
Niclosamide Attenuates Inflammation-Associated Profibrotic Responses in Human Subepithelial Lung Myofibroblasts
 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Chemicals
2.3. Subepithelial Lung Myofibroblast Isolation
2.4. Subepithelial Lung Myofibroblast Culture
2.5. Wound-Healing Assay
2.6. Collagen Production
2.7. Immunofluorescence
2.8. Human Fibronectin Enzyme-Linked Immunosorbent Assay
2.9. Caspase-3 Activity Assay
2.10. Total RNA Isolation and Purification
2.11. cDNA Synthesis and Real-Time RT-PCR

{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer | Reference |
|---|---|---|---|
| GAPDH | GACATCAAGAAGGTGGTGAA | TGTCATACCAGGAAATGAGC | [27] |
| Collagen Type I (COL1) | CCCTGGAAAGAATGGAGATGAT | ACTGAAACCTCTGTGTCCCTTCA | |
| Collagen Type III (COL3) | GCTCTGCTTCATCCCACTATTA | TGCGAGTCCTCCTACTGCTAC | |
| Fibronectin (FN) | CCAGTCCACAGCTATTCCTG | ACAACCACGGATGAGCTG | |
| α-sma (ACTA-2) | AATGCAGAAGGAGATCACGG | TCCTGTTTGCTGATCCACATC | |
| CD90 | CGCTCTCCTGCTAACAGTCTT | CAGGCTGAACTCGTACTGGA | [30] |
2.12. Statistics
3. Results
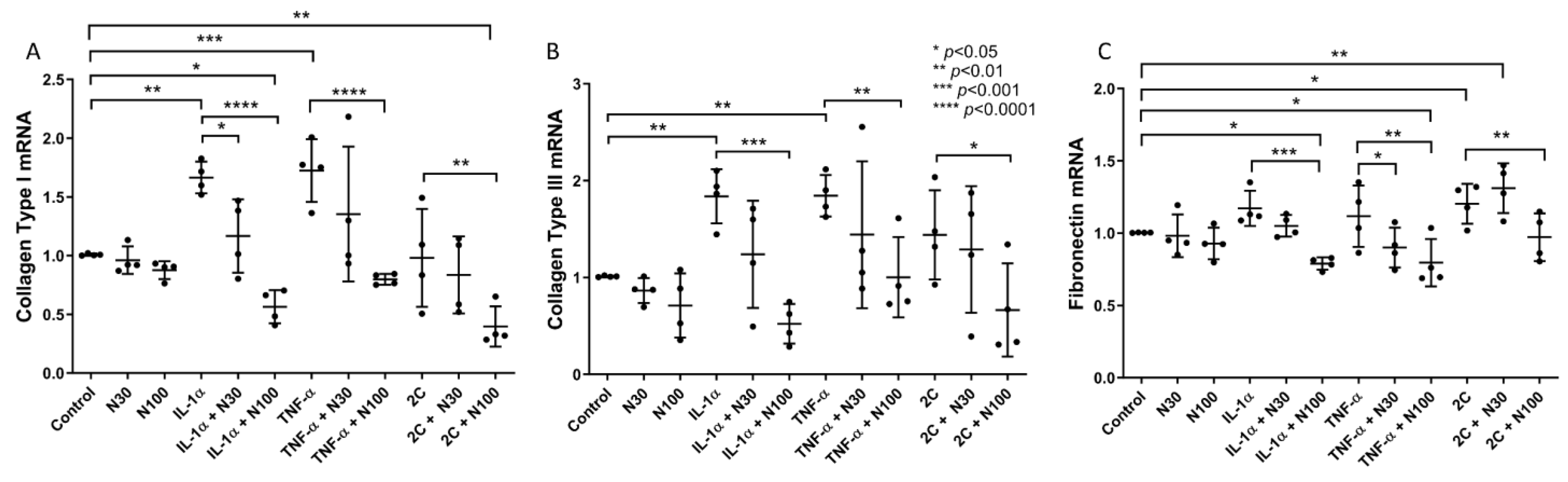
3.1. Niclosamide Treatment Attenuates SELM Fibrotic mRNA Expression in Response to Inflammatory Stimuli
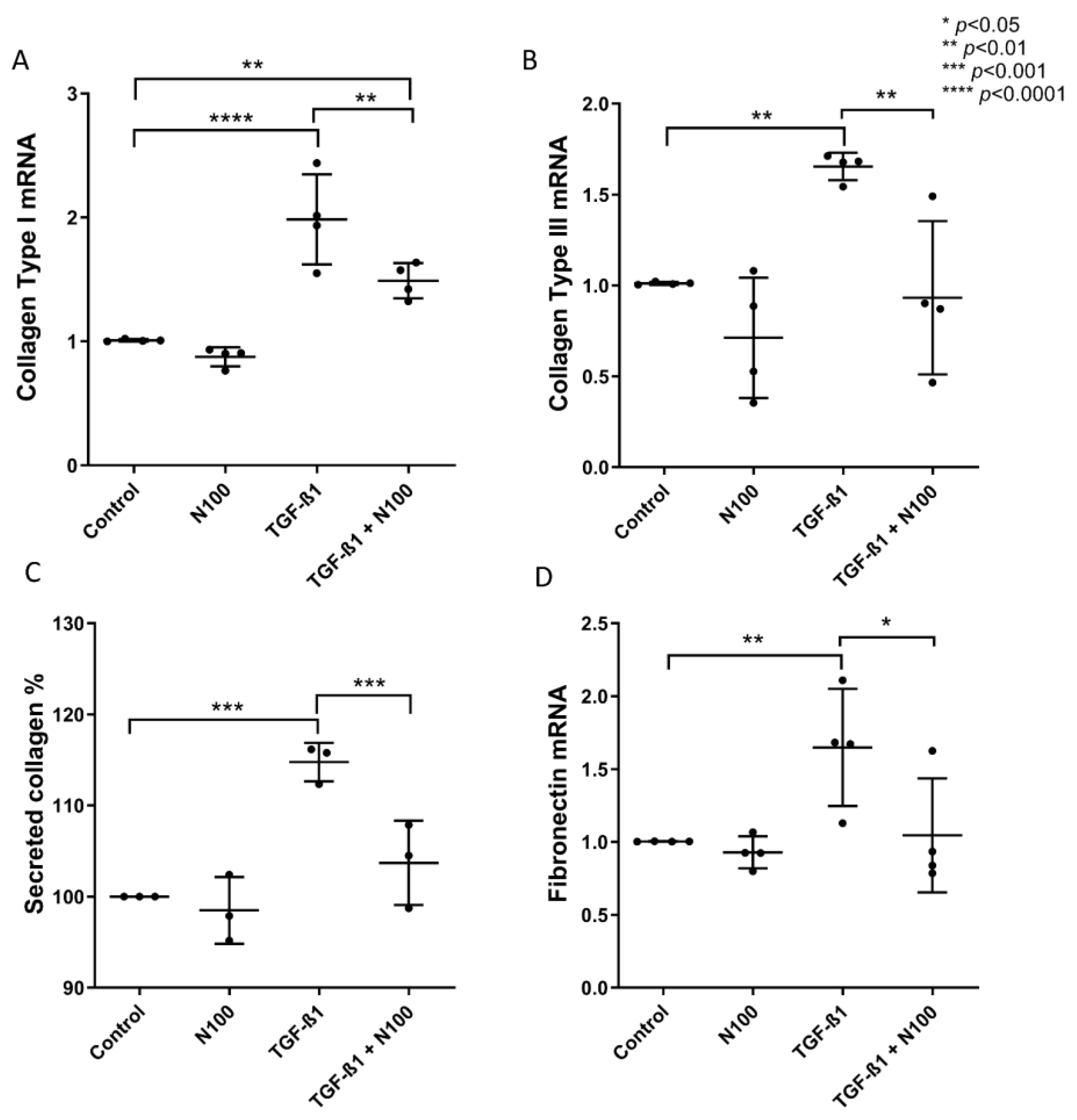
3.2. Niclosamide Treatment Attenuates SELM Fibrotic Expression in Response to Fibrotic Stimuli
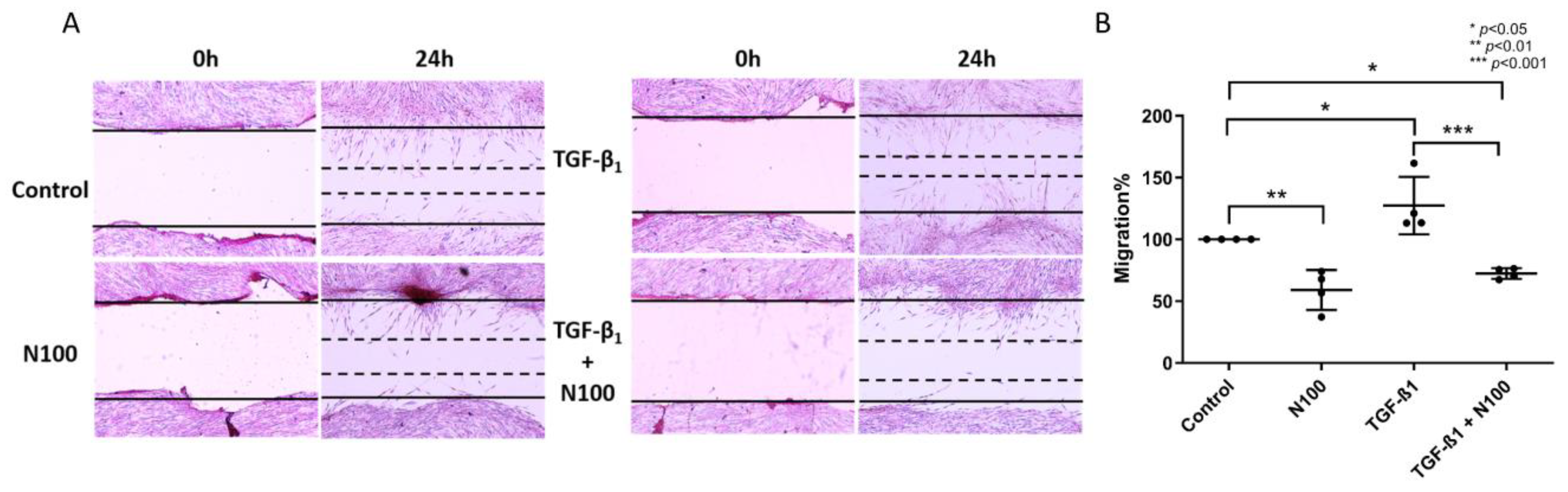
3.3. Niclosamide Inhibits SELM Migration
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meyer, K.C. Pulmonary Fibrosis, Part I: Epidemiology, Pathogenesis, and Diagnosis. Expert Rev. Respir. Med. 2017, 11, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Distler, O.; Ryerson, C.J.; Tzouvelekis, A.; Lee, J.S.; Bonella, F.; Bouros, D.; Hoffmann-Vold, A.M.; Crestani, B.; Matteson, E.L. Mechanisms of Progressive Fibrosis in Connective Tissue Disease (CTD)-Associated Interstitial Lung Diseases (ILDs). Ann. Rheum. Dis. 2021, 80, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From Mechanisms to Medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic Pulmonary Fibrosis: Pathogenesis and Management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic Pulmonary Fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Ryu, J.H.; Moua, T.; Daniels, C.E.; Hartman, T.E.; Yi, E.S.; Utz, J.P.; Limper, A.H. Idiopathic Pulmonary Fibrosis: Evolving Concepts. Mayo Clin. Proc. 2014, 89, 1130–1142. [Google Scholar] [CrossRef]
- Bringardner, B.D.; Baran, C.P.; Eubank, T.D.; Marsh, C.B. The Role of Inflammation in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Antioxid. Redox Signal. 2008, 10, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Bolourani, S.; Brenner, M.; Wang, P. The Interplay of DAMPs, TLR4, and Proinflammatory Cytokines in Pulmonary Fibrosis. J. Mol. Med. 2021, 99, 1373–1384. [Google Scholar] [CrossRef]
- Terlizzi, M.; Molino, A.; Colarusso, C.; Donovan, C.; Imitazione, P.; Somma, P.; Aquino, R.P.; Hansbro, P.M.; Pinto, A.; Sorrentino, R. Activation of the Absent in Melanoma 2 Inflammasome in Peripheral Blood Mononuclear Cells from Idiopathic Pulmonary Fibrosis Patients Leads to the Release of Pro-Fibrotic Mediators. Front. Immunol. 2018, 9, 670. [Google Scholar] [CrossRef] [Green Version]
- Nigdelioglu, R.; Hamanaka, R.B.; Meliton, A.Y.; O’Leary, E.; Witt, L.J.; Cho, T.; Sun, K.; Bonham, C.; Wu, D.; Woods, P.S.; et al. Transforming Growth Factor (TGF)-β Promotes de Novo Serine Synthesis for Collagen Production. J. Biol. Chem. 2016, 291, 27239–27251. [Google Scholar] [CrossRef] [Green Version]
- Pilling, D.; Vakil, V.; Cox, N.; Gomer, R.H.; Herzog, E. TNF-α-Stimulated Fibroblasts Secrete Lumican to Promote Fibrocyte Differentiation. Proc. Natl. Acad. Sci. USA 2015, 112, 11929–11934. [Google Scholar] [CrossRef]
- Kadri, H.; Lambourne, O.A.; Mehellou, Y. Niclosamide, a Drug with Many (Re)Purposes. ChemMedChem 2018, 13, 1088–1091. [Google Scholar] [CrossRef]
- Chen, W.; Mook, R.A.; Premont, R.T.; Wang, J. Niclosamide: Beyond an Antihelminthic Drug. Cell Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef]
- Barbosa, E.J.; Löbenberg, R.; de Araujo, G.L.B.; Bou-Chacra, N.A. Niclosamide Repositioning for Treating Cancer: Challenges and Nano-Based Drug Delivery Opportunities. Eur. J. Pharm. Biopharm. 2019, 141, 58–69. [Google Scholar] [CrossRef]
- Xu, J.; Shi, P.Y.; Li, H.; Zhou, J. Broad Spectrum Antiviral Agent Niclosamide and Its Therapeutic Potential. ACS Infect. Dis. 2020, 6, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Cabrita, I.; Benedetto, R.; Schreiber, R.; Kunzelmann, K. Niclosamide Repurposed for the Treatment of Inflammatory Airway Disease. JCI Insight 2019, 4, e128414. [Google Scholar] [CrossRef]
- Guo, J.; Tao, H.; Alasadi, A.; Huang, Q.; Jin, S. Niclosamide Piperazine Prevents High-Fat Diet-Induced Obesity and Diabetic Symptoms in Mice. Eat. Weight Disord. 2019, 24, 91–96. [Google Scholar] [CrossRef]
- Esmail, M.M.; Saeed, N.M.; Michel, H.E.; El-Naga, R.N. The Ameliorative Effect of Niclosamide on Bile Duct Ligation Induced Liver Fibrosis via Suppression of NOTCH and Wnt Pathways. Toxicol. Lett. 2021, 347, 23–35. [Google Scholar] [CrossRef]
- Chang, X.; Zhen, X.; Liu, J.; Ren, X.; Hu, Z.; Zhou, Z.; Zhu, F.; Ding, K.; Nie, J. The Antihelmenthic Phosphate Niclosamide Impedes Renal Fibrosis by Inhibiting Homeodomain-Interacting Protein Kinase 2 Expression. Kidney Int. 2017, 92, 612–624. [Google Scholar] [CrossRef]
- Milani, M.; Mammarella, E.; Rossi, S.; Miele, C.; Lattante, S.; Sabatelli, M.; Cozzolino, M.; D’Ambrosi, N.; Apolloni, S. Targeting S100A4 with Niclosamide Attenuates Inflammatory and Profibrotic Pathways in Models of Amyotrophic Lateral Sclerosis. J. Neuroinflamm. 2021, 18, 132. [Google Scholar] [CrossRef]
- Morin, F.; Kavian, N.; Nicco, C.; Cerles, O.; Chéreau, C.; Batteux, F. Improvement of Sclerodermatous Graft-Versus-Host Disease in Mice by Niclosamide. J. Investig. Dermatol. 2016, 136, 2158–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karatzas, E.; Bourdakou, M.M.; Kolios, G.; Spyrou, G.M. Drug Repurposing in Idiopathic Pulmonary Fibrosis Filtered by a Bioinformatics-Derived Composite Score. Sci. Rep. 2017, 7, 12569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Ohno, S.; Steer, B.; Klee, S.; Staab-Weijnitz, C.A.; Wagner, D.; Lehmann, M.; Stoeger, T.; Königshoff, M.; Adler, H. S100a4 Is Secreted by Alternatively Activated Alveolar Macrophages and Promotes Activation of Lung Fibroblasts in Pulmonary Fibrosis. Front. Immunol. 2018, 9, 1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyapally, R.; Pulivendala, G.; Bale, S.; Godugu, C. Niclosamide Alleviates Pulmonary Fibrosis in Vitro and in Vivo by Attenuation of Epithelial-to-Mesenchymal Transition, Matrix Proteins & Wnt/β-Catenin Signaling: A Drug Repurposing Study. Life Sci. 2019, 220, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Bouros, E.; Filidou, E.; Arvanitidis, K.; Mikroulis, D.; Steiropoulos, P.; Bamias, G.; Bouros, D.; Kolios, G. Lung Fibrosis-Associated Soluble Mediators and Bronchoalveolar Lavage from Idiopathic Pulmonary Fibrosis Patients Promote the Expression of Fibrogenic Factors in Subepithelial Lung Myofibroblasts. Pulm. Pharmacol. Ther. 2017, 46, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Drygiannakis, I.; Valatas, V.; Sfakianaki, O.; Bourikas, L.; Manousou, P.; Kambas, K.; Ritis, K.; Kolios, G.; Kouroumalis, E. Proinflammatory Cytokines Induce Crosstalk between Colonic Epithelial Cells and Subepithelial Myofibroblasts: Implication in Intestinal Fibrosis. J. Crohns Colitis 2013, 7, 286–300. [Google Scholar] [CrossRef] [Green Version]
- Filidou, E.; Valatas, V.; Drygiannakis, I.; Arvanitidis, K.; Vradelis, S.; Kouklakis, G.; Kolios, G.; Bamias, G. Cytokine Receptor Profiling in Human Colonic Subepithelial Myofibroblasts: A Differential Effect of Th Polarization-Associated Cytokines in Intestinal Fibrosis. Inflamm. Bowel Dis. 2018, 24, 2224–2241. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of Gene Expression and Chemoresistance of CD133+ Cancer Stem Cells in Glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Filidou, E.; Kandilogiannakis, L.; Tarapatzi, G.; Spathakis, M.; Steiropoulos, P.; Mikroulis, D.; Arvanitidis, K.; Paspaliaris, V.; Kolios, G. Anti-Inflammatory and Anti-Fibrotic Effect of Immortalized Mesenchymal-Stem-Cell-Derived Conditioned Medium on Human Lung Myofibroblasts and Epithelial Cells. Int. J. Mol. Sci. 2022, 23, 4570. [Google Scholar] [CrossRef]
- Baron, R.M.; Choi, A.J.S.; Owen, C.A.; Choi, A.M.K. Genetically Manipulated Mouse Models of Lung Disease: Potential and Pitfalls. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 157–179. [Google Scholar] [CrossRef] [Green Version]
- Heukels, P.; Moor, C.C.; von der Thüsen, J.H.; Wijsenbeek, M.S.; Kool, M. Inflammation and Immunity in IPF Pathogenesis and Treatment. Respir. Med. 2019, 147, 79–91. [Google Scholar] [CrossRef]
- Kinoshita, T.; Goto, T. Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review. Int. J. Mol. Sci. 2019, 20, 1461. [Google Scholar] [CrossRef] [Green Version]
- Klingberg, F.; Hinz, B.; White, E.S. The Myofibroblast Matrix: Implications for Tissue Repair Andfibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Philp, A.M.; Corte, T.; Travis, M.A.; Schilter, H.; Hansbro, N.G.; Burns, C.J.; Eapen, M.S.; Sohal, S.S.; Burgess, J.K.; et al. Therapeutic Targets in Lung Tissue Remodelling and Fibrosis. Pharmacol. Ther. 2021, 225, 107839. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xiao, L.; Sun, L.; Liu, F. Wnt/β-Catenin Signaling: A Promising New Target for Fibrosis Diseases. Physiol. Res. 2012, 61, 337–346. [Google Scholar] [CrossRef]
- Königshoff, M.; Balsara, N.; Pfaff, E.M.; Kramer, M.; Chrobak, I.; Seeger, W.; Eickelberg, O. Functional Wnt Signaling Is Increased in Idiopathic Pulmonary Fibrosis. PLoS ONE 2008, 3, e2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Ashmawy, N.E.; Al-Ashmawy, G.M.; Fakher, H.E.; Khedr, N.F. The Role of WNT/β-Catenin Signaling Pathway and Glutamine Metabolism in the Pathogenesis of CCl4-Induced Liver Fibrosis: Repositioning of Niclosamide and Concerns about Lithium. Cytokine 2020, 136, 155250. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.-S.; Xin, H.-R.; Qiu, R.-L.; Deng, Z.-L.; Deng, F.; Yan, Z.-J. Niclosamide: Drug Repurposing for Human Chondrosarcoma Treatment via the Caspase-Dependent Mitochondrial Apoptotic Pathway. Am. J. Transl. Res. 2020, 12, 3688–3701. [Google Scholar]
- Lam, S.K.; Yan, S.; Lam, J.S.M.; Feng, Y.; Khan, M.; Chen, C.; Ko, F.C.F.; Ho, J.C.M. Disturbance of the Warburg Effect by Dichloroacetate and Niclosamide Suppresses the Growth of Different Sub-Types of Malignant Pleural Mesothelioma in Vitro and in Vivo. Front. Pharmacol. 2022, 13, 1020343. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, T.H.; Lee, K.; Kang, M.A.; Jang, H.J.; Ryu, H.W.; Oh, S.R.; Lee, H.J. Kurarinone Attenuates Blm-Induced Pulmonary Fibrosis via Inhibiting Tgf-b Signaling Pathways. Int. J. Mol. Sci. 2021, 22, 8388. [Google Scholar] [CrossRef]
- Wang, X.; Song, W.; Zhang, F.; Huang, R. Dihydroartemisinin Inhibits Tgf-β-Induced Fibrosis in Human Tenon Fibroblasts via Inducing Autophagy. Drug. Des. Devel Ther. 2021, 15, 973–981. [Google Scholar] [CrossRef]
- Ba, Y.-D.; Sun, J.-H.; Zhao, X.-X. Evogliptin Attenuates Bleomycin-Induced Lung Fibrosis via Inhibiting TGF-β/Smad Signaling in Fibroblast. Eur. Rev. Med. Pharmacol. Sci. 2020, 20, 10790–10798. [Google Scholar] [CrossRef]
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The Myofibroblast at a Glance. J. Cell Sci. 2020, 133, jcs227900. [Google Scholar] [CrossRef]
- Li, B.; Wang, J.H.C. Fibroblasts and Myofibroblasts in Wound Healing: Force Generation and Measurement. J. Tissue Viability 2011, 20, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of Fibrosis: Therapeutic Translation for Fibrotic Disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Kis, K.; Liu, X.; Hagood, J.S. Myofibroblast Differentiation and Survival in Fibrotic Disease. Expert. Rev. Mol. Med. 2011, 13, e27. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase Functions in Cell Death and Disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Liu, H.; Yuan, J.; Yao, Y. Targeting Wnt/β-Catenin by Anthelmintic Drug Niclosamide Overcomes Paclitaxel Resistance in Esophageal Cancer. Fundam. Clin. Pharmacol. 2021, 35, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zeng, S.; Qiu, Q.; Xiao, Y.; Shi, M.; Zou, Y.; Yang, X.; Xu, H.; Liang, L. Niclosamide Induces Apoptosis in Human Rheumatoid Arthritis Fibroblast-like Synoviocytes. Int. Immunopharmacol. 2016, 31, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.; Zheng, F.; Li, Y.; Lin, Z.; Han, X.; Feng, Y.; Tian, Z.; Ren, D.; Cao, K.; Li, C. Niclosamide Ethanolamine Salt Alleviates Idiopathic Pulmonary Fibrosis by Modulating the PI3K-MTORC1 Pathway. Cells 2022, 11, 346. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Alkhatib, A.; Kolls, J.K.; Kondoh, Y.; Lasky, J.A. Pharmacotherapy and Adjunctive Treatment for Idiopathic Pulmonary Fibrosis (IPF). J. Thorac. Dis. 2019, 11, S1740–S1754. [Google Scholar] [CrossRef]
- Finnerty, J.P.; Ponnuswamy, A.; Dutta, P.; Abdelaziz, A.; Kamil, H. Efficacy of Antifibrotic Drugs, Nintedanib and Pirfenidone, in Treatment of Progressive Pulmonary Fibrosis in Both Idiopathic Pulmonary Fibrosis (IPF) and Non-IPF: A Systematic Review and Meta-Analysis. BMC Pulm. Med. 2021, 21, 411. [Google Scholar] [CrossRef]
- Schaefer, C.J.; Ruhrmund, D.W.; Pan, L.; Seiwert, S.D.; Kossen, K. Antifibrotic Activities of Pirfenidone in Animal Models. Eur. Respir. Rev. 2011, 20, 85–97. [Google Scholar] [CrossRef]
- Ying, H.; Fang, M.; Hang, Q.Q.; Chen, Y.; Qian, X.; Chen, M. Pirfenidone Modulates Macrophage Polarization and Ameliorates Radiation-Induced Lung Fibrosis by Inhibiting the TGF-Β1/Smad3 Pathway. J. Cell Mol. Med. 2021, 25, 8662–8675. [Google Scholar] [CrossRef]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of Action of Nintedanib in the Treatment of Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [Green Version]



| Identifier | Age | Gender |
|---|---|---|
| 1 | 59 | Male |
| 2 | 51 | Female |
| 3 | 64 | Male |
| 4 | 70 | Male |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spathakis, M.; Tarapatzi, G.; Filidou, E.; Kandilogiannakis, L.; Karatzas, E.; Steiropoulos, P.; Mikroulis, D.; Spyrou, G.M.; Manolopoulos, V.G.; Kolios, G.; et al. Niclosamide Attenuates Inflammation-Associated Profibrotic Responses in Human Subepithelial Lung Myofibroblasts. Biomedicines 2023, 11, 2032. https://doi.org/10.3390/biomedicines11072032
Spathakis M, Tarapatzi G, Filidou E, Kandilogiannakis L, Karatzas E, Steiropoulos P, Mikroulis D, Spyrou GM, Manolopoulos VG, Kolios G, et al. Niclosamide Attenuates Inflammation-Associated Profibrotic Responses in Human Subepithelial Lung Myofibroblasts. Biomedicines. 2023; 11(7):2032. https://doi.org/10.3390/biomedicines11072032
Chicago/Turabian StyleSpathakis, Michail, Gesthimani Tarapatzi, Eirini Filidou, Leonidas Kandilogiannakis, Evangelos Karatzas, Paschalis Steiropoulos, Dimitrios Mikroulis, George M. Spyrou, Vangelis G. Manolopoulos, George Kolios, and et al. 2023. "Niclosamide Attenuates Inflammation-Associated Profibrotic Responses in Human Subepithelial Lung Myofibroblasts" Biomedicines 11, no. 7: 2032. https://doi.org/10.3390/biomedicines11072032