
High Rate of Mutations of Adhesion Molecules and Extracellular Matrix Glycoproteins in Patients with Adult-Onset Focal and Segmental Glomerulosclerosis

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Controls
2.2. Identification and Annotation of Genes of Cellular Adhesion Molecules
2.3. Sample Preparation and Mutational Analysis
2.3.1. Design of the Targeted Sequencing Panel
2.3.2. Library Preparation
2.3.3. Sequencing
2.3.4. Sequencing Data Collection
2.3.5. Bioinformatic Analysis
3. Results
3.1. Clinical and Pathological Characteristics
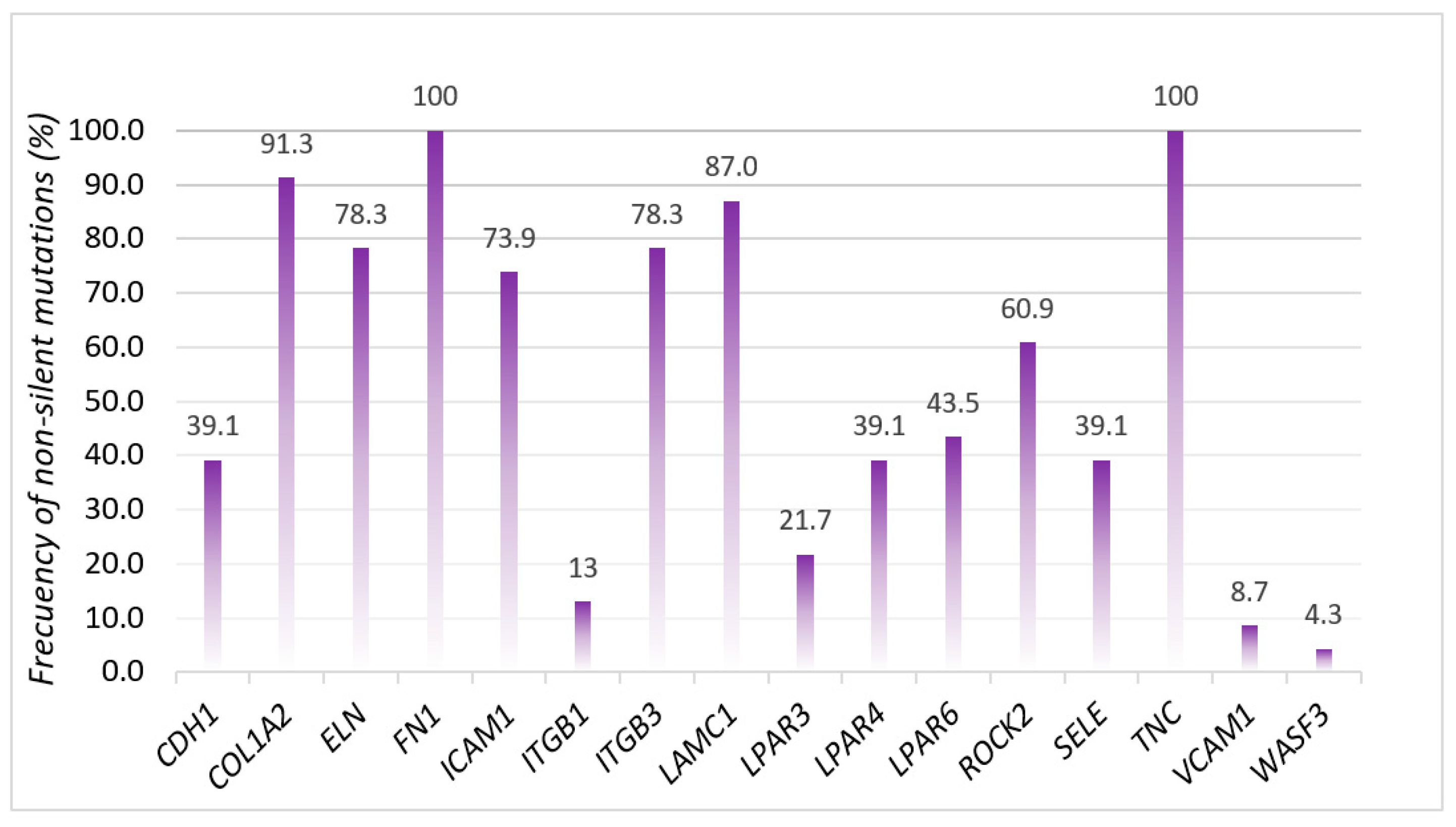
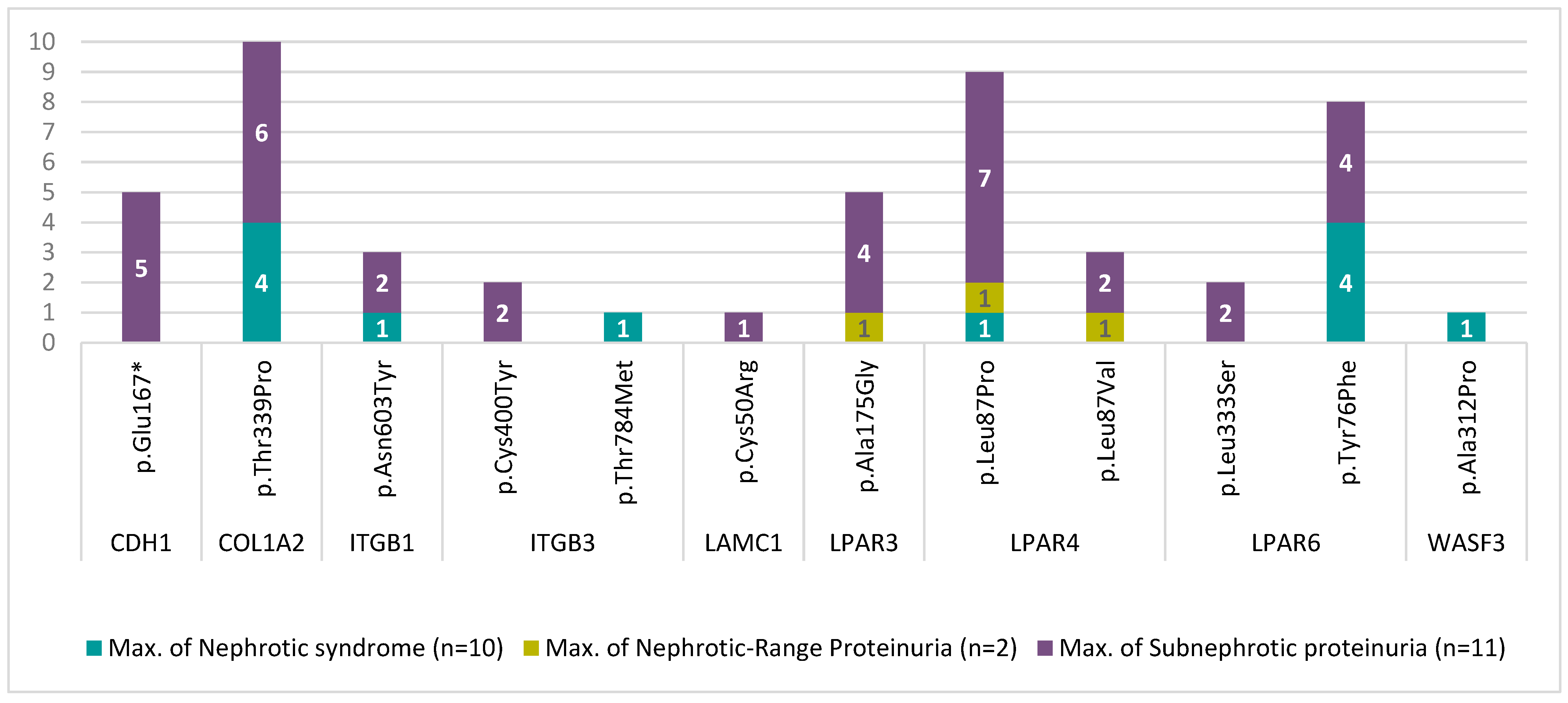
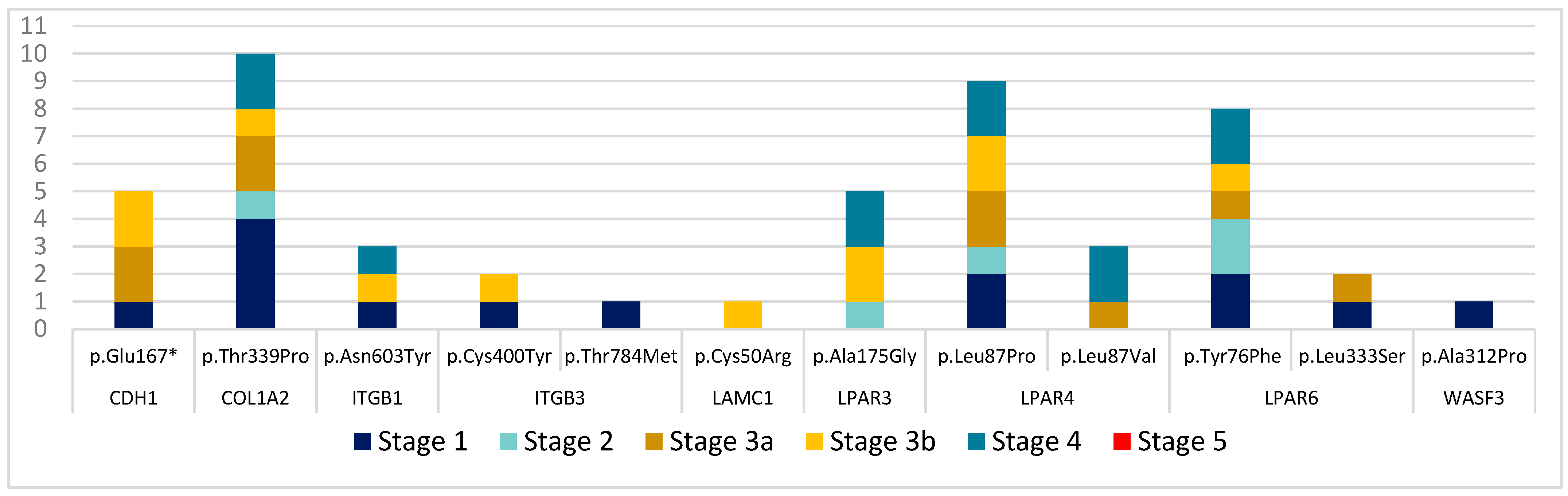
3.2. Mutational Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braden, G.L.; Mulhern, J.G.; O’Shea, M.H.; Nash, S.V.; Ucci, A.A.; Germain, M.J. Changing incidence of glomerular diseases in adults. Am. J. Kidney Dis. 2000, 35, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Kitiyakara, C.; Eggers, P.; Kopp, J.B. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am. J. Kidney Dis. 2004, 44, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Stirling, C.M.; Mathieson, P.; Boulton-Jones, J.; Feehally, J.; Jayne, D.; Murray, H.; Adu, D. Treatment and outcome of adult patients with primary focal segmental glomerulosclerosis in five UK renal units. QJM-Mon. J. Assoc. Physicians 2005, 98, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Roles for microRNAs in the regulation of cell adhesion molecules. J. Cell Sci. 2011, 124 Pt 7, 999–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef]
- Sharif, B.; Barua, M. Advances in molecular diagnosis and therapeutics in nephrotic syndrome and focal and segmental glomerulosclerosis. Curr. Opin. Nephrol. Hypertens. 2018, 27, 194–200. [Google Scholar] [CrossRef]
- Kopp, J.B.; Nelson, G.W.; Sampath, K.; Johnson, R.C.; Genovese, G.; An, P.; Friedman, D.; Briggs, W.; Dart, R.; Korbet, S.; et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 2011, 22, 2129–2137. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Artomov, M.; Brähler, S.; Stander, M.C.; Shamsan, G.; Sampson, M.G.; White, J.M.; Kretzler, M.; Miner, J.H.; Jain, S.; et al. A role for genetic susceptibility in sporadic focal segmental glomerulosclerosis. J. Clin. Investig. 2016, 126, 1067–1078. [Google Scholar] [CrossRef] [Green Version]
- Wiggins, R.C. The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 2007, 71, 1205–1214. [Google Scholar] [CrossRef] [Green Version]
- Lennon, R.; Randles, M.J.; Humphries, M.J. The importance of podocyte adhesion for a healthy glomerulus. Front. Endocrinol. 2014, 14, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schell, C.; Huber, T.B. The Evolving Complexity of the Podocyte Cytoskeleton. J. Am. Soc. Nephrol. 2017, 28, 3166–3174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Has, C.; Spartà, G.; Kiritsi, D.; Weibel, L.; Moeller, A.; Vega-Warner, V.; Waters, A.; He, Y.; Anikster, Y.; Esser, P.; et al. Integrin α 3 Mutations with Kidney, Lung, and Skin Disease. N. Engl. J. Med. 2012, 366, 1508–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.-C.; Fornoni, A.; Weins, A.; Hakroush, S.; Maiguel, D.; Sageshima, J.; Chen, L.; Ciancio, G.; Faridi, M.H.; Behr, D.; et al. Abatacept in B7-1–Positive Proteinuric Kidney Disease. N. Engl. J. Med. 2013, 369, 2416–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansrivijit, P.; Puthenpura, M.M.; Ghahramani, N. Efficacy of abatacept treatment for focal segmental glomerulosclerosis and minimal change disease: A systematic review of case reports, case series, and observational studies. Clin. Nephrol. 2020, 94, 117–126. [Google Scholar] [CrossRef]
- Renkema, K.Y.; Stokman, M.F.; Giles, R.H.; Knoers, N.V.A.M. Next-generation sequencing for research and diagnostics in kidney disease. Nat. Rev. Nephrol. 2014, 10, 433–444. [Google Scholar] [CrossRef]
- Bullich, G.; Trujillano, D.; Santín, S.; Ossowski, S.; Mendizábal, S.; Fraga, G.; Madrid, A.; Ariceta, G.; Ballarín, J.; Torra, R.; et al. Targeted next-generation sequencing in steroid-resistant nephrotic syndrome: Mutations in multiple glomerular genes may influence disease severity. Eur. J. Hum. Genet. 2015, 23, 1192–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierzynska, A.; Soderquest, K.; Dean, P.; Colby, E.; Rollason, R.; Jones, C.; Inward, C.D.; McCarthy, H.J.; Simpson, M.A.; Lord, G.M.; et al. MAGI2 mutations cause congenital nephrotic syndrome. J. Am. Soc. Nephrol. 2017, 28, 1614–1621. [Google Scholar] [CrossRef] [Green Version]
- Landini, S.; Mazzinghi, B.; Becherucci, F.; Allinovi, M.; Provenzano, A.; Palazzo, V.; Ravaglia, F.; Artuso, R.; Bosi, E.; Stagi, S.; et al. Reverse phenotyping after whole-Exome sequencing in steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2020, 15, 89–100. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovin, B.H.; Adler, S.G.; Barratt, J.; Bridoux, F.; Burdge, K.A.; Chan, T.M.; Cook, H.T.; Fervenza, F.C.; Gibson, K.L.; Glassock, R.J.; et al. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, S1–S276. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Schlöndorff, J.S.; Becker, D.J.; Tsukaguchi, H.; Tonna, S.J.; Uscinski, A.L.; Higgs, H.N.; Henderson, J.M.; Pollak, M.R. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat. Genet. 2010, 42, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Hao, X.; Azeloglu, E.U.; Ren, H.; Wang, Z.; Ma, J.; Liu, J.; Ma, X.; Wang, W.; Pan, X.; et al. Novel mutations in the inverted formin 2 gene of Chinese families contribute to focal segmental glomerulosclerosis. Kidney Int. 2015, 88, 593–604. [Google Scholar] [CrossRef]
- Marx, D.; Caillard, S.; Olagne, J.; Moulin, B.; Hannedouche, T.; Touchard, G.; Dupuis, A.; Gachet, C.; Molitor, A.; Bahram, S.; et al. Atypical focal segmental glomerulosclerosis associated with a new PODXL nonsense variant. Mol. Genet. Genom. Med. 2021, 9, e1658. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Udwan, K.; John, R.; Rana, A.; Haghighi, A.; Xu, L.; Hack, S.; Reich, H.N.; Hladunewich, M.A.; Cattran, D.C.; et al. Integration of genetic testing and pathology for the diagnosis of adults with FSGS. Clin. J. Am. Soc. Nephrol. 2019, 14, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunisch, M.C.; Riedhammer, K.M.; Herr, P.-M.; Draut, S.; Günthner, R.; Wagner, M.; Weidenbusch, M.; Lungu, A.; Alhaddad, B.; Renders, L.; et al. Identification of disease-causing variants by comprehensive genetic testing with exome sequencing in adults with suspicion of hereditary FSGS. Eur. J. Hum. Genet. 2021, 29, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, C.E.; Lovric, S.; Ashraf, S.; Pabst, W.L.; Gee, H.Y.; Kohl, S.; Engelmann, S.; Vega-Warner, V.; Fang, H.; Halbritter, J.; et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 2015, 26, 1279–1289. [Google Scholar] [CrossRef] [Green Version]
- Potter, A.S.; Drake, K.; Brunskill, E.W.; Steven Potter, S. A bigenic mouse model of FSGS reveals perturbed pathways in podocytes, mesangial cells and endothelial cells. PLoS ONE 2019, 14, e0216261. [Google Scholar] [CrossRef] [Green Version]
- Kliewe, F.; Kaling, S.; Lötzsch, H.; Artelt, N.; Schindler, M.; Rogge, H.; Schröder, S.; Scharf, C.; Amann, K.; Daniel, C.; et al. Fibronectin is up-regulated in podocytes by mechanical stress. FASEB J. 2019, 33, 14450–14460. [Google Scholar] [CrossRef] [Green Version]
- Madne, T.H.; Dockrell, M.E.C. TGFβ1-mediated PI3K/Akt and p38 MAP kinase dependent alternative splicing of fibronectin extra domain A in human podocyte culture. Cell. Mol. Biol. 2018, 64, 127–135. [Google Scholar] [CrossRef]
- Chan, G.C.; Eng, D.G.; Miner, J.H.; Alpers, C.E.; Hudkins, K.L.; Chang, A.; Pippin, J.W.; Shankland, S.J. Differential expression of parietal epithelial cell and podocyte extracellular matrix proteins in focal segmental glomerulosclerosis and diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2019, 317, F1680–F1694. [Google Scholar] [CrossRef]
- Wei, C.; El Hindi, S.; Li, J.; Fornoni, A.; Goes, N.; Sageshima, J.; Maiguel, D.; Karumanchi, S.A.; Yap, H.-K.; Saleem, M.; et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med 2011, 17, 952–960. [Google Scholar] [CrossRef] [Green Version]
- Lim, B.J.; Yang, J.W.; Do, W.S.; Fogo, A.B. Pathogenesis of focal segmental glomerulosclerosis. J. Pathol. Transl. Med. 2016, 50, 405–410. [Google Scholar] [CrossRef]
- Sohrabi-Jahromi, S.; Marashi, S.A.; Kalantari, S. A kidney-specific genome-scale metabolic network model for analyzing focal segmental glomerulosclerosis. Mamm. Genome 2016, 27, 158–167. [Google Scholar] [CrossRef]
- Sachs, N.; Sonnenberg, A. Cell-matrix adhesion of podocytes in physiology and disease. Nat. Rev. Nephrol. 2013, 9, 200–210. [Google Scholar] [CrossRef]
- Uchio-Yamada, K.; Yasuda, K.; Monobe, Y.; Akagi, K.I.; Suzuki, O.; Manabe, N. Tensin2 is important for podocyte-glomerular basement membrane interaction and integrity of the glomerular filtration barrier. Am. J. Physiol. Ren. Physiol. 2020, 318, F1520–F1530. [Google Scholar] [CrossRef]
- Stotter, B.R.; Talbot, B.E.; Capen, D.E.; Artelt, N.; Zeng, J.; Matsumoto, Y.; Endlich, N.; Cummings, R.D.; Schlondorff, J.S. Cosmc-dependent mucin-type O-linked glycosylation is essential for podocyte function. Am. J. Physiol. Ren. Physiol. 2020, 318, F518–F530. [Google Scholar] [CrossRef]
- Sugar, T.; Wassenhove-McCarthy, D.J.; Wayne Orr, A.; Green, J.; Van Kuppevelt, T.H.; McCarthy, K.J. N-sulfation of heparan sulfate is critical for syndecan-4-mediated podocyte cell-matrix interactions. Am. J. Physiol. Ren. Physiol. 2016, 310, F1123–F1135. [Google Scholar] [CrossRef] [Green Version]
- Bukosza, E.N.; Kratochwill, K.; Kornauth, C.; Schachner, H.; Aufricht, C.; Gebeshuber, C.A. Podocyte RNA sequencing reveals Wnt- And ECM-associated genes as central in FSGS. PLoS ONE 2020, 15, e0231898. [Google Scholar] [CrossRef]
- Matejas, V.; Hinkes, B.; Alkandari, F.; Al-Gazali, L.; Annexstad, E.; Aytac, M.B.; Barrow, M.; Bláhová, K.; Bockenhauer, D.; Cheong, H.I.; et al. Mutations in the human laminin β2 (LAMB2) gene and the associated phenotypic spectrum. Hum. Mutat. 2010, 31, 992–1002. [Google Scholar] [CrossRef] [Green Version]
- Kambham, N.; Tanji, N.; Seigle, R.L.; Markowitz, G.S.; Pulkkinen, L.; Uitto, J.; D’Agati, V.D. Congenital focal segmental glomerulosclerosis associated with β4 integrin mutation and epidermolysis bullosa. Am. J. Kidney Dis. 2000, 36, 190–196. [Google Scholar] [CrossRef]
- Chen, Y.M.; Kikkawa, Y.; Miner, J.H. A missense LAMB2 mutation causes congenital nephrotic syndrome by impairing laminin secretion. J. Am. Soc. Nephrol. 2011, 22, 849–858. [Google Scholar] [CrossRef] [Green Version]
- Merchant, M.L.; Barati, M.T.; Caster, D.J.; Hata, J.L.; Hobeika, L.; Coventry, S.; Brier, M.E.; Wilkey, D.W.; Li, M.; Rood, I.M.; et al. Proteomic analysis identifies distinct glomerular extracellular matrix in collapsing focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. 2020, 31, 1883–1904. [Google Scholar] [CrossRef]
- Lepori, N.; Zand, L.; Sethi, S.; Fernandez-Juarez, G.; Fervenza, F.C. Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clin. Kidney J. 2018, 11, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Frese, J.; Kettwig, M.; Zappel, H.; Hofer, J.; Gröne, H.-J.; Nagel, M.; Sunder-Plassmann, G.; Kain, R.; Neuweiler, J.; Gross, O. Kidney injury by variants in the COL4A5 gene aggravated by polymorphisms in slit diaphragm genes causes focal segmental glomerulosclerosis. Int. J. Mol. Sci. 2019, 20, 519. [Google Scholar] [CrossRef] [Green Version]
- Assad, L.; Schwartz, M.M.; Virtanen, I.; Gould, V.E. Immunolocalization of tenascin and cellular fibronectins in diverse glomerulopathies. Virchows Arch B Cell Pathol Incl Mol Pathol. 1993, 63, 307–316. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wang, Y. Genetic studies of IgA nephropathy. Nephron-Exp Nephrol 2006, 102, 76–80. [Google Scholar] [CrossRef]
- Adler, S.; Brady, H.R. Cell adhesion molecules and the glomerulopathies. Am. J. Med. 1999, 107, 371–386. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Patients | Clinical Characteristics | Pathological Characteristics | Clinical Onset | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gender | Age | Morphological Variants | Proteinuria (g/Day) | Serum Albumin (g/dL) | HBP | Creatinine (mg/dL) | Creatinine Clearance (mL/min) | CKD Stage | |
| 1 | M | 52 | Perihiliar | 2.2 | 3.1 | Yes | 1.9 | 40 | 3b |
| 2 | F | 68 | NOS | 3.0 | 3.7 | Yes | 0.7 | 91 | 1 |
| 3 | M | 82 | NOS | 2.1 | 3.4 | Yes | 0.9 | 80 | 2 |
| 4 | M | 26 | Perihiliar | 4.3 | 3.1 | No | 1.5 | 110 | 1 |
| 5 | M | 68 | NOS | 1.3 | 4.2 | Yes | 1.5 | 46 | 3a |
| 6 | M | 52 | NOS | 2.6 | 3.2 | Yes | 1.8 | 42 | 3b |
| 7 | M | 38 | NOS | 0.4 | 3.8 | No | 2.2 | 37 | 3b |
| 8 | M | 70 | NOS | 6.5 | 3.8 | Yes | 1.9 | 35 | 3b |
| 9 | F | 37 | NOS | 1.3 | 4.1 | Yes | 1.3 | 49 | 3a |
| 10 | M | 79 | NOS | 1.2 | 3.6 | Yes | 2.4 | 28 | 4 |
| 11 | M | 65 | NOS | 6.5 | 3.6 | Yes | 3.8 | 17 | 4 |
| 12 | M | 56 | NOS | 3.8 | 3.4 | Yes | 1.0 | 60 | 2 |
| 13 | M | 62 | NOS | 6.6 | 3.1 | Yes | 2.8 | 25 | 4 |
| 14 | M | 43 | NOS | 6.3 | 3.0 | Yes | 1.8 | 60 | 2 |
| 15 | M | 56 | NOS | 5.5 | 3.3 | No | 0.9 | 100 | 1 |
| 16 | F | 74 | Tip lesion | 1.6 | 3.8 | No | 1.0 | 90 | 1 |
| 17 | M | 54 | NOS | 12.9 | 2.8 | Yes | 0.9 | 100 | 1 |
| 18 | M | 73 | Cellular | 1.6 | 2.8 | Yes | 3.3 | 30 | 3b |
| 19 | M | 53 | NOS | 6.5 | 3.0 | Yes | 0.9 | 75 | 2 |
| 20 | F | 67 | NOS | 32.0 | 1.9 | Yes | 1.4 | 37 | 3b |
| 21 | F | 29 | Perihiliar | 6.2 | 3.5 | No | 2.3 | 35 | 3b |
| 22 | F | 18 | NOS | 1.9 | 4.0 | No | 1.2 | 100 | 1 |
| 23 | M | 19 | Perihiliar | 15.0 | 2.7 | Yes | 0.8 | 100 | 1 |
| Gene | Variant | Frequency in Patients (n = 23) | Frequency in European Population (dbSNP, 1000 G) | Zygosity | ACMG |
|---|---|---|---|---|---|
| CDH1 | c.499 G>T (p.Glu167 *) | (5) 0.2174 | Het | LP | |
| COL1A2 | c.1015 A>C (p.Thr339Pro) | (9) 0.3913 | Het | US | |
| ITGB1 | c.1807 A>T (p.Asn603Tyr) | (3) 0.1304 | Het | US | |
| ITGB3 | c.1199 G>A (p.Cys400Tyr) | (2) 0.0869 | 0.0000 | Het | P |
| c.2351 C>T (p.Thr784Met) | (1) 0.0435 | 0.0010 | Het | US | |
| LAMC1 | c.148 T>C (p.Cys50Arg) | (1) 0.0435 | Het | US | |
| LPAR3 | c.524 C>G (p.Ala175Gly) | (5) 0.2174 | Het | US | |
| LPAR4 | c.260 T>C (p.Leu87Pro) | (9) 0.3913 | Het | US | |
| c.259 C>G (p.Leu87Val) | (3) 0.1304 | Het | US | ||
| LPAR6 | c.227 A>T ( p.Tyr76Phe) | (7) 0.3043 | Het | US | |
| c.998 T>C (p.Leu333Ser) | (2) 0.0869 | Het | US | ||
| WASF3 | c.934 G>C (p.Ala312Pro) | (1) 0.0435 | Het | US |
| Gene | rs | Nucleotide Change | Amino Acid Change | Type of Alteration | ACMG |
|---|---|---|---|---|---|
| CDH1 | - | c.500 A>G | p.Glu167Gly | Missense | LB |
| - | c.499 G>T | p.Glu167 * | Nonsense | LP | |
| COL1A2 | - | c.1015 A>C | p.Thr339Pro | Missense | US |
| ELN | - | c.1435 G>A | p.Val479Met | Missense | LB |
| ICAM1 | - | Exon 3 + 1 G>A | - | Essential splice | - |
| ITGB1 | - | c.1807 A>T | p.Asn603Tyr | Missense | US |
| LAMC1 | - | c.148 T>C | p.Cys50Arg | Missense | US |
| LPAR3 | - | c.524 C>G | p.Ala175Gly | Missense | US |
| LPAR4 | - | c.259 C>G | p.Leu87Val | Missense | US |
| - | c.260 T>C | p.Leu87Pro | US | ||
| LPAR6 | - | c.227 A>T | p.Tyr76Phe | Missense | US |
| - | c.998 T>C | p.Leu333Ser | US | ||
| TNC | - | c.4241 G>C | p.Arg1414Thr | Missense | LB |
| - | Exon 28—2 A>T | - | Essential splice | - | |
| WASF3 | - | c.934 G>C | p.Ala312Pro | Missense | US |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcos González, S.; Rodrigo Calabia, E.; Varela, I.; Červienka, M.; Freire Salinas, J.; Gómez Román, J.J. High Rate of Mutations of Adhesion Molecules and Extracellular Matrix Glycoproteins in Patients with Adult-Onset Focal and Segmental Glomerulosclerosis. Biomedicines 2023, 11, 1764. https://doi.org/10.3390/biomedicines11061764
Marcos González S, Rodrigo Calabia E, Varela I, Červienka M, Freire Salinas J, Gómez Román JJ. High Rate of Mutations of Adhesion Molecules and Extracellular Matrix Glycoproteins in Patients with Adult-Onset Focal and Segmental Glomerulosclerosis. Biomedicines. 2023; 11(6):1764. https://doi.org/10.3390/biomedicines11061764
Chicago/Turabian StyleMarcos González, Sara, Emilio Rodrigo Calabia, Ignacio Varela, Michal Červienka, Javier Freire Salinas, and José Javier Gómez Román. 2023. "High Rate of Mutations of Adhesion Molecules and Extracellular Matrix Glycoproteins in Patients with Adult-Onset Focal and Segmental Glomerulosclerosis" Biomedicines 11, no. 6: 1764. https://doi.org/10.3390/biomedicines11061764