.; Semenkovich, C.F. Fatty acid synthase modulates intestinal barrier function through palmitoylation of mucin 2. Cell Host Microbe. 2012, 11, 140–152. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.K.; Miyoshi, H.; Beatty, W.L.; Head, R.D.; Malvin, N.P.; Cadwell, K.; Guan, J.L.; Saitoh, T.; Akira, S.; Seglen, P.O.; et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J. 2013, 32, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12 (Suppl. 2), 1528–1534. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.S.; Zimmermann, E.M.; Chuang, B.M.; Song, B.; Nwokoye, A.; Wilkinson, J.E.; Eaton, K.A.; Kaufman, R.J. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 2013, 144, 989–1000.e6. [Google Scholar] [CrossRef] [Green Version]
- Kalvakolanu, D.V.; Gade, P. IFNG and autophagy: A critical role for the ER-stress mediator ATF6 in controlling bacterial infections. Autophagy 2012, 8, 1673–1674. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Upton, J.P.; Hagen, A.; Callahan, J.; Oakes, S.A.; Papa, F.R. A kinase inhibitor activates the IRE1alpha RNase to confer cytoprotection against ER stress. Biochem. Biophys. Res. Commun. 2008, 365, 777–783. [Google Scholar] [CrossRef]
- Wang, L.; Perera, B.G.; Hari, S.B.; Bhhatarai, B.; Backes, B.J.; Seeliger, M.A.; Schurer, S.C.; Oakes, S.A.; Papa, F.R.; Maly, D.J. Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 2012, 8, 982–989. [Google Scholar] [CrossRef]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef]
- Voorhees, P.M.; Orlowski, R.Z. The proteasome and proteasome inhibitors in cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 189–213. [Google Scholar] [CrossRef]
- Deka, D.; D’Inca, R.; Sturniolo, G.C.; Das, A.; Pathak, S.; Banerjee, A. Role of ER Stress Mediated Unfolded Protein Responses and ER Stress Inhibitors in the Pathogenesis of Inflammatory Bowel Disease. Dig. Dis. Sci. 2022, 67, 5392–5406. [Google Scholar] [CrossRef]
- Crespo, I.; San-Miguel, B.; Prause, C.; Marroni, N.; Cuevas, M.J.; Gonzalez-Gallego, J.; Tunon, M.J. Glutamine treatment attenuates endoplasmic reticulum stress and apoptosis in TNBS-induced colitis. PLoS ONE 2012, 7, e50407. [Google Scholar] [CrossRef]
- Okazaki, T.; Nishio, A.; Takeo, M.; Sakaguchi, Y.; Fukui, T.; Uchida, K.; Okazaki, K. Inhibition of the dephosphorylation of eukaryotic initiation factor 2alpha ameliorates murine experimental colitis. Digestion 2014, 90, 167–178. [Google Scholar] [CrossRef]
- Yin, S.; Yang, H.; Tao, Y.; Wei, S.; Li, L.; Liu, M.; Li, J. Artesunate ameliorates DSS-induced ulcerative colitis by protecting intestinal barrier and inhibiting inflammatory response. Inflammation 2020, 43, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Li, L.; Tao, Y.; Yu, J.; Wei, S.; Liu, M.; Li, J. The Inhibitory Effect of Artesunate on Excessive Endoplasmic Reticulum Stress Alleviates Experimental Colitis in Mice. Front. Pharmacol. 2021, 12, 629798. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, Z.; Liu, K.; Liu, J.; Chai, S.; Chen, G.; Wen, S.; Ming, T.; Wang, J.; Ma, Y.; et al. Activation of the G Protein-Coupled Estrogen Receptor Prevented the Development of Acute Colitis by Protecting the Crypt Cell. J. Pharmacol. Exp. Ther. 2021, 376, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, Z.; Song, G.; Tang, X.; Song, H.; Deng, A.; Wang, W.; Wu, L.; Qin, H. Development of an XBP1 agonist, HLJ2, as a potential therapeutic agent for ulcerative colitis. Eur. J. Pharm. Sci. 2017, 109, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, K.J.; Royal, J.M.; Kouokam, J.C.; Haribabu, B.; Jala, V.R.; Yaddanapudi, K.; Hamorsky, K.T.; Dryden, G.W.; Matoba, N. Oral administration of a recombinant cholera toxin B subunit promotes mucosal healing in the colon. Mucosal Immunol. 2017, 10, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Royal, J.M.; Oh, Y.J.; Grey, M.J.; Lencer, W.I.; Ronquillo, N.; Galandiuk, S.; Matoba, N. A modified cholera toxin B subunit containing an ER retention motif enhances colon epithelial repair via an unfolded protein response. FASEB J. 2019, 33, 13527–13545. [Google Scholar] [CrossRef] [Green Version]
- Koutsoumpli, G.; Ip, P.P.; Schepel, I.; Hoogeboom, B.N.; Boerma, A.; Daemen, T. Alphavirus-based hepatitis C virus therapeutic vaccines: Can universal helper epitopes enhance HCV-specific cytotoxic T lymphocyte responses? Ther. Adv. Vaccines Immunother. 2019, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Puente, D.H.; Perez-Trujillo, J.J.; Gutierrez-Puente, Y.; Rodriguez-Rocha, H.; Garcia-Garcia, A.; Saucedo-Cardenas, O.; Montes-de-Oca-Luna, R.; Loera-Arias, M.J. Targeting HPV-16 antigens to the endoplasmic reticulum induces an endoplasmic reticulum stress response. Cell Stress Chaperones 2019, 24, 149–158. [Google Scholar] [CrossRef]
- Hamorsky, K.T.; Kouokam, J.C.; Bennett, L.J.; Baldauf, K.J.; Kajiura, H.; Fujiyama, K.; Matoba, N. Rapid and scalable plant-based production of a cholera toxin B subunit variant to aid in mass vaccination against cholera outbreaks. PLoS Neglected Trop Dis. 2013, 7, e2046. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.A.; Jobling, M.G.; Saslowsky, D.E.; Kern, E.; Drake, K.R.; Kenworthy, A.K.; Holmes, R.K.; Lencer, W.I. Attenuated endocytosis and toxicity of a mutant cholera toxin with decreased ability to cluster ganglioside GM1 molecules. Infect. Immun. 2008, 76, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Jobling, M.G.; Yang, Z.; Kam, W.R.; Lencer, W.I.; Holmes, R.K. A single native ganglioside GM1-binding site is sufficient for cholera toxin to bind to cells and complete the intoxication pathway. mBio 2012, 3, e00401-12. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Yue, X.; Zhu, L.; Jing, S.; Wang, Y.; Gim, B.; Qian, Y.; Lee, I. KDEL receptor is a cell surface receptor that cycles between the plasma membrane and the Golgi via clathrin-mediated transport carriers. Cell. Mol. Life Sci. 2021, 78, 1085–1100. [Google Scholar] [CrossRef]
- Beck, P.L.; Rosenberg, I.M.; Xavier, R.J.; Koh, T.; Wong, J.F.; Podolsky, D.K. Transforming growth factor-beta mediates intestinal healing and susceptibility to injury in vitro and in vivo through epithelial cells. Am. J. Pathol. 2003, 162, 597–608. [Google Scholar] [CrossRef]
- Royal, J.M.; Reeves, M.A.; Matoba, N. Repeated Oral Administration of a KDEL-tagged Recombinant Cholera Toxin B Subunit Effectively Mitigates DSS Colitis Despite a Robust Immunogenic Response. Toxins 2019, 11, 678. [Google Scholar] [CrossRef] [Green Version]
- Verjan Garcia, N.; Santisteban Celis, I.C.; Dent, M.; Matoba, N. Characterization and utility of two monoclonal antibodies to cholera toxin B subunit. Sci. Rep. 2023, 13, 4305. [Google Scholar] [CrossRef]
- Yang, J.; Liu, H.; Li, L.; Liu, H.; Shi, W.; Yuan, X.; Wu, L. Structural Insights into IRE1 Functions in the Unfolded Protein Response. Curr. Med. Chem. 2016, 23, 4706–4716. [Google Scholar] [CrossRef]
- Lam, M.; Marsters, S.A.; Ashkenazi, A.; Walter, P. Misfolded proteins bind and activate death receptor 5 to trigger apoptosis during unresolved endoplasmic reticulum stress. Elife 2020, 9, e52291. [Google Scholar] [CrossRef]
- Zhang, Z.; Venditti, R.; Ran, L.; Liu, Z.; Vivot, K.; Schurmann, A.; Bonifacino, J.S.; De Matteis, M.A.; Ricci, R. Distinct changes in endosomal composition promote NLRP3 inflammasome activation. Nat. Immunol. 2023, 24, 30–41. [Google Scholar] [CrossRef]

{kind=link}
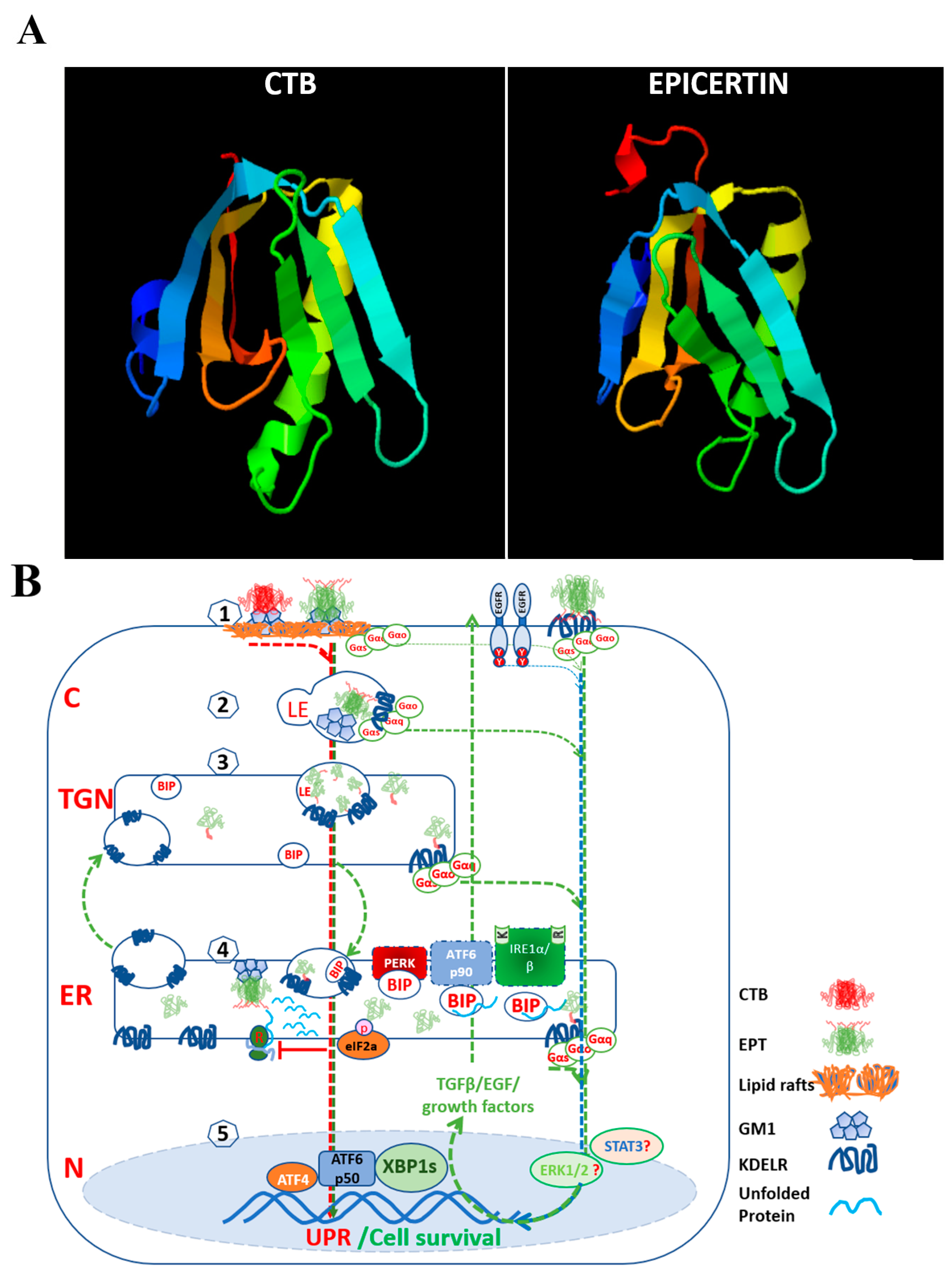{kind=link}
| In Vivo/In Vitro Model | ER Stress Inducer | Targeted UPR Sensor | Drug Candidate | Mode of Action | References |
|---|---|---|---|---|---|
| Mouse model of DSS-induced colitis. P58 (IPK−/−), ATF6a−/−, IL10−/− mice, and IEC-6 cells | DSS-induced colitis/cytokine cocktail (TNF-α, MCP-1, IL-1β) | Induction of ATF6α- and chaperone P58; reduce ER stress | Phenyl butyric acid (PBA), Taurine-conjugated ursodeoxycholic acid (TUDCA) | Promote protein folding; reduce features of acute and chronic colitis; reduce BiP, peIF2a, CHOP, and cleaved caspase 3/12. | Cao et al., 2013 [101] |
| Rat model of TNBS-induced colitis; Caco2 cells | TNBS-induced colitis. Brefeldin A and Tunicamyin-induced ER stress | PERK-CHOP, ATF4, ATF6, XBP1s | Glutamine | Reduce CHOP, BiP, Caspases 12, 9, 8, 3. calpain-1, and pJNK. | Crespo et al., 2012 [109] |
| Mouse model of DSS-induced colitis | DSS-induced colitis | Phosphatase inhibitor Increase eIF2α phosphorylation | Salubrinal | Increase BiP, ATF4, and HSP70; reduce CHOP; suppress MPO, TNF-α, and IL-1β. | Okazaki et al., 2014 [110] |
| Mouse model of DSS-induced colitis | DSS-induced colitis. ER stress | Prevents activation of PERK-eIF2α-ATF4-CHOP and IRE1α-XBP1 signaling pathways | Artesunate | Increase Bcl-2/Bax ratio; suppress NF-κB p65 and IκBα, IL-1β, IL-6, and TNF-α; increase IL-10; inhibit expression of cleaved-caspase 3. | Yin et al., 2020, 2021 [111,112] |
| Mouse model of DSS-induced colitis; CCD841 cell line | DSS induced colon ER stress/Thapsigargin | Inhibited PERK, IRE1α, ATF6, reduced BiP, CHOP | G-1 | Inhibit apoptosis, increased Ki-67+, and BrdU+ crypt cells; reverse the decrease in cyclin D1 and B1. | Wang et al., 2021 [113] |
| Mouse model of DSS colitis model | DSS-induced colitis. ER stress | XBP1 agonist, Coptisine-derivative | HLJ2 | Decrease MPO, TNFα, IL-1β, and IL-6; increase the expression of ZO-1 and claudin-1. | Zhang et al., 2017 [114] |
| Mouse model of DSS-induced acute and chronic colitis. Caco2 cells scratch assay | DSS-induced ER stress | Induce IRE1-XBP1s axis. | EPICERTIN | Binds to KDELRs and may promote vesicle trafficking; promotes mucosal wound healing mediated by TGFβ, CDH1, and WNT5. | Baldauf et al., 2017 [115]; Royal et al., 2019 [116] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verjan Garcia, N.; Hong, K.U.; Matoba, N. The Unfolded Protein Response and Its Implications for Novel Therapeutic Strategies in Inflammatory Bowel Disease. Biomedicines 2023, 11, 2066. https://doi.org/10.3390/biomedicines11072066
Verjan Garcia N, Hong KU, Matoba N. The Unfolded Protein Response and Its Implications for Novel Therapeutic Strategies in Inflammatory Bowel Disease. Biomedicines. 2023; 11(7):2066. https://doi.org/10.3390/biomedicines11072066
Chicago/Turabian StyleVerjan Garcia, Noel, Kyung U. Hong, and Nobuyuki Matoba. 2023. "The Unfolded Protein Response and Its Implications for Novel Therapeutic Strategies in Inflammatory Bowel Disease" Biomedicines 11, no. 7: 2066. https://doi.org/10.3390/biomedicines11072066