
N-Glycoprofiling of SLC35A2-CDG: Patient with a Novel Hemizygous Variant
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Participants
2.2. Isoelectric Focusing of Transferrin
2.3. Analysis of Serum N-Glycans
2.4. Analysis of Transferrin N-Glycans
2.5. Analysis of Apolipoprotein C-III
2.6. DNA Isolation, Whole Exome Sequencing (WES), and Data Analysis
2.7. Sanger Sequencing
2.8. Bioinformatics
2.9. Statistics
3. Results
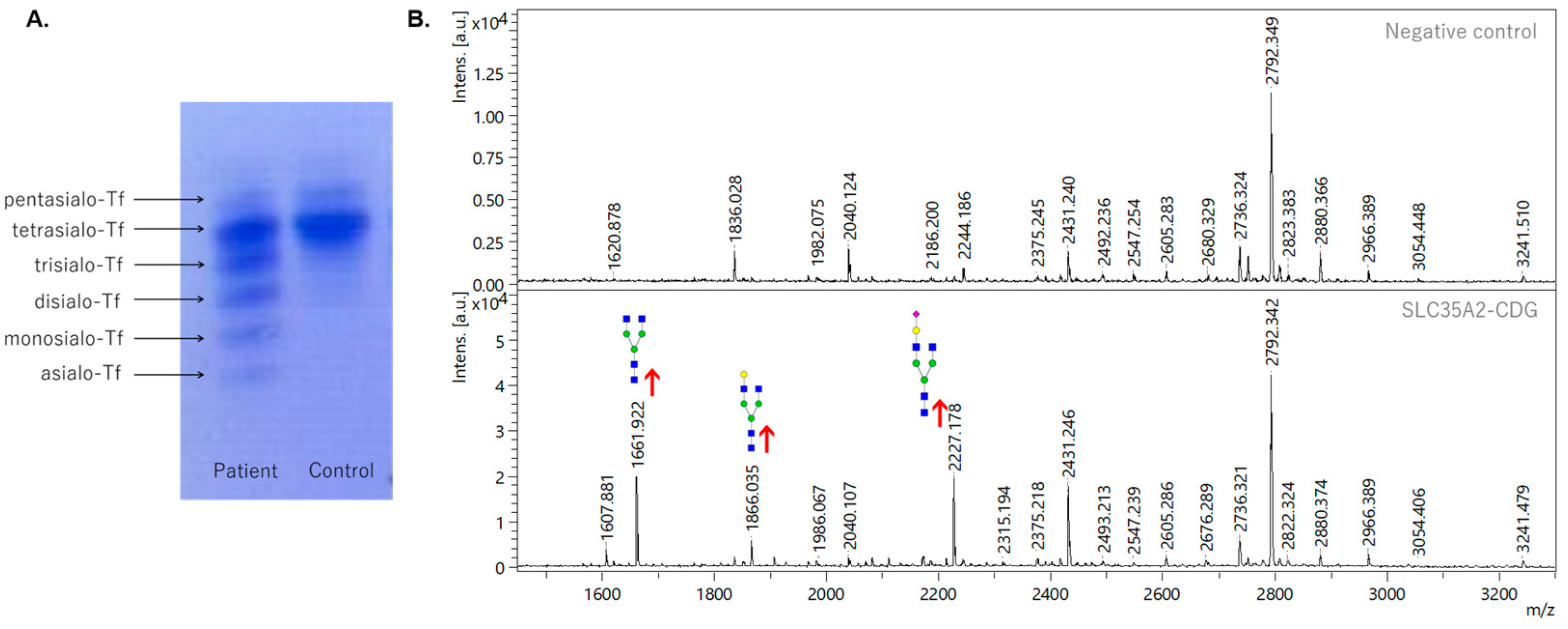
3.1. Analysis of Transferrin Glycosylation by Isoelectric Focusing and Mass Spectrometry
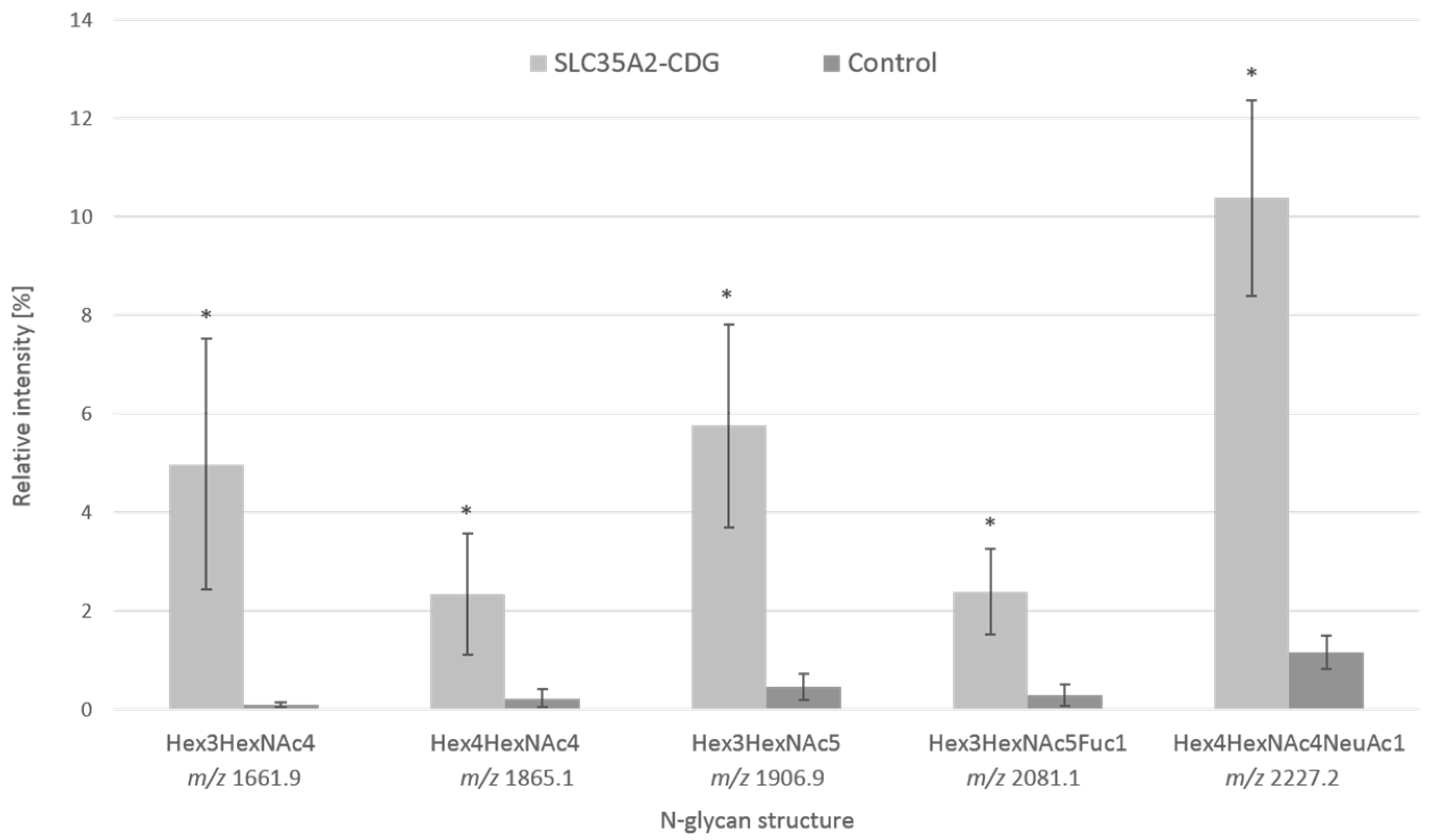
3.2. Analysis of Serum Protein Glycosylation by Mass Spectrometry
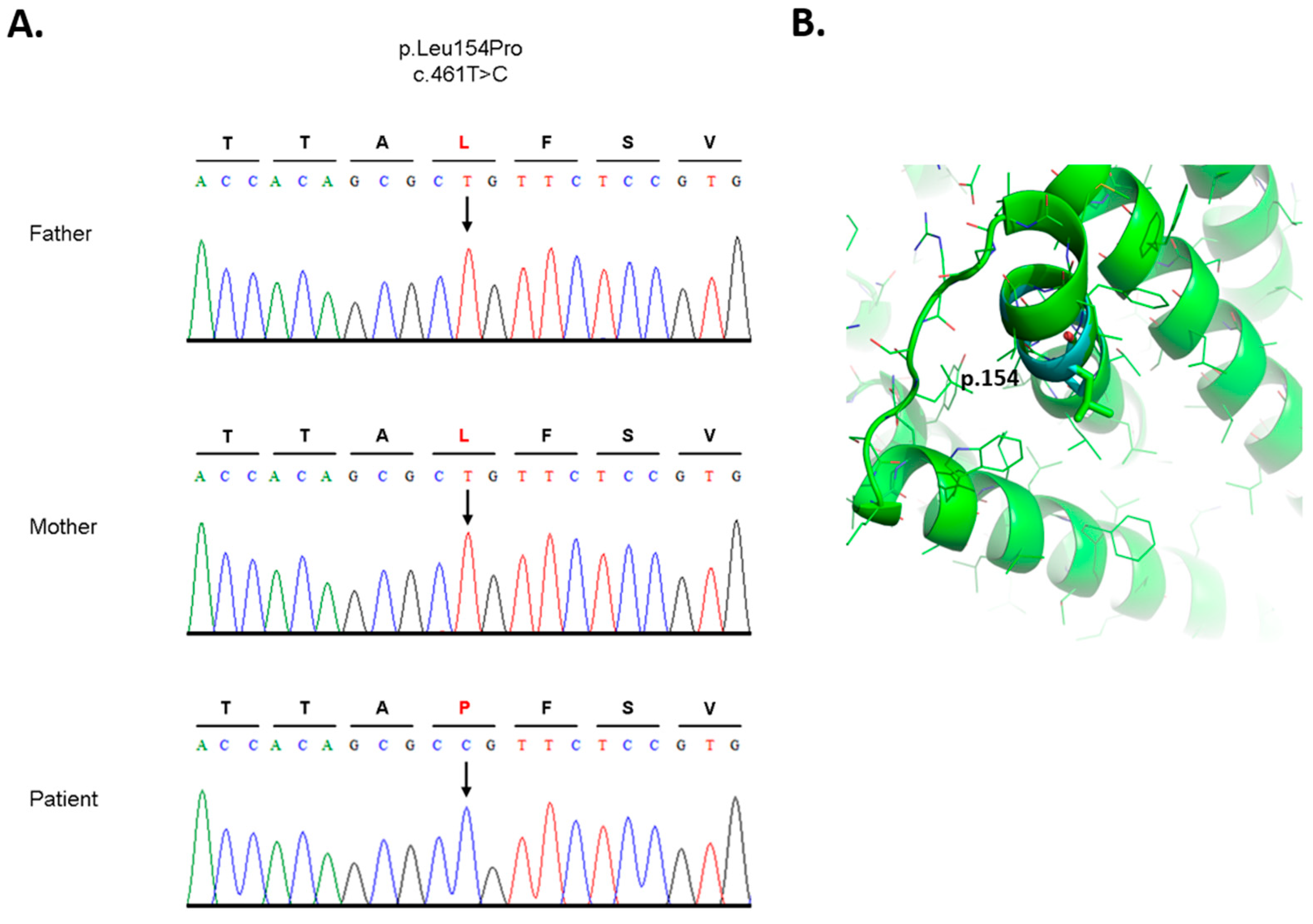
3.3. Analysis of DNA and Bioinformatic Processing
4. Discussion
- mutation at position chr1: 63648630 T > C (dbSNP ID: rs11208257, ClinVar ID: 297885) is homozygous in the patient and one of the parents, the other parent is heterozygous (Supplementary Figure S3),
- mutation chr1:63631761 C > T (dbSNP ID: rs1126728; ClinVar ID: 297874) is heterozygous in the patient and one of the parents, and homozygous in the other parent (Supplementary Figure S4) [38].
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Péanne, R.; de Lonlay, P.; Foulquier, F.; Kornak, U.; Lefeber, D.J.; Morava, E.; Pérez, B.; Seta, N.; Thiel, C.; Van Schaftingen, E.; et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur. J. Med. Genet. 2018, 61, 643–663. [Google Scholar] [CrossRef]
- Chang, I.J.; He, M.; Lam, C.T. Congenital disorders of glycosylation. Ann. Transl. Med. 2018, 6, 477. [Google Scholar] [CrossRef]
- Paprocka, J.; Jezela-Stanek, A.; Tylki-Szymańska, A.; Grunewald, S. Congenital Disorders of Glycosylation from a Neu-rological Perspective. Brain Sci. 2021, 11, 88. [Google Scholar] [CrossRef]
- Quelhas, D.; Correia, J.; Jaeken, J.; Azevedo, L.; Lopes-Marques, M.; Bandeira, A.; Keldermans, L.; Matthijs, G.; Sturiale, L.; Martins, E. SLC35A2-CDG: Novel variant and review. Mol. Genet. Metab. Rep. 2021, 26, 100717. [Google Scholar] [CrossRef] [PubMed]
- Kodera, H.; Nakamura, K.; Osaka, H.; Maegaki, Y.; Haginoya, K.; Mizumoto, S.; Kato, M.; Okamoto, N.; Iai, M.; Kondo, Y.; et al. De Novo Mutations in SLC35A2 Encoding a UDP-Galactose Transporter Cause Early-Onset Epileptic Encephalopathy. Hum. Mutat. 2013, 34, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Vals, M.-A.; Ashikov, A.; Ilves, P.; Loorits, D.; Zeng, Q.; Barone, R.; Huijben, K.; Sykut-Cegielska, J.; Diogo, L.; Elias, A.F.; et al. Clinical, neuroradiological, and biochemical features of SLC35A2-CDG patients. J. Inherit. Metab. Dis. 2019, 42, 553–564. [Google Scholar] [CrossRef]
- Witters, P.; Tahata, S.; Barone, R.; Õunap, K.; Salvarinova, R.; Grønborg, S.; Hoganson, G.; Scaglia, F.; Lewis, A.M.; Mori, M.; et al. Clinical and biochemical improvement with galactose supplementation in SLC35A2-CDG. Genet. Med. 2020, 22, 1102–1107. [Google Scholar] [CrossRef]
- Westenfield, K.; Sarafoglou, K.; Speltz, L.C.; Pierpont, E.I.; Steyermark, J.; Nascene, D.; Bower, M.; Pierpont, M.E. Mo-saicism of the UDP-Galactose transporter SLC35A2 in a female causing a congenital disorder of glycosylation: A case report. BMC Med. Genet. 2018, 19, 100. [Google Scholar] [CrossRef]
- Ng, B.G.; Sosicka, P.; Agadi, S.; Almannai, M.; Bacino, C.A.; Barone, R.; Botto, L.D.; Burton, J.E.; Carlston, C.; Chung, B.H.; et al. SLC35A2-CDG: Functional characterization, expanded molecular, clinical, and biochemical phenotypes of 30 unreported Individuals. Hum. Mutat. 2019, 40, 908–925. [Google Scholar] [CrossRef] [PubMed]
- de Jong, G.; van Eijk, H.G. Microheterogeneity of human serum transferrin: A biological phenomenon studied by isoelectric focusing in immobilized pH gradients. Electrophoresis 1988, 9, 589–598. [Google Scholar] [CrossRef]
- Hackler, R.; Arndt, T.; Kleine, T.O.; Gressner, A.M. Effect of Separation Conditions on Automated Isoelectric Focusing of Carbohydrate-Deficient Transferrin and Other Human Isotransferrins Using the PhastSystem. Anal. Biochem. 1995, 230, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Ziburová, J.; Nemčovič, M.; Šesták, S.; Bellová, J.; Pakanová, Z.; Siváková, B.; Šalingová, A.; Šebová, C.; Ostrožlíková, M.; Lekka, D.E.; et al. A novel homozygous mutation in the human ALG12 gene results in an aberrant profile of oligomannose N-Glycans in patient’s serum. Am. J. Med. Genet. A 2021, 185, 3494–3501. [Google Scholar] [CrossRef]
- Ceroni, A.; Maass, K.; Geyer, H.; Geyer, R.; Dell, A.; Haslam, S.M. GlycoWorkbench: A Tool for the Computer-Assisted Annotation of Mass Spectra of Glycans. J. Proteome Res. 2008, 7, 1650–1659. [Google Scholar] [CrossRef] [Green Version]
- Ondruskova, N.; Honzik, T.; Kolarova, H.; Pakanova, Z.; Mucha, J.; Zeman, J.; Hansikova, H. Aberrant apolipoprotein C-III glycosylation in glycogen storage disease type III and IX. Metabolism 2018, 82, 135–141. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 8 October 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.-C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017, 201178. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Patel, V.M.; Coon, M.; Nguyen, T.; Land, S.J.; Ruden, D.M.; Lu, X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012, 3, 35. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2019, 47, D158–D169. [Google Scholar] [CrossRef] [Green Version]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Goto, N.K.; A Williams, K.; Deber, C.M. Alpha-helical, but not beta-sheet, propensity of proline is determined by peptide environment. Proc. Natl. Acad. Sci. USA 1996, 93, 6676–6681. [Google Scholar] [CrossRef] [Green Version]
- Van Scherpenzeel, M.; Steenbergen, G.; Morava, E.; Wevers, R.A.; Lefeber, D. J High resolution mass spectrometry gly-coprofiling of intact transferrin for diagnosis and subtype identification in the congenital disorders of glycosylation. Transl. Res. 2015, 166, 639–649.e1. [Google Scholar] [CrossRef] [PubMed]
- Dörre, K.; Olczak, M.; Wada, Y.; Sosicka, P.; Grüneberg, M.; Reunert, J.; Kurlemann, G.; Fiedler, B.; Biskup, S.; Hörtnagel, K.; et al. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): Molecular basis, clinical phenotype, and therapeutic approach. J. Inherit. Metab. Dis. 2015, 38, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xia, B.; Gleason, T.J.; Castañeda, U.; He, M.; Berry, G.T.; Fridovich-Keil, J.L. N- and O-linked glycosylation of total plasma glycoproteins in galactosemia. Mol. Genet. Metab. 2012, 106, 442–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillard, M.; Morava, E.; van Delft, F.L.; Hague, R.; Körner, C.; Adamowicz, M.; Wevers, R.A.; Lefeber, D.J. Plasma N-Glycan Profiling by Mass Spectrometry for Congenital Disorders of Glycosylation Type II. Clin. Chem. 2011, 57, 593–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staretz-Chacham, O.; Noyman, I.; Wormser, O.; Abu Quider, A.; Hazan, G.; Morag, I.; Hadar, N.; Raymond, K.; Birk, O.S.; Ferreira, C.R.; et al. B4GALT1-congenital disorders of glycosylation: Expansion of the phenotypic and molecular spectrum and review of the literature. Clin. Genet. 2020, 97, 920–926. [Google Scholar] [CrossRef]
- Nilsson, I.; Sääf, A.; Whitley, P.; Gafvelin, G.; Waller, C.; von Heijne, G. Proline-induced disruption of a transmembrane α-helix in its natural environment. J. Mol. Biol. 1998, 284, 1165–1175. [Google Scholar] [CrossRef] [Green Version]
- Foulquier, F.; Amyere, M.; Jaeken, J.; Zeevaert, R.; Schollen, E.; Race, V.; Bammens, R.; Morelle, W.; Rosnoblet, C.; Legrand, D.; et al. TMEM165 Deficiency Causes a Congenital Disorder of Glycosylation. Am. J. Hum. Genet. 2012, 91, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Xia, B.; Zhang, W.; Li, X.; Jiang, R.; Harper, T.; Liu, R.; Cummings, R.D.; He, M. Serum N-glycan and O-glycan analysis by mass spectrometry for diagnosis of congenital disorders of glycosylation. Anal. Biochem. 2013, 442, 178–185. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Abuduxikuer, K.; Wang, J.-S. Four New Cases of SLC35A2-CDG With Novel Mutations and Clinical Features. Front. Genet. 2021, 12, 658786. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kodríková, R.; Pakanová, Z.; Krchňák, M.; Šedivá, M.; Šesták, S.; Květoň, F.; Beke, G.; Šalingová, A.; Skalická, K.; Brennerová, K.; et al. N-Glycoprofiling of SLC35A2-CDG: Patient with a Novel Hemizygous Variant. Biomedicines 2023, 11, 580. https://doi.org/10.3390/biomedicines11020580
Kodríková R, Pakanová Z, Krchňák M, Šedivá M, Šesták S, Květoň F, Beke G, Šalingová A, Skalická K, Brennerová K, et al. N-Glycoprofiling of SLC35A2-CDG: Patient with a Novel Hemizygous Variant. Biomedicines. 2023; 11(2):580. https://doi.org/10.3390/biomedicines11020580
Chicago/Turabian StyleKodríková, Rebeka, Zuzana Pakanová, Maroš Krchňák, Mária Šedivá, Sergej Šesták, Filip Květoň, Gábor Beke, Anna Šalingová, Katarína Skalická, Katarína Brennerová, and et al. 2023. "N-Glycoprofiling of SLC35A2-CDG: Patient with a Novel Hemizygous Variant" Biomedicines 11, no. 2: 580. https://doi.org/10.3390/biomedicines11020580