
Phosphatidic Acid Stimulates Lung Cancer Cell Migration through Interaction with the LPA1 Receptor and Subsequent Activation of MAP Kinases and STAT3
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Delivery of PA to Cells in Culture
2.4. Cell Viability Assay (Crystal Violet Method)
2.5. Determination of Cell Migration
2.6. Phosphatidic and Lysophosphatidic Acid Analyses
2.7. Western Blotting Analysis
2.8. Statistical Analysis
3. Results
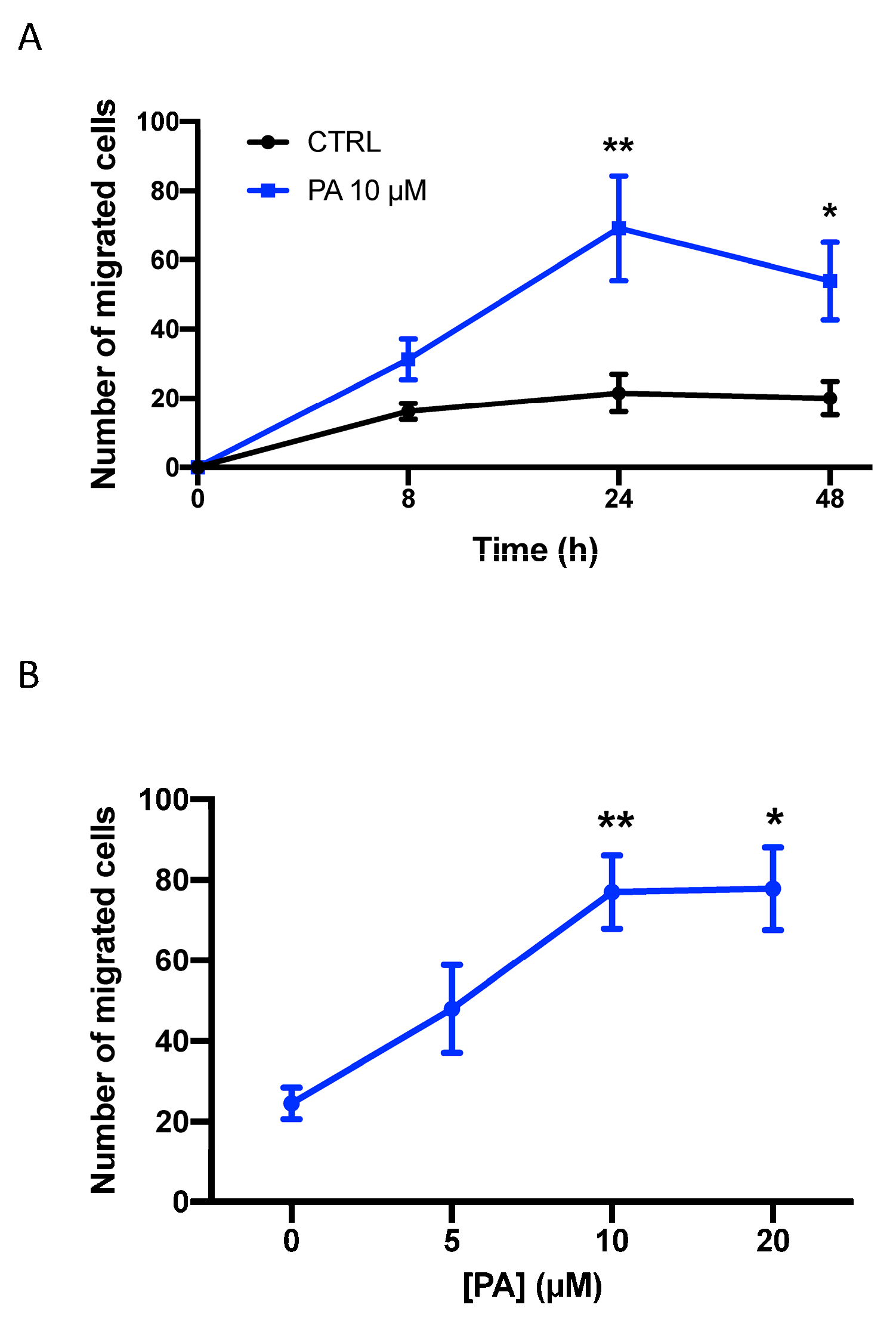
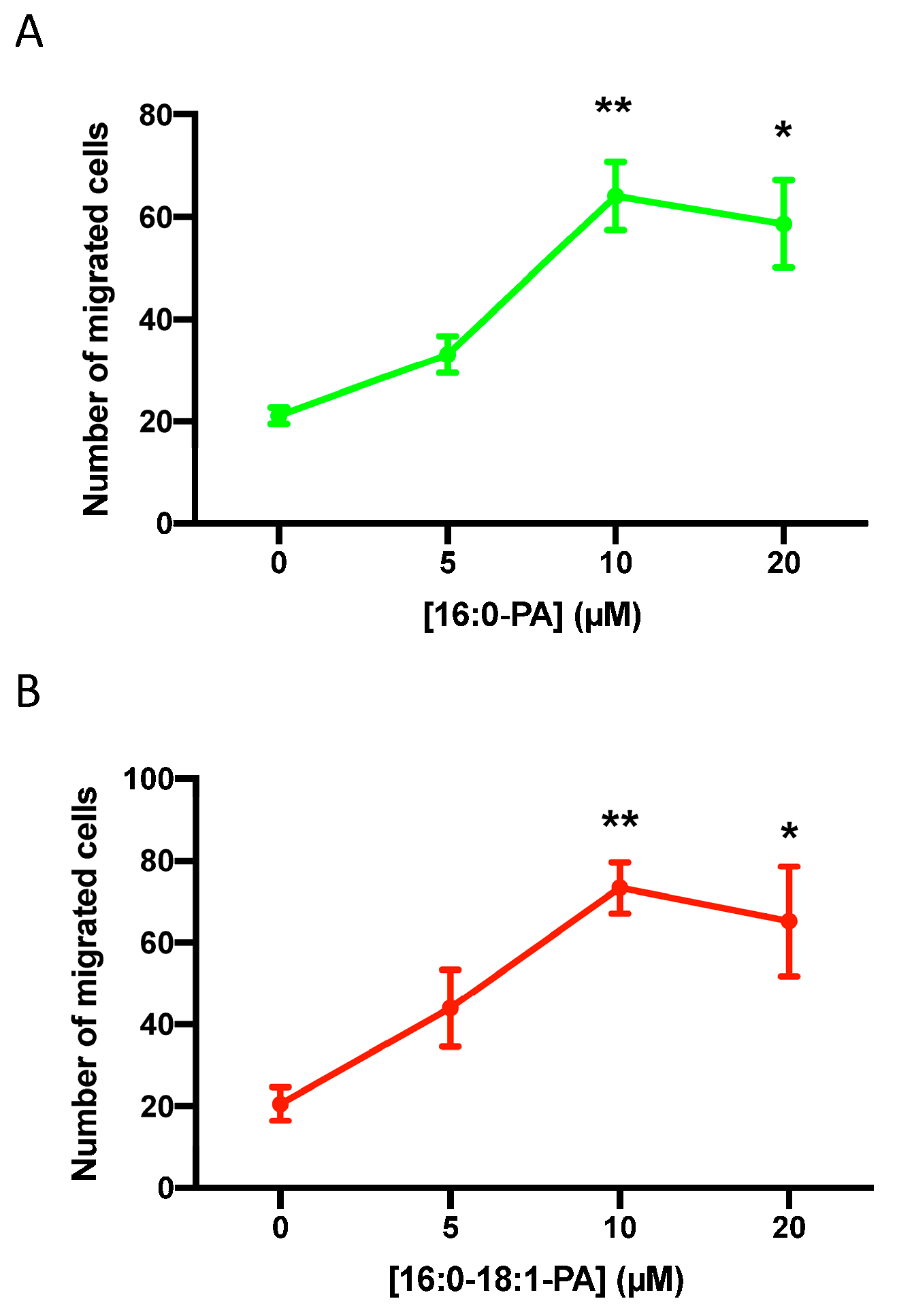
3.1. Phosphatidic Acid Stimulates Human Lung Adenocarcinoma Cell Migration
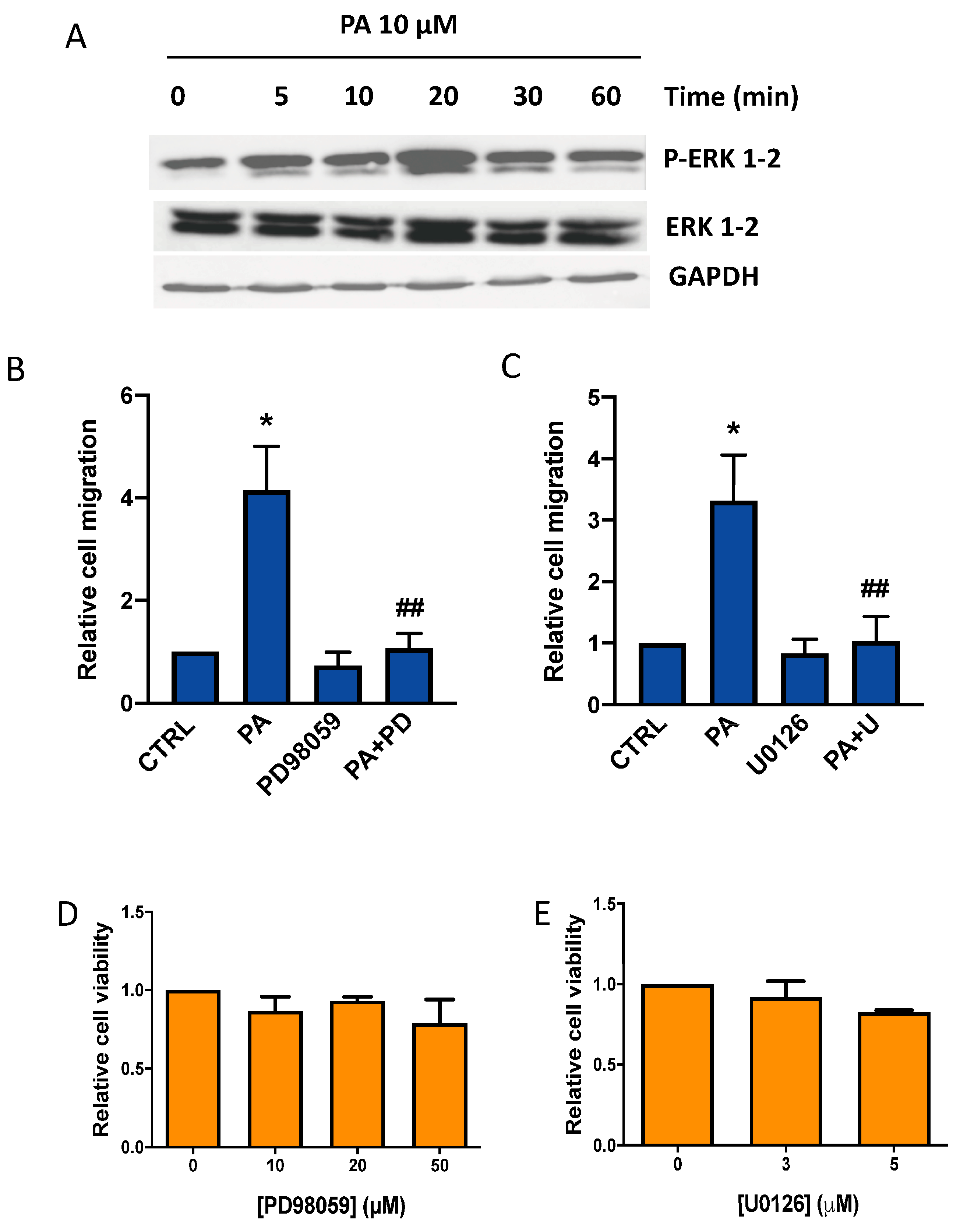
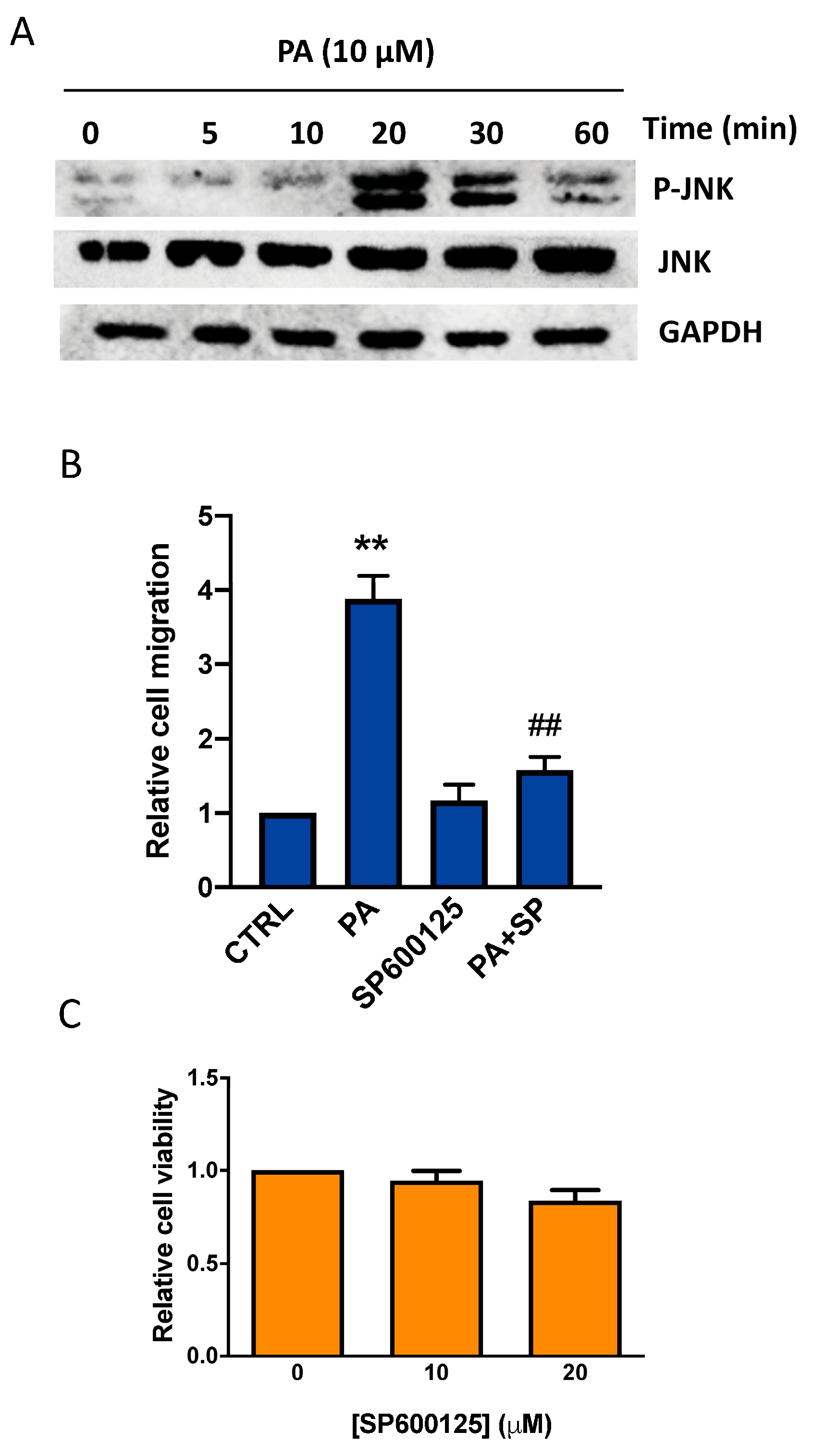
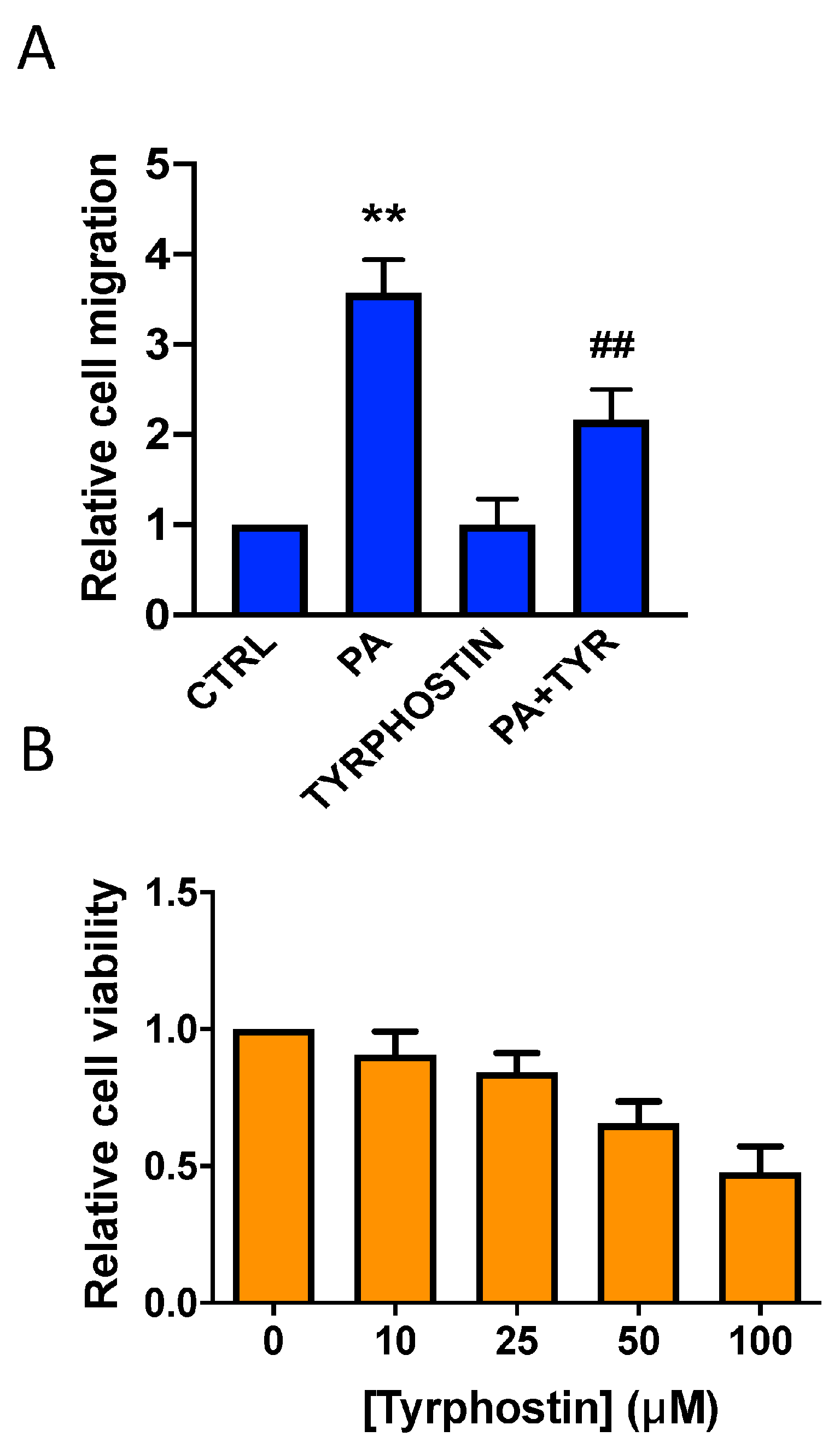
3.2. Phosphatidic Acid Induces Phosphorylation of the MAPKs ERK1-2, p38, and JNK. Implication in PA-Stimulated Lung Cancer Cell Migration
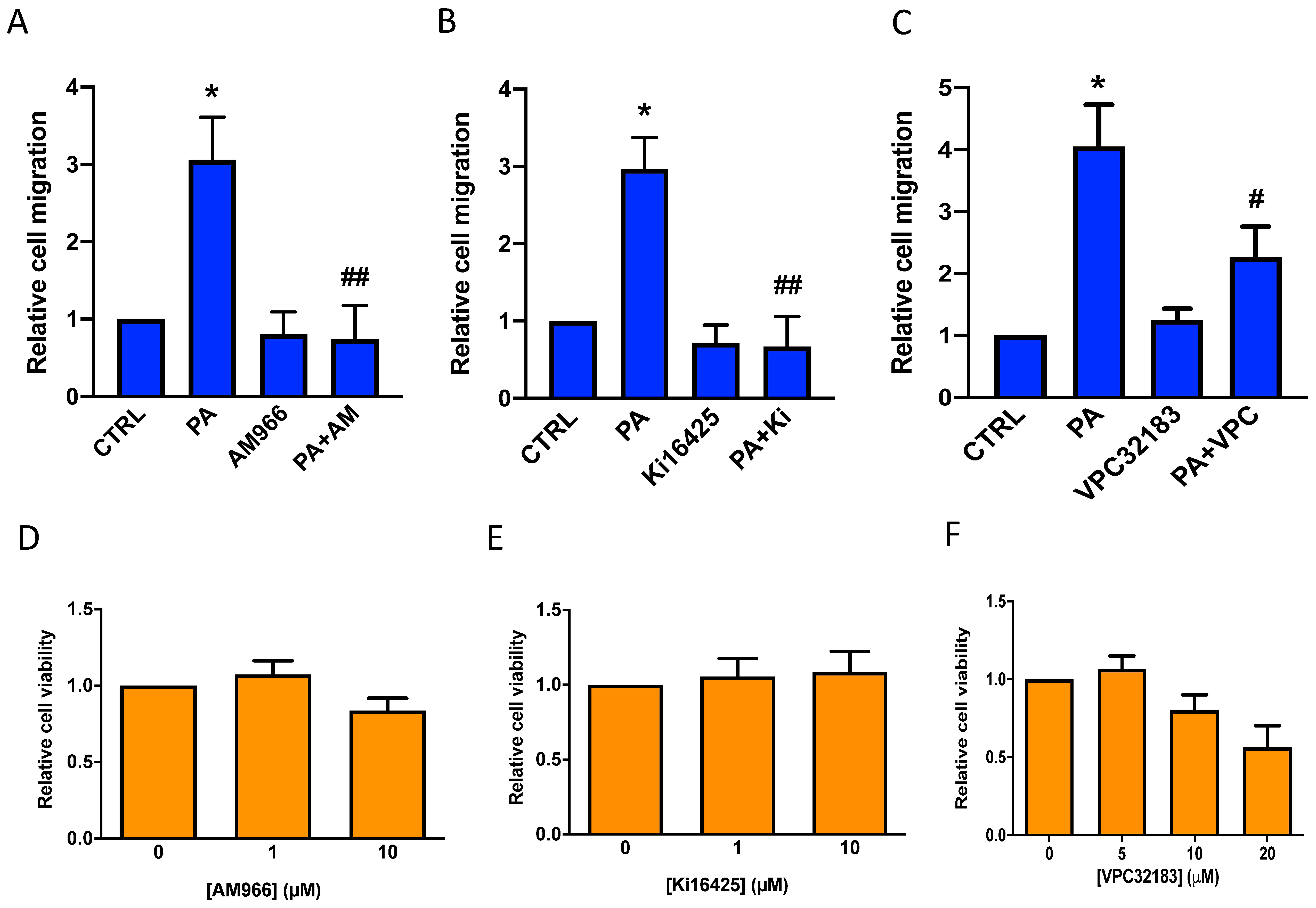
3.3. Phosphatidic Acid Stimulates Lung Cancer Cell Migration through Interaction with LPA Receptor 1
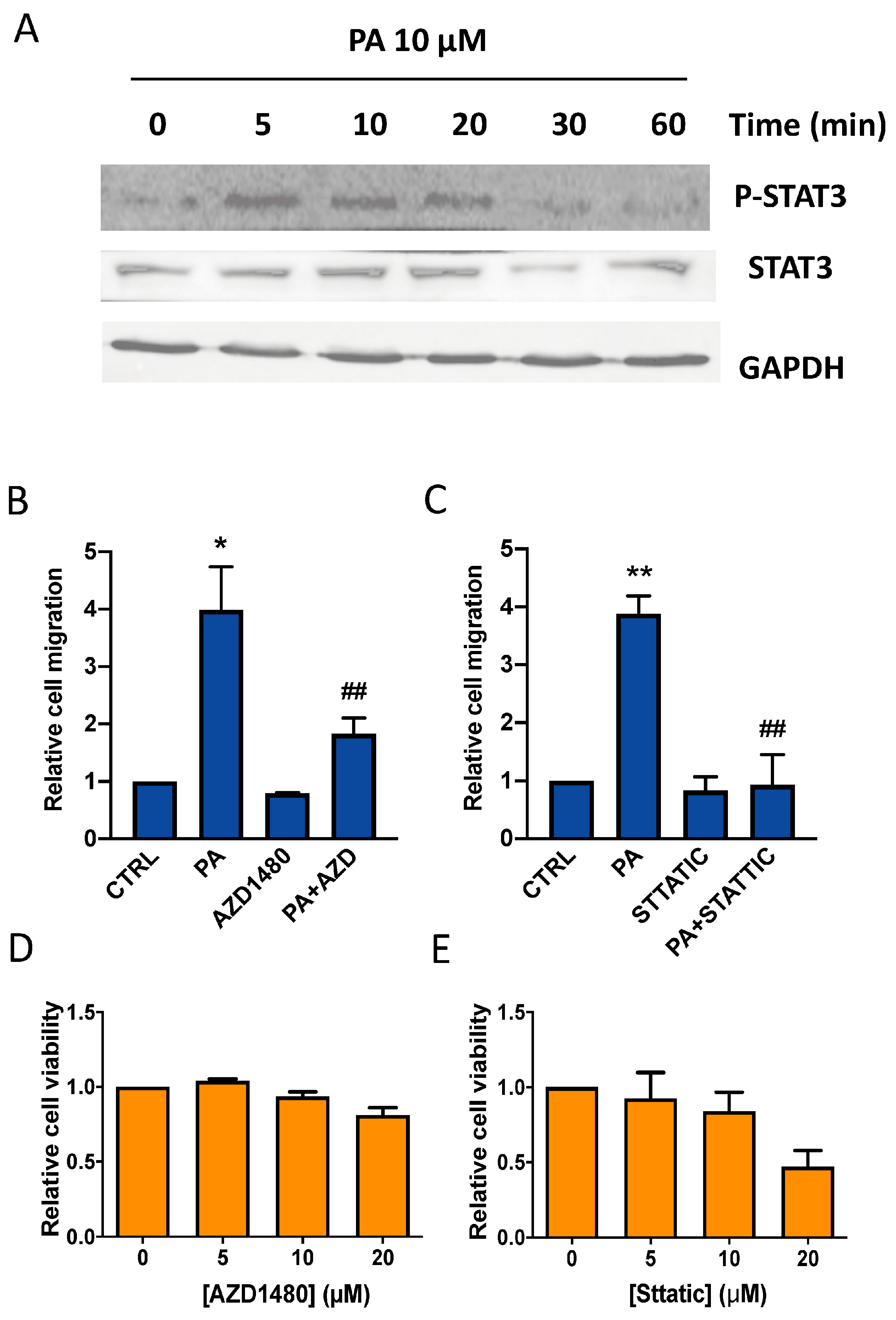
3.4. Implication of the JAK2/STAT3 Pathway in the Stimulation of Lung Cancer Cell Migration by Phosphatidic Acid
3.5. The Stimulation of Lung Cancer Cell Migration by PA Is Independent of Conversion to LPA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greenberg, A.K.; Basu, S.; Hu, J.; Yie, T.-A.; Tchou-Wong, K.M.; Rom, W.N.; Lee, T.C. Selective p38 Activation in Human Non-Small Cell Lung Cancer. Am. J. Respir. Cell Mol. Biol. 2002, 26, 558–564. [Google Scholar] [CrossRef]
- Devreotes, P.N.; Horwitz, A.R. Signaling Networks that Regulate Cell Migration. Cold Spring Harb. Perspect. Biol. 2015, 7, a005959. [Google Scholar] [CrossRef] [Green Version]
- Van Meeteren, L.A.; Ruurs, P.; Stortelers, C.; Bouwman, P.; van Rooijen, M.A.; Pradère, J.P.; Pettit, T.R.; Wakelam, M.J.O.; Saulnier-Blache, J.S.; Mummery, C.L.; et al. Autotaxin, a Secreted Lysophospholipase D, Is Essential for Blood Vessel Formation during Development. Mol. Cell. Biol. 2006, 26, 5015–5022. [Google Scholar] [CrossRef] [Green Version]
- Brindley, D.N. Lysophosphatidic Acid Signaling in Cancer. Cancers 2020, 12, 3791. [Google Scholar] [CrossRef] [PubMed]
- Hama, K.; Aoki, J.; Fukaya, M.; Kishi, Y.; Sakai, T.; Suzuki, R.; Ohta, H.; Yamori, T.; Watanabe, M.; Chun, J.; et al. Lysophosphatidic Acid and Autotaxin Stimulate Cell Motility of Neoplastic and Non-neoplastic Cells through LPA1. J. Biol. Chem. 2004, 279, 17634–17639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmings, D.G.; Brindley, D.N. Signalling by lysophosphatidate and its health implications. Essays Biochem. 2020, 64, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Ha, J.M.; Kim, Y.W.; Jin, S.Y.; Ha, H.K.; Bae, S.S. Inhibitory role of polyunsaturated fatty acids on lysophosphatidic acid-induced cancer cell migration and adhesion. FEBS Lett. 2014, 588, 2971–2977. [Google Scholar] [CrossRef] [Green Version]
- Malchinkhuu, E.; Sato, K.; Horiuchi, Y.; Mogi, C.; Ohwada, S.; Ishiuchi, S.; Saito, N.; Kurose, H.; Tomura, H.; Okajima, F. Role of p38 mitogen-activated kinase and c-Jun terminal kinase in migration response to lysophosphatidic acid and sphingosine-1-phosphate in glioma cells. Oncogene 2005, 24, 6676–6688. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.-F.; Wu, S.; Li, Y.; Bao, G.; Pei, J.-Y.; Wang, Y.-W.; Ma, Q.; Sun, H.-J.; Damirin, A. LPA receptor1 antagonists as anticancer agents suppress human lung tumours. Eur. J. Pharmacol. 2020, 868, 172886. [Google Scholar] [CrossRef]
- Zhou, Z.B.; Niu, J.P.; Zhang, Z.J. Receptor-mediated vascular smooth muscle migration induced by LPA involves p38 mitogen-activated protein kinase pathway activation. Int. J. Mol. Sci. 2009, 10, 3194–3208. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E.; Vance, D.E. Phospholipid biosynthesis in mammalian cells. Biochem. Cell Biol. 2004, 82, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Tei, R.; Baskin, J.M. Spatiotemporal control of phosphatidic acid signaling with optogenetic, engineered phospholipase Ds. J. Cell Biol. 2020, 219, e201907013. [Google Scholar] [CrossRef] [Green Version]
- Pelech, S.L.; Sanghera, J.S. MAP Kinases: Charting the Regulatory Pathways. Science 1992, 257, 1355–1356. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, T.; Bayer, H.; Myrtek, D.; Ferrari, D.; Sorichter, S.; Ziegenhagen, M.W.; Zissel, G.; Virchow, J.C., Jr.; Luttmann, W.; Norgauer, J.; et al. The P2Y14 receptor of airway epithelial cells: Coupling to intracellular Ca2+ and IL-8 secretion. Am. J. Respir. Cell Mol. Biol. 2005, 33, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Larrauri, A.; Gangoiti, P.; Presa, N.; Dominguez-Herrera, A.; Donati, C.; Bruni, P.; Trueba, M.; Gomez-Muñoz, A.; Ouro, A. Phosphatidic Acid Stimulates Myoblast Proliferation through Interaction with LPA1 and LPA2 Receptors. Int. J. Mol. Sci. 2021, 22, 1452. [Google Scholar] [CrossRef]
- Carmona-Rosas, G.; Alfonzo-Méndez, M.A.; Hernández-Espinosa, D.A.; Romero-Ávila, M.T.; García-Sáinz, J.A. A549 cells as a model to study endogenous LPA 1 receptor signaling and regulation. Eur. J. Pharmacol. 2017, 815, 258–265. [Google Scholar] [CrossRef]
- Swaney, J.S.; Chapman, C.; Correa, L.D.; Stebbins, K.J.; Bundey, R.A.; Prodanovich, P.C.; Fagan, P.; Baccei, C.S.; Santini, A.M.; Hutchinson, J.H.; et al. A novel, orally active LPA(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 2010, 160, 1699–1713. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-J.; Kim, Y.-L.; Lee, Y.-K.; Sacket, S.J.; Kim, K.; Kim, H.-L.; Han, M.; Bae, Y.-S.; Okajima, F.; Im, D.-S. Dioleoyl phosphatidic acid increases intracellular Ca2+ through endogenous LPA receptors in C6 glioma and L2071 fibroblasts. Prostaglandins Other Lipid Mediat. 2007, 83, 268–276. [Google Scholar] [CrossRef]
- Heasley, B.H.; Jarosz, R.; Lynch, K.R.; Macdonald, T.L. Initial structure-activity relationships of lysophosphatidic acid receptor antagonists: Discovery of a high-affinity LPA1/LPA3 receptor antagonist. Bioorg. Med. Chem. Lett. 2004, 14, 2735–2740. [Google Scholar] [CrossRef]
- Tveteraas, I.H.; Aasrum, M.; Brusevold, I.J.; Ødegård, J.; Christoffersen, T.; Sandnes, D. Lysophosphatidic acid induces both EGFR-dependent and EGFR-independent effects on DNA synthesis and migration in pancreatic and colorectal carcinoma cells. Tumor Biol. 2016, 37, 2519–2526. [Google Scholar] [CrossRef] [PubMed]
- Albers, H.M.H.G.; Dong, A.; van Meeteren, L.A.; Egan, D.A.; Sunkara, M.; van Tilburg, E.W.; Schuurman, K.; van Tellingen, O.; Morris, A.J.; Smyth, S.S.; et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. USA 2010, 107, 7257–7262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houben, A.J.S.; Moolenaar, W.H. Autotaxin and LPA receptor signaling in cancer. Cancer Metastasis Rev. 2011, 30, 557–565. [Google Scholar] [CrossRef]
- Okudaira, S.; Yukiura, H.; Aoki, J. Biological roles of lysophosphatidic acid signaling through its production by autotaxin. Biochimie 2010, 92, 698–706. [Google Scholar] [CrossRef]
- Valdés-Rives, S.A.; González-Arenas, A. Autotaxin-Lysophosphatidic Acid: From Inflammation to Cancer Development. Mediat. Inflamm. 2017, 2017, 9173090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.-C.M.; Krummel, M.F.; Rosen, S.D. Autotaxin through Lysophosphatidic Acid Stimulates Polarization, Motility, and Transendothelial Migration of Naive T Cells. J. Immunol. 2012, 189, 3914–3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.; Duffy, P.A.; Liossis, C.; Gomez-Muñoz, A.; O’Brien, L.; Stone, J.C.; Brindley, D.N. Increased concentrations of phosphatidate, di-acylglycerol and ceramide in ras- and tyrosine kinase (fps)-transformed fibroblasts. Oncogene 1997, 14, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Muñoz, A.; Martin, A.; O’Brien, L.; Brindley, D. Cell-permeable ceramides inhibit the stimulation of DNA synthesis and phospholipase D activity by phosphatidate and lysophosphatidate in rat fibroblasts. J. Biol. Chem. 1994, 269, 8937–8943. [Google Scholar] [CrossRef]
- Lim, H.-K.; Choi, Y.-A.; Park, W.; Lee, T.; Ryu, S.H.; Kim, S.-Y.; Kim, J.-R.; Kim, J.-H.; Baek, S.-H. Phosphatidic Acid Regulates Systemic Inflammatory Responses by Modulating the Akt-Mammalian Target of Rapamycin-p70 S6 Kinase 1 Pathway. J. Biol. Chem. 2003, 278, 45117–45127. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, F.G.; Wang, D.; Bargiacchi, F.; DuBois, R.N. Prostaglandin E2 Regulates Cell Migration via the Intracellular Activation of the Epidermal Growth Factor Receptor. J. Biol. Chem. 2003, 278, 35451–35457. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, M.; Vicovac, L. Interleukin-6 stimulates cell migration, invasion and integrin expression in HTR-8/SVneo cell line. Placenta 2009, 30, 320–328. [Google Scholar] [CrossRef]
- Razidlo, G.L.; Burton, K.M.; McNiven, M.A. Interleukin-6 promotes pancreatic cancer cell migration by rapidly activating the small GTPase CDC42. J. Biol. Chem. 2018, 293, 11143–11153. [Google Scholar] [CrossRef] [Green Version]
- Wolczyk, D.; Zaremba-Czogalla, M.; Hryniewicz-Jankowska, A.; Tabola, R.; Grabowski, K.; Sikorski, A.F.; Augoff, K. TNF-α promotes breast cancer cell migration and enhances the concentration of membrane-associated proteases in lipid rafts. Cell. Oncol. 2016, 39, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Zhan, R.; He, W.; Wang, F.; Yao, Z.; Tan, J.; Xu, R.; Zhou, J.; Wang, Y.; Li, H.; Wu, J.; et al. Nitric oxide promotes epidermal stem cell migration via cGMP-Rho GTPase signalling. Sci. Rep. 2016, 6, 30687. [Google Scholar] [CrossRef]
- Bond, P. Phosphatidic acid: Biosynthesis, pharmacokinetics, mechanisms of action and effect on strength and body composition in resistance-trained individuals. Nutr. Metab. 2017, 14, 12. [Google Scholar] [CrossRef] [Green Version]
- Michalczyk, A.; Budkowska, M.; Dołęgowska, B.; Chlubek, D.; Safranow, K. Lysophosphatidic acid plasma concentrations in healthy subjects: Circadian rhythm and associations with demographic, anthropometric and biochemical parameters. Lipids Health Dis. 2017, 16, 140. [Google Scholar] [CrossRef] [Green Version]
- Kurz, T.; Schneider, I.; Tölg, R.; Richardt, G. Alpha 1-adrenergic receptor-mediated increase in the mass of phosphatidic acid and 1,2-diacylglycerol in ischemic rat heart. Cardiovasc. Res. 1999, 42, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Noh, J.-Y.; Lim, K.-M.; Bae, O.-N.; Chung, S.-M.; Lee, S.-W.; Joo, K.-M.; Lee, S.-D.; Chung, J.-H. Procoagulant and prothrombotic activation of human erythrocytes by phosphatidic acid. Am. J. Physiol. Circ. Physiol. 2010, 299, H347–H355. [Google Scholar] [CrossRef] [Green Version]
- Egea-Jimenez, A.L.; Zimmermann, P. Phospholipase D and phosphatidic acid in the biogenesis and cargo loading of extracellular vesicles. J. Lipid Res. 2018, 59, 1554–1560. [Google Scholar] [CrossRef] [Green Version]
- Record, M.; Silvente-Poirot, S.; Poirot, M.; Wakelam, M.J. Extracellular vesicles: Lipids as key components of their biogenesis and functions. J. Lipid Res. 2018, 59, 1316–1324. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Ha, K.S.; Exton, J.H. Activation of actin polymerization by phosphatidic acid derived from phosphatidylcholine in IIC9 fibroblasts. J. Cell Biol. 1993, 123, 1789–1796. [Google Scholar] [CrossRef]
- Litosch, I.; Pujari, R.; Lee, S.J. Phosphatidic acid regulates signal output by G protein coupled receptors through direct interaction with phospholipase C-beta(1). Cell Signal. 2009, 21, 1379–1384. [Google Scholar] [CrossRef]
- Pearce, B.; Jakobson, K.; Morrow, C.; Murphy, S. Phosphatidic acid promotes phosphoinositide metabolism and DNA synthesis in cultured cortical astrocytes. Neurochem. Int. 1994, 24, 165–171. [Google Scholar] [CrossRef]
- Ryder, N.S.; Talwar, H.S.; Reynolds, N.J.; Voorhees, J.J.; Fisher, G.J. Phosphatidic acid and phospholipase D both stimulate phospho-inositide turnover in cultured human keratinocytes. Cell Signal. 1993, 5, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Yamaji, H.; Sakai, K.; Joho, T.; Izumoto, E.; Fukuda, H. Cell cycle analysis of Chinese hamster ovary cells stimulated by phosphatidic acid in serum-free culture. J. Biosci. Bioeng. 2004, 98, 487–489. [Google Scholar] [CrossRef]
- Zhou, D.; Luini, W.; Bernasconi, S.; Diomede, L.; Salmona, M.; Mantovani, A.; Sozzani, S. Phosphatidic Acid and Lysophosphatidic Acid Induce Haptotactic Migration of Human Monocytes. J. Biol. Chem. 1995, 270, 25549–25556. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Kamimura, A.; Hamazono-Matsuoka, T.; Honda, S. Phosphatidic Acid Has a Potential to Promote Hair Growth In Vitro and In Vivo, and Activates Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Kinase in Hair Epithelial Cells. J. Investig. Dermatol. 2003, 121, 448–456. [Google Scholar] [CrossRef]
- Frondorf, K.; Henkels, K.M.; Frohman, M.A.; Gomez-Cambronero, J. Phosphatidic Acid Is a Leukocyte Chemoattractant That Acts through S6 Kinase Signaling. J. Biol. Chem. 2010, 285, 15837–15847. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.J.; Wilson, G.C.; Keitsch, S.; Soddemann, M.; Wilker, B.; Müller, C.P.; Kornhuber, J.; Gulbins, E. Molecular targets of endothelial phosphatidic acid regulating major depressive disorder. J. Neurochem. 2022, 163, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A. Smad signalling network. J. Cell Sci. 2002, 115, 3355–3356. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [LPA] (µM) | Number of Migrated Cells (Mean ± SD) | Statistical Significance |
|---|---|---|
| 0 | 19.2 ± 4.1 | |
| 0.1 | 22.7 ± 7.7 | N.S. |
| 1 | 36.6 ± 9.2 | p < 0.05 |
| 10 | 72.4 ± 10.0 | p < 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez-Larrauri, A.; Gangoiti, P.; Camacho, L.; Presa, N.; Martin, C.; Gomez-Muñoz, A. Phosphatidic Acid Stimulates Lung Cancer Cell Migration through Interaction with the LPA1 Receptor and Subsequent Activation of MAP Kinases and STAT3. Biomedicines 2023, 11, 1804. https://doi.org/10.3390/biomedicines11071804
Gomez-Larrauri A, Gangoiti P, Camacho L, Presa N, Martin C, Gomez-Muñoz A. Phosphatidic Acid Stimulates Lung Cancer Cell Migration through Interaction with the LPA1 Receptor and Subsequent Activation of MAP Kinases and STAT3. Biomedicines. 2023; 11(7):1804. https://doi.org/10.3390/biomedicines11071804
Chicago/Turabian StyleGomez-Larrauri, Ana, Patricia Gangoiti, Laura Camacho, Natalia Presa, Cesar Martin, and Antonio Gomez-Muñoz. 2023. "Phosphatidic Acid Stimulates Lung Cancer Cell Migration through Interaction with the LPA1 Receptor and Subsequent Activation of MAP Kinases and STAT3" Biomedicines 11, no. 7: 1804. https://doi.org/10.3390/biomedicines11071804