
Leukocyte Imbalances in Mucopolysaccharidoses Patients

 , , , , , , ,
, , , , , , ,  , , and
, , and 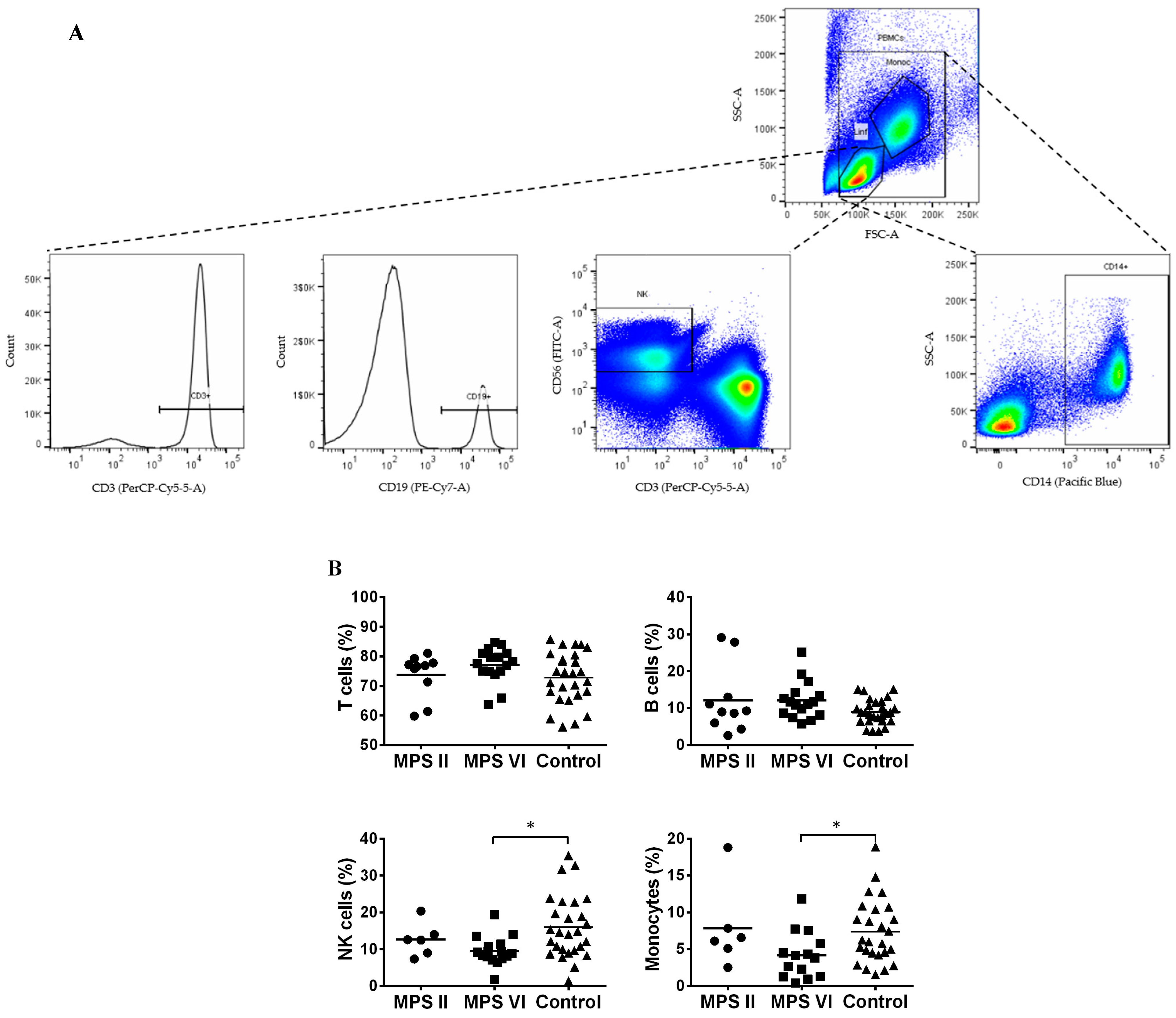{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Population Studied
2.2. Patient Population
2.3. Control Population
2.4. Isolation of Mononuclear Cells from Human Peripheral Blood and Flow Cytometry
2.5. Statistical Analysis
3. Results
3.1. Major Leukocyte Populations in MPS II and VI Disease Patients
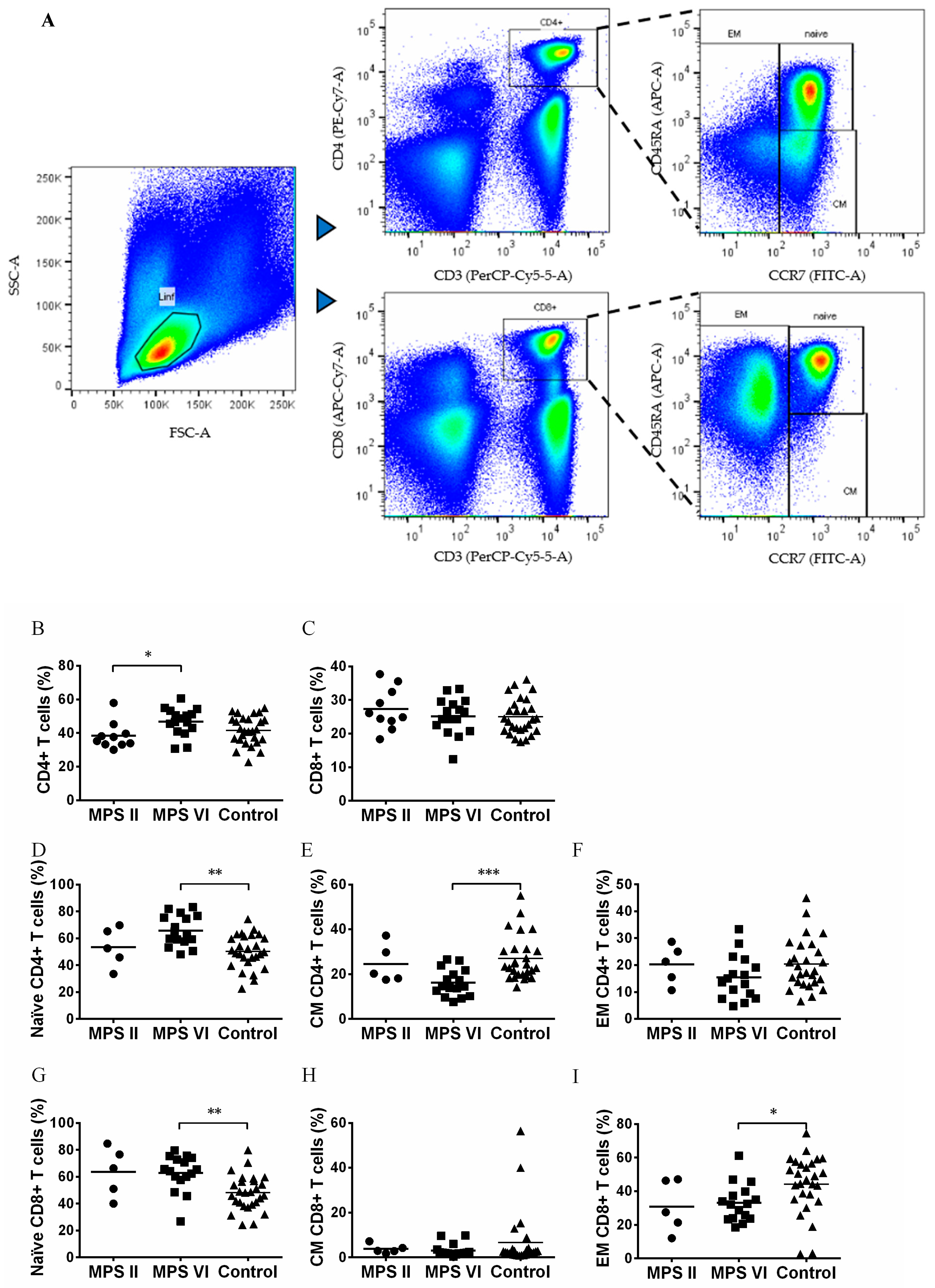
3.2. Naïve vs. Memory T Cell Pool in MPS II and VI Disease Patients
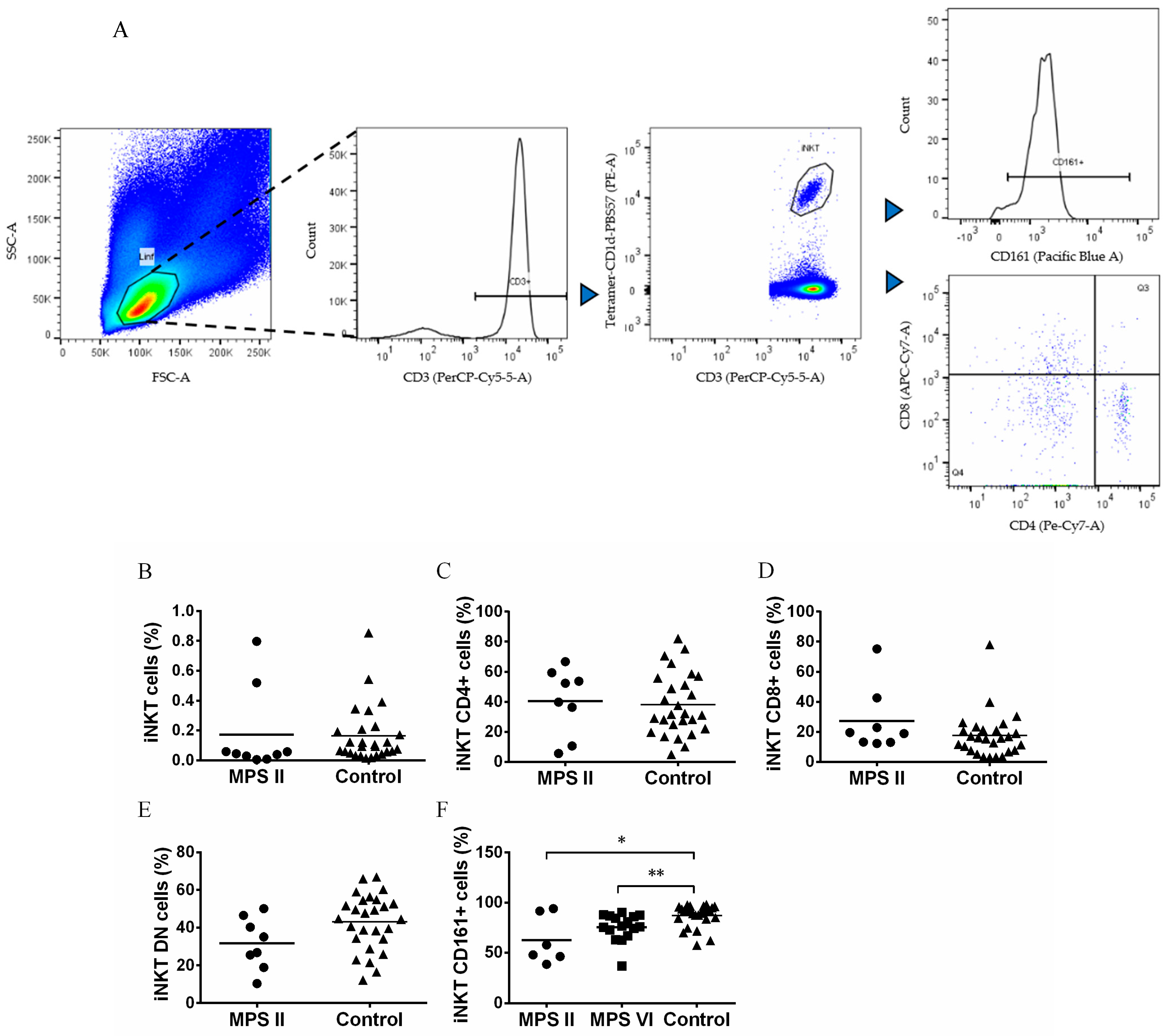
3.3. iNKT Cells Are Normal in Number but Phenotypically Altered in MPS II and MPS VI Disease Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coutinho, M.F.; Lacerda, L.; Alves, S. Glycosaminoglycan Storage Disorders: A Review. Biochem. Res. Int. 2012, 2012, 471325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Quiagen Site. QIAGEN Digital Insights. Available online: https://my.qiagendigitalinsights.com/ (accessed on 9 February 2023).
- Giugliani, R.; Federhen, A.; Rojas, M.V.M.; Vieira, T.; Artigalás, O.; Pinto, L.L.; Azevedo, A.C.; Acosta, A.; Bonfim, C.; Lourenço, C.M.; et al. Mucopolysaccharidosis I, II, and VI: Brief review and guidelines for treatment. Genet. Mol. Biol. 2010, 33, 589–604. [Google Scholar] [CrossRef]
- Parker, H.; Bigger, B.W. The role of innate immunity in mucopolysaccharide diseases. J. Neurochem. 2019, 148, 639–651. [Google Scholar] [CrossRef] [Green Version]
- Celik, B.; Tomatsu, S.C.; Tomatsu, S.; Khan, S.A. Epidemiology of Mucopolysaccharidoses Update. Diagnostics 2021, 11, 273. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.R.; Caseiro, C.; Lemos, M.; Lopes, L.; Fontes, A.; Ribeiro, H.; Pinto, E.; Silva, E.; Rocha, S.; Marcão, A.; et al. Prevalence of lysosomal storage diseases in Portugal. Eur. J. Hum. Genet. 2004, 12, 87–92. [Google Scholar] [CrossRef] [Green Version]
- D’avanzo, F.; Zanetti, A.; De Filippis, C.; Tomanin, R. Mucopolysaccharidosis Type VI, an Updated Overview of the Disease. Int. J. Mol. Sci. 2021, 22, 13456. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Wijburg, F.A. Therapy for the mucopolysaccharidoses. Rheumatology 2011, 50 (Suppl. S5), v49–v59. [Google Scholar] [CrossRef] [Green Version]
- Burton, B.K.; Whiteman, D.A.H. Incidence and timing of infusion-related reactions in patients with mucopolysaccharidosis type II (Hunter syndrome) on idursulfase therapy in the real-world setting: A perspective from the Hunter Outcome Survey (HOS). Mol. Genet. Metab. 2011, 103, 113–120. [Google Scholar] [CrossRef]
- Harmatz, P.; Giugliani, R.D.; Schwartz, I.V.; Guffon, N.; Teles, E.L.; Miranda, M.C.S.; Wraith, J.E.; Beck, M.; Arash, L.; Scarpa, M.; et al. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: Final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol. Genet. Metab. 2008, 94, 469–475. [Google Scholar] [CrossRef]
- Broomfield, A.; Jones, S.; Hughes, S.M.; Bigger, B. The impact of the immune system on the safety and efficiency of enzyme replacement therapy in lysosomal storage disorders. J. Inherit. Metab. Dis. 2016, 39, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Concolino, D.; Deodato, F.; Parini, R. Enzyme replacement therapy: Efficacy and limitations. Ital. J. Pediatr. 2018, 44, 120. [Google Scholar] [CrossRef]
- Donida, B.; Marchetti, D.P.; Biancini, G.B.; Deon, M.; Manini, P.R.; da Rosa, H.T.; Moura, D.J.; Saffi, J.; Bender, F.; Burin, M.G.; et al. Oxidative stress and inflammation in mucopolysaccharidosis type IVA patients treated with enzyme replacement therapy. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Viana, G.M.; Priestman, D.A.; Platt, F.M.; Khan, S.; Tomatsu, S.; Pshezhetsky, A.V. Brain Pathology in Mucopolysaccharidoses (MPS) Patients with Neurological Forms. J. Clin. Med. 2020, 9, 396. [Google Scholar] [CrossRef] [Green Version]
- Bigger, B.W.; Begley, D.J.; Virgintino, D.; Pshezhetsky, A.V. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2018, 125, 322–331. [Google Scholar] [CrossRef]
- Archer, L.D.; Langford-Smith, K.J.; Bigger, B.W.; Fildes, J.E. Mucopolysaccharide diseases: A complex interplay between neuroinflammation, microglial activation and adaptive immunity. J. Inherit. Metab. Dis. 2014, 37, 1–12. [Google Scholar] [CrossRef]
- Poli, E.F.; Zalfa, C.; D’avanzo, F.; Tomanin, R.; Carlessi, L.; Bossi, M.; Nodari, L.R.; Binda, E.; Marmiroli, P.; Scarpa, M.; et al. Murine neural stem cells model Hunter disease in vitro: Glial cell-mediated neurodegeneration as a possible mechanism involved. Cell Death Dis. 2013, 4, e906. [Google Scholar] [CrossRef] [Green Version]
- Simonaro, C.M.; Haskins, M.E.; Schuchman, E.H. Articular Chondrocytes from Animals with a Dermatan Sulfate Storage Disease Undergo a High Rate of Apoptosis and Release Nitric Oxide and Inflammatory Cytokines: A Possible Mechanism Underlying Degenerative Joint Disease in the Mucopolysaccharidoses. Lab. Investig. 2001, 81, 1319–1328. [Google Scholar] [CrossRef] [Green Version]
- Mandolfo, O.; Parker, H.; Bigger, B. Innate Immunity in Mucopolysaccharide Diseases. Int. J. Mol. Sci. 2022, 23, 1999. [Google Scholar] [CrossRef] [PubMed]
- Parini, R.; Deodato, F. Intravenous Enzyme Replacement Therapy in Mucopolysaccharidoses: Clinical Effectiveness and Limitations. Int. J. Mol. Sci. 2020, 21, 2975. [Google Scholar] [CrossRef] [PubMed]
- Campo, G.M.; Avenoso, A.; Campo, S.; D’Ascola, A.; Traina, P.; Samà, D.; Calatroni, A. Glycosaminoglycans modulate inflammation and apoptosis in LPS-treated chondrocytes. J. Cell. Biochem. 2009, 106, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Naur, O.M.; Anggraini, A.; Indraswari, B.W.; Wandita, S.; Wibowo, T.; Haksari, E.L. Immunisation issues in patient with mucopolysaccharidosis: A case report. Med. J. Malays. 2020, 75, 51–52. [Google Scholar]
- Opoka-Winiarska, V.; Jurecka, A.; Emeryk, A.; Tylki-Szymańska, A. Osteoimmunology in mucopolysaccharidoses type I, II, VI and VII. Immunological regulation of the osteoarticular system in the course of metabolic inflammation. Osteoarthr. Cartil. 2013, 21, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Davis, D.A.S.; Parish, C.R. Heparan Sulfate: A Ubiquitous Glycosaminoglycan with Multiple Roles in Immunity. Front. Immunol. 2013, 4, 470. [Google Scholar] [CrossRef] [Green Version]
- Johnson, G.B.; Brunn, G.J.; Kodaira, Y.; Platt, J.L. Receptor-Mediated Monitoring of Tissue Well-Being Via Detection of Soluble Heparan Sulfate by Toll-Like Receptor 4. J. Immunol. 2002, 168, 5233–5239. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, N.S.; Mancera, R.L. The Structure of Glycosaminoglycans and their Interactions with Proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef]
- Tessitore, A.; Pirozzi, M.; Auricchio, A. Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI. Pathogenetics 2009, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Archer, L.D.; Langford-Smith, K.J.; Critchley, W.R.; Bigger, B.W.; Fildes, J.E. Characterisation of the T cell and dendritic cell repertoire in a murine model of mucopolysaccharidosis I (MPS I). J. Inherit. Metab. Dis. 2013, 36, 257–262. [Google Scholar] [CrossRef]
- Daly, T.M.; Lorenz, R.G.; Sands, M.S. Abnormal Immune Function In Vivo in a Murine Model of Lysosomal Storage Disease. Pediatr. Res. 2000, 47, 757–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiRosario, J.; Divers, E.; Wang, C.; Etter, J.; Charrier, A.; Jukkola, P.; Auer, H.; Best, V.; Newsom, D.L.; McCarty, D.M.; et al. Innate and adaptive immune activation in the brain of MPS IIIB mouse model. J. Neurosci. Res. 2009, 87, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Kilavuz, S.; Kor, D.; Bulut, F.; Serbes, M.; Karagoz, D.; Altıntas, D.; Bisgin, A.; Seydaoğlu, G.; Mungan, H. Real-world patient data on immunity and COVID-19 status of patients with MPS, Gaucher, and Pompe diseases from Turkey. Arch. Pédiatrie 2022, 29, 415–423. [Google Scholar] [CrossRef]
- Pereira, C.S.; Pérez-Cabezas, B.; Ribeiro, H.; Maia, M.L.; Cardoso, M.T.; Dias, A.F.; Azevedo, O.; Ferreira, M.F.; Garcia, P.; Rodrigues, E.; et al. Lipid Antigen Presentation by CD1b and CD1d in Lysosomal Storage Disease Patients. Front. Immunol. 2019, 10, 1264. [Google Scholar] [CrossRef] [PubMed]
- Gadola, S.D.; Silk, J.D.; Jeans, A.; Illarionov, P.A.; Salio, M.; Besra, G.S.; Dwek, R.; Butters, T.D.; Platt, F.M.; Cerundolo, V. Impaired selection of invariant natural killer T cells in diverse mouse models of glycosphingolipid lysosomal storage diseases. J. Exp. Med. 2006, 203, 2293–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macedo, M.F.; Quinta, R.; Pereira, C.S.; Miranda, M.C.S. Enzyme replacement therapy partially prevents invariant Natural Killer T cell deficiency in the Fabry disease mouse model. Mol. Genet. Metab. 2012, 106, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.S.; Ribeiro, H.; Macedo, M.F. From Lysosomal Storage Diseases to NKT Cell Activation and Back. Int. J. Mol. Sci. 2017, 18, 502. [Google Scholar] [CrossRef] [Green Version]
- Melum, E.; Jiang, X.; Baker, K.D.; Macedo, M.F.; Fritsch, J.; Dowds, C.M.; Wang, J.; Pharo, A.; Kaser, A.; Tan, C.; et al. Control of CD1d-restricted antigen presentation and inflammation by sphingomyelin. Nat. Immunol. 2019, 20, 1644–1655. [Google Scholar] [CrossRef]
- Zahran, A.M.; Saad, K.; Elsayh, K.I.; Abdou, M.A.A.; Abo-Elgheet, A.M.; Eloseily, E.M.; Khalaf, S.M.; Sror, S.; Ahmad, F.-A.; Elhoufey, A.; et al. Upregulation of Cytotoxic T-cells in pediatric patients with Gaucher disease. Sci. Rep. 2022, 12, 4977. [Google Scholar] [CrossRef]
- Burstein, Y.; Zakuth, V.; Rechavi, G.; Spirer, Z. Abnormalities of cellular immunity and natural killer cells in Gaucher’s disease. J. Clin. Lab. Immunol. 1987, 23, 149–151. [Google Scholar]
- Speak, A.O.; Vruchte, D.T.; Davis, L.C.; Morgan, A.J.; Smith, D.A.; Yanjanin, N.M.; Simmons, L.; Hartung, R.; Runz, H.; Mengel, E.; et al. Altered distribution and function of natural killer cells in murine and human Niemann-Pick disease type C1. Blood 2014, 123, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Rozenfeld, P.; Agriello, E.; De Francesco, N.; Martinez, P.; Fossati, C. Leukocyte perturbation associated with Fabry disease. J. Inherit. Metab. Dis. 2009, 32, 67–77. [Google Scholar] [CrossRef]
- Bettman, N.; Avivi, I.; Rosenbaum, H.; Bisharat, L.; Katz, T. Impaired migration capacity in monocytes derived from patients with Gaucher disease. Blood Cells Mol. Dis. 2015, 55, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Zahran, A.M.; Eltayeb, A.A.; Elsayh, K.I.; Saad, K.; Ahmad, F.-A.; Ibrahim, A.I.M. Activated and Memory T Lymphocytes in Children with Gaucher Disease. Arch. Immunol. Ther. Exp. 2017, 65, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Rigante, D.; Cipolla, C.; Basile, U.; Gulli, F.; Savastano, M.C. Overview of immune abnormalities in lysosomal storage disorders. Immunol. Lett. 2017, 188, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Kieseier, B.C.; Wisniewski, K.E.; Goebel, H.H. The monocyte-macrophage system is affected in lysosomal storage diseases: An immunoelectron microscopic study. Acta Neuropathol. 1997, 94, 359–362. [Google Scholar] [CrossRef]
- Orange, J.S. Natural killer cell deficiency. J. Allergy Clin. Immunol. 2013, 132, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Orange, J.S. Human natural killer cell deficiencies. Curr. Opin. Allergy Clin. Immunol. 2006, 6, 399–409. [Google Scholar] [CrossRef]
- Small, C.-L.; McCormick, S.; Gill, N.; Kugathasan, K.; Santosuosso, M.; Donaldson, N.; Heinrichs, D.E.; Ashkar, A.; Xing, Z. NK Cells Play a Critical Protective Role in Host Defense against Acute Extracellular Staphylococcus aureus Bacterial Infection in the Lung. J. Immunol. 2008, 180, 5558–5568. [Google Scholar] [CrossRef] [Green Version]
- Valayannopoulos, V.; Nicely, H.; Harmatz, P.; Turbeville, S. Mucopolysaccharidosis VI. Orphanet J. Rare Dis. 2010, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Malm, D.; Halvorsen, D.S.; Tranebjærg, L.; Sjursen, H. Immunodeficiency in alpha-mannosidosis: A matched case-control study on immunoglobulins, complement factors, receptor density, phagocytosis and intracellular killing in leucocytes. Eur. J. Pediatr. 2000, 159, 699–703. [Google Scholar] [CrossRef]
- Otomo, T.; Schweizer, M.; Kollmann, K.; Schumacher, V.; Muschol, N.; Tolosa, E.; Mittrücker, H.-W.; Braulke, T. Mannose 6 phosphorylation of lysosomal enzymes controls B cell functions. J. Cell Biol. 2015, 208, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Sagiv, Y.; Hudspeth, K.; Mattner, J.; Schrantz, N.; Stern, R.K.; Zhou, D.; Savage, P.B.; Teyton, L.; Bendelac, A. Cutting Edge: Impaired Glycosphingolipid Trafficking and NKT Cell Development in Mice Lacking Niemann-Pick Type C1 Protein. J. Immunol. 2006, 177, 26–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrantz, N.; Sagiv, Y.; Liu, Y.; Savage, P.B.; Bendelac, A.; Teyton, L. The Niemann-Pick type C2 protein loads isoglobotrihexosylceramide onto CD1d molecules and contributes to the thymic selection of NKT cells. J. Exp. Med. 2007, 204, 841–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schümann, J.; Facciotti, F.; Panza, L.; Michieletti, M.; Compostella, F.; Collmann, A.; Mori, L.; De Libero, G. Differential alteration of lipid antigen presentation to NKT cells due to imbalances in lipid metabolism. Eur. J. Immunol. 2007, 37, 1431–1441. [Google Scholar] [CrossRef]
- McNab, F.W.; Berzins, S.P.; Pellicci, D.G.; Kyparissoudis, K.; Field, K.; Smyth, M.J.; Godfrey, D.I. The Influence of CD1d in Postselection NKT Cell Maturation and Homeostasis. J. Immunol. 2005, 175, 3762–3768. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, N.; Maia, M.L.; Pereira, C.S.; Mondragão-Rodrigues, I.; Martins, E.; Ribeiro, R.; Gaspar, A.; Aguiar, P.; Garcia, P.; Cardoso, M.T.; et al. Leukocyte Imbalances in Mucopolysaccharidoses Patients. Biomedicines 2023, 11, 1699. https://doi.org/10.3390/biomedicines11061699
Lopes N, Maia ML, Pereira CS, Mondragão-Rodrigues I, Martins E, Ribeiro R, Gaspar A, Aguiar P, Garcia P, Cardoso MT, et al. Leukocyte Imbalances in Mucopolysaccharidoses Patients. Biomedicines. 2023; 11(6):1699. https://doi.org/10.3390/biomedicines11061699
Chicago/Turabian StyleLopes, Nuno, Maria L. Maia, Cátia S. Pereira, Inês Mondragão-Rodrigues, Esmeralda Martins, Rosa Ribeiro, Ana Gaspar, Patrício Aguiar, Paula Garcia, Maria Teresa Cardoso, and et al. 2023. "Leukocyte Imbalances in Mucopolysaccharidoses Patients" Biomedicines 11, no. 6: 1699. https://doi.org/10.3390/biomedicines11061699