


NLRP3 Inflammasome’s Activation in Acute and Chronic Brain Diseases—An Update on Pathogenetic Mechanisms and Therapeutic Perspectives with Respect to Other Inflammasomes


Abstract
:
1. Introduction
1.1. An Overall Picture
NLRs Assembly and Signaling Activation
1.2. Brain NLRP3 Inflammasome
1.2.1. NLRP3 Inflammasome Priming and Canonical Activation
1.2.2. Noncanonical NLRP3 Activation
1.3. Brain NLRP3 Inflammasome’s Modulation by RNAs
1.4. Brain NLRP3 Inflammasome’s Modulation by Extracellular Vesicles (EVs) and Exosomes (Exos)
1.5. Other Brain NLRP3 Inflammasome Regulators
1.6. Brain NLRP3 Inflammasome Inhibitors
1.7. Brain NLRP3 Downregulation by Officinal Plant Agents/Herbal Extracts
2. NLRP3 Inflammasome in Brain Acute Injuries
3. NLRP3 Inflammasome in Chronic Neurodegenerative Disease
3.1. Alzheimer’s Disease (AD)
3.2. Parkinson’s Disease (PD)
3.3. Multiple Sclerosis (MS) and Experimental Autoimmune (or Allergic) Encephalomyelitis (EAE)
3.4. Amyotrophic Lateral Sclerosis (ALS)
3.5. Huntington’s Disease (HD)
4. Brain NLRP3 and Neurotropic Viruses Infections
4.1. Zika Virus (ZIKV) Encephalitis
4.2. West Nile Virus (WNV) Encephalitis
4.3. Japanese Encephalitis Virus (JEV)
4.4. Human Immunodeficiency Virus-1 (HIV-1) Encephalitis
4.5. Viroporin Proteins
4.6. Encephalomyocarditis Virus (EMCV)
4.7. SARS-CoV-2 Encephalitis
5. Comments and Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization endorses global action plan on rising incidence of dementia. Nurs. Older People 2017, 29, 7. [CrossRef]
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O′Connor, K.; Keene, C.D. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol. Psychiatry 2022, 91, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celsius, A.C. De Medicina, Volume 3, Passim; Spencer WG Loeb Classical Library, Translator; Harvard University Press: Cambridge, MA, USA, 1935; ISBN 978-067-499-370-9. [Google Scholar]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.M., 3rd; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Cao, Z.; Wang, Y.; Long, Z.; He, G. Interaction between autophagy and the NLRP3 inflammasome. Acta Biochim. Biophys. Sin. 2019, 51, 1087–1095. [Google Scholar] [CrossRef]
- Bellut, M.; Papp, L.; Bieber, M.; Kraft, P.; Stoll, G.; Schuhmann, M.K. NLPR3 Inflammasome Inhibition Alleviates Hypoxic Endothelial Cell Death in Vitro and Protects Blood–Brain Barrier Integrity in Murine Stroke. Cell Death Dis. 2021, 13, 20. [Google Scholar] [CrossRef]
- Kreher, C.; Favret, J.; Maulik, M.; Shin, D. Lysosomal Functions in Glia Associated with Neurodegeneration. Biomolecules 2021, 11, 400. [Google Scholar] [CrossRef]
- Chiarini, A.; Dal Pra, I.; Gottardo, R.; Bortolotti, F.; Whitfield, J.F.; Armato, U. BH(4) (tetrahydrobiopterin)-dependent activation, but not the expression, of inducible NOS (nitric oxide synthase)-2 in proinflammatory cytokine-stimulated, cultured normal human astrocytes is mediated by MEK-ERK kinases. J. Cell. Biochem. 2005, 94, 731–743. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- de Alba, E. Structure, interactions and self-assembly of ASC-dependent inflammasomes. Arch. Biochem. Biophys. 2019, 670, 15–31. [Google Scholar] [CrossRef]
- Stehlik, C.; Lee, S.H.; Dorfleutner, A.; Stassinopoulos, A.; Sagara, J.; Reed, J.C. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J. Immunol. 2003, 171, 6154–6163. [Google Scholar] [CrossRef] [Green Version]
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Gambin, Y.; Giles, N.; O′Carroll, A.; Polinkovsky, M.; Hunter, D.; Sierecki, E. Single-molecule fluorescence reveals the oligomerization and folding steps driving the prion-like behavior of ASC. J. Mol. Biol. 2018, 430, 491–508. [Google Scholar] [CrossRef] [Green Version]
- Kesavardhana, S.; Kanneganti, T.D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int. Immunol. 2017, 29, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Lupfer, C.; Kanneganti, T.D. Unsolved mysteries in NLR biology. Front. Immunol. 2013, 4, 285. [Google Scholar] [CrossRef] [Green Version]
- Devi, S.; Stehlik, C.; Dorfleutner, A. An update on CARD only proteins (COPs) and PYD only proteins (POPs) as inflammasome regulators. Int. J. Mol. Sci. 2020, 21, 6901. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Fabi, C.; Bellet, M.M.; Costantini, C.; Nunziangeli, L.; Romani, L.; Brancorsini, S. Epigenetic mechanisms of inflammasome regulation. Int. J. Mol. Sci. 2020, 21, 5758. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Armato, U.; Gui, L.; Dal Prà, I. “Other Than NLRP3” Inflammasomes: Multiple Roles in Brain Disease. Neuroscientist 2022, 11, 10738584221106114. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71–76. [Google Scholar] [CrossRef]
- Chiarini, A.; Armato, U.; Hu, P.; Dal Prà, I. Danger-sensing/pattern recognition receptors and neuroinflammation in Alzheimer′s disease. Int. J. Mol. Sci. 2020, 21, 9036. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer′s Disease. Neurochem. Res. 2020, 45, 2560–2572. [Google Scholar] [CrossRef]
- Holbrook, J.A.; Jarosz-Griffiths, H.H.; Caseley, E.; Lara-Reyna, S.; Poulter, J.A.; Williams-Gray, C.H.; Peckham, D.; McDermott, M.F. Neurodegenerative Disease and the NLRP3 Inflammasome. Front. Pharmacol. 2021, 12, 643254. [Google Scholar] [CrossRef]
- Mészáros, Á.; Molnár, K.; Nógrádi, B.; Hernádi, Z.; Nyúl-Tóth, Á.; Wilhelm, I.; Krizbai, I.A. Neurovascular Inflammaging in Health and Disease. Cells 2020, 9, 1614. [Google Scholar] [CrossRef]
- Lee, G.S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflamm. 2018, 15, 242. [Google Scholar] [CrossRef] [Green Version]
- Su, S.H.; Wu, Y.F.; Wang, D.P.; Hai, J. Inhibition of excessive autophagy and mitophagy mediates neuroprotective effects of URB597 against chronic cerebral hypoperfusion. Cell Death Dis. 2018, 9, 733. [Google Scholar] [CrossRef] [Green Version]
- Su, S.H.; Wu, Y.F.; Lin, Q.; Wang, D.P.; Hai, J. URB597 protects against NLRP3 inflammasome activation by inhibiting autophagy dysfunction in a rat model of chronic cerebral hypoperfusion. J. Neuroinflamm. 2019, 16, 260. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.J.; Yu, B.Y.; Huang, X.K.; He, M.Z.; Chen, B.W.; Chen, T.T.; Fang, H.Y.; Chen, S.Q.; Fu, X.Q.; Li, P.J.; et al. Neferine Protects against Hypoxic-Ischemic Brain Damage in Neonatal Rats by Suppressing NLRP3-Mediated Inflammasome Activation. Oxid. Med. Cell. Longev. 2021, 2021, 6654954. [Google Scholar] [CrossRef]
- Franke, M.; Bieber, M.; Kraft, P.; Weber, A.N.R.; Stoll, G.; Schuhmann, M.K. The NLRP3 inflammasome drives inflammation in ischemia/reperfusion injury after transient middle cerebral artery occlusion in mice. Brain Behav. Immun. 2021, 92, 223–233. [Google Scholar] [CrossRef]
- Xu, Q.; Zhao, B.; Ye, Y.; Li, Y.; Zhang, Y.; Xiong, X.; Gu, L. Relevant mediators involved in and therapies targeting the inflammatory response induced by activation of the NLRP3 inflammasome in ischemic stroke. J. Neuroinflamm. 2021, 18, 123. [Google Scholar] [CrossRef]
- Chen, S.H.; Scott, X.O.; Ferrer Marcelo, Y.; Almeida, V.W.; Blackwelder, P.L.; Yavagal, D.R.; Peterson, E.C.; Starke, R.M.; Dietrich, W.D.; Keane, R.W.; et al. Netosis and Inflammasomes in Large Vessel Occlusion Thrombi. Front. Pharmacol. 2021, 11, 607287. [Google Scholar] [CrossRef]
- Xiao, L.; Zheng, H.; Li, J.; Wang, Q.; Sun, H. Neuroinflammation Mediated by NLRP3 Inflammasome after Intracerebral Hemorrhage and Potential Therapeutic Targets. Mol. Neurobiol. 2020, 57, 5130–5149. [Google Scholar] [CrossRef]
- Yang, S.J.; Shao, G.F.; Chen, J.L.; Gong, J. The NLRP3 Inflammasome: An Important Driver of Neuroinflammation in Hemorrhagic Stroke. Cell. Mol. Neurobiol. 2018, 38, 595–603. [Google Scholar] [CrossRef]
- Cristina de Brito Toscano, E.; Leandro Marciano Vieira, É.; Boni Rocha Dias, B.; Vidigal Caliari, M.; Paula Gonçalves, A.; Varela Giannetti, A.; Maurício Siqueira, J.; Kimie Suemoto, C.; Elaine Paraizo Leite, R.; Nitrini, R.; et al. NLRP3 and NLRP1 inflammasomes are up-regulated in patients with mesial temporal lobe epilepsy and may contribute to overexpression of caspase-1 and IL-β in sclerotic hippocampi. Brain Res. 2021, 1752, 147230. [Google Scholar] [CrossRef]
- Wang, S.; He, H.; Long, J.; Sui, X.; Yang, J.; Lin, G.; Wang, Q.; Wang, Y.; Luo, Y. TRPV4 Regulates Soman-Induced Status Epilepticus and Secondary Brain Injury via NMDA Receptor and NLRP3 Inflammasome. Neurosci. Bull. 2021, 37, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, J.; Jiang, W.; Cao, Z.; Zhao, F.; Cai, T.; Aschner, M.; Luo, W. The role of NLRP3-CASP1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability. Autophagy 2017, 13, 914–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Rokad, D.; Malovic, E.; Luo, J.; Harischandra, D.S.; Jin, H.; Anantharam, V.; Huang, X.; Lewis, M.; Kanthasamy, A.; et al. Manganese activates NLRP3 inflammasome signaling and propagates exosomal release of ASC in microglial cells. Sci. Signal. 2019, 12, eaat9900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, P.; Wang, D.; Cao, Z.; Chen, J.; Zhang, J. The role of NLRP3 in lead-induced neuroinflammation and possible underlying mechanism. Environ. Pollut. 2021, 287, 117520. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wang, X.; Xu, C.; Gao, M.; Wang, S.; Zhang, J.; Tong, H.; Wang, L.; Han, Y.; Cheng, N.; et al. Inhibiting NLRP3 inflammasome activation prevents copper-induced neuropathology in a murine model of Wilson′s disease. Cell Death Dis. 2021, 12, 87. [Google Scholar] [CrossRef]
- Cai, J.; Guan, H.; Jiao, X.; Yang, J.; Chen, X.; Zhang, H.; Zheng, Y.; Zhu, Y.; Liu, Q.; Zhang, Z. NLRP3 inflammasome mediated pyroptosis is involved in cadmium exposure-induced neuroinflammation through the IL-1β/IkB-α-NF-κB-NLRP3 feedback loop in swine. Toxicology 2021, 453, 152720. [Google Scholar] [CrossRef]
- Brewer, G.J. Divalent Copper as a Major Triggering Agent in Alzheimer′s Disease. J. Alzheimer’s Dis. 2015, 46, 593–604. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, Y.; Lu, L.; Zhang, H.; Zhao, C.; Pu, Y.; Yin, L. Copper induces microglia-mediated neuroinflammation through ROS/NF-κB pathway and mitophagy disorder. Food Chem. Toxicol. 2022, 16, 113369. [Google Scholar] [CrossRef]
- Quandt, D.; Rothe, K.; Baerwald, C.; Rossol, M. GPRC6A mediates Alum-induced Nlrp3 inflammasome activation but limits Th2 type antibody responses. Sci. Rep. 2015, 5, 16719. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Pi, M.; Nooh, M.M.; Bahout, S.W.; Quarles, L.D. Human GPRC6A Mediates Testosterone-Induced Mitogen-Activated Protein Kinases and mTORC1 Signaling in Prostate Cancer Cells. Mol. Pharmacol. 2019, 95, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shu, Q.; Liu, Z.; Gao, C.; Wang, Z.; Xing, Z.; Song, J. Recombinant osteopontin provides protection for cerebral infarction by inhibiting the NLRP3 inflammasome in microglia. Brain Res. 2021, 1751, 147170. [Google Scholar] [CrossRef]
- Chen, Y.; Meng, J.; Bi, F.; Li, H.; Chang, C.; Ji, C.; Liu, W. NEK7 Regulates NLRP3 Inflammasome Activation and Neuroinflammation Post-traumatic Brain Injury. Front. Mol. Neurosci. 2019, 12, 202, Erratum in Front. Mol. Neurosci. 2019, 12, 247. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Song, Z.; He, J.; Guo, S.; Chen, Y.; Wang, H.; Zhang, J.; Xu, X.; Liu, J. NIMA-related kinase 7 amplifies NLRP3 inflammasome pro-inflammatory signaling in microglia/macrophages and mice models of spinal cord injury. Exp. Cell Res. 2021, 398, 112418. [Google Scholar] [CrossRef]
- O′Brien, W.T.; Pham, L.; Symons, G.F.; Monif, M.; Shultz, S.R.; McDonald, S.J. The NLRP3 inflammasome in traumatic brain injury: Potential as a biomarker and therapeutic target. J. Neuroinflamm. 2020, 17, 104. [Google Scholar] [CrossRef]
- Irrera, N.; Russo, M.; Pallio, G.; Bitto, A.; Mannino, F.; Minutoli, L.; Altavilla, D.; Squadrito, F. The Role of NLRP3 Inflammasome in the Pathogenesis of Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 6204. [Google Scholar] [CrossRef]
- Albalawi, F.; Lu, W.; Beckel, J.M.; Lim, J.C.; McCaughey, S.A.; Mitchell, C.H. The P2X7 Receptor Primes IL-1β and the NLRP3 Inflammasome in Astrocytes Exposed to Mechanical Strain. Front. Cell. Neurosci. 2017, 11, 227. [Google Scholar] [CrossRef]
- Ding, H.; Li, Y.; Wen, M.; Liu, X.; Han, Y.; Zeng, H. Elevated intracranial pressure induces IL1β and IL18 overproduction via activation of the NLRP3 inflammasome in microglia of ischemic adult rats. Int. J. Mol. Med. 2021, 47, 183–194. [Google Scholar] [CrossRef]
- Chi, W.; Chen, H.; Li, F.; Zhu, Y.; Yin, W.; Zhuo, Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-κB pathway in acute glaucoma. J. Neuroinflamm. 2015, 12, 137. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Maphis, N.M.; Binder, J.; Chisholm, D.; Weston, L.; Duran, W.; Peterson, C.; Zimmerman, A.; Mandell, M.A.; Jett, S.D.; et al. Proteopathic tau primes and activates interleukin-1β via myeloid-cell-specific MyD88- and NLRP3-ASC-inflammasome pathway. Cell Rep. 2021, 36, 109720. [Google Scholar] [CrossRef]
- Shi, F.; Yang, L.; Kouadir, M.; Yang, Y.; Wang, J.; Zhou, X.; Yin, X.; Zhao, D. The NALP3 inflammasome engages in neurotoxic prion peptide-induced microglial activation. J. Neuroinflamm. 2012, 9, 73. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.; Yao, H.; Shah, S.Z.A.; Wu, W.; Wang, D.; Zhao, Y.; Wang, L.; Zhou, X.; Zhao, D.; Yang, L. The NLRP3-Caspase 1 Inflammasome Negatively Regulates Autophagy via TLR4-TRIF in Prion Peptide-Infected Microglia. Front. Aging Neurosci. 2018, 10, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, M.T.; Maddugoda, M.; Götz, J.; Burgener, S.S.; Schroder, K. The NLRP3 inflammasome triggers sterile neuroinflammation and Alzheimer′s disease. Curr. Opin. Immunol. 2021, 68, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Pike, A.F.; Varanita, T.; Herrebout, M.A.C.; Plug, B.C.; Kole, J.; Musters, R.J.P.; Teunissen, C.E.; Hoozemans, J.J.M.; Bubacco, L.; Veerhuis, R. α-Synuclein evokes NLRP3 inflammasome-mediated IL-1β secretion from primary human microglia. Glia 2021, 69, 1413–1428. [Google Scholar] [CrossRef] [PubMed]
- Deora, V.; Lee, J.D.; Albornoz, E.A.; McAlary, L.; Jagaraj, C.J.; Robertson, A.A.B.; Atkin, J.D.; Cooper, M.A.; Schroder, K.; Yerbury, J.J.; et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 2020, 68, 407–421, Erratum in Glia 2020, 68, 2167–2168. [Google Scholar] [CrossRef] [PubMed]
- Ismael, S.; Nasoohi, S.; Li, L.; Aslam, K.S.; Khan, M.M.; El-Remessy, A.B.; McDonald, M.P.; Liao, F.F.; Ishrat, T. Thioredoxin interacting protein regulates age-associated neuroinflammation. Neurobiol. Dis. 2021, 156, 105399. [Google Scholar] [CrossRef]
- Ismael, S.; Wajidunnisa; Sakata, K.; McDonald, M.P.; Liao, F.F.; Ishrat, T. ER stress associated TXNIP-NLRP3 inflammasome activation in hippocampus of human Alzheimer′s disease. Neurochem. Int. 2021, 148, 105104. [Google Scholar] [CrossRef]
- Shen, H.; Guan, Q.; Zhang, X.; Yuan, C.; Tan, Z.; Zhai, L.; Hao, Y.; Gu, Y.; Han, C. New mechanism of neuroinflammation in Alzheimer′s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 100, 109884. [Google Scholar] [CrossRef]
- Shukla, P.K.; Delotterie, D.F.; Xiao, J.; Pierre, J.F.; Rao, R.; McDonald, M.P.; Khan, M.M. Alterations in the Gut-Microbial-Inflammasome-Brain Axis in a Mouse Model of Alzheimer′s Disease. Cells 2021, 10, 779. [Google Scholar] [CrossRef]
- Yi, W.; Cheng, J.; Wei, Q.; Pan, R.; Song, S.; He, Y.; Tang, C.; Liu, X.; Zhou, Y.; Su, H. Effect of temperature stress on gut-brain axis in mice: Regulation of intestinal microbiome and central NLRP3 inflammasomes. Sci. Total Environ. 2021, 772, 144568. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Wang, B.R.; Shi, J.Q.; Ge, N.N.; Ou, Z.; Tian, Y.Y.; Jiang, T.; Zhou, J.S.; Xu, J.; Zhang, Y.D. PM2.5 exposure aggravates oligomeric amyloid beta-induced neuronal injury and promotes NLRP3 inflammasome activation in an in vitro model of Alzheimer′s disease. J. Neuroinflamm. 2018, 15, 132. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.Q.; Wang, B.R.; Jiang, T.; Gao, L.; Zhang, Y.D.; Xu, J. NLRP3 Inflammasome: A Potential Therapeutic Target in Fine Particulate Matter-Induced Neuroinflammation in Alzheimer′s Disease. J. Alzheimers Dis. 2020, 77, 923–934. [Google Scholar] [CrossRef]
- Yuan, L.; Zhu, Y.; Huang, S.; Lin, L.; Jiang, X.; Chen, S. NF-κB/ROS and ERK pathways regulate NLRP3 inflammasome activation in Listeria monocytogenes infected BV2 microglia cells. J. Microbiol. 2021, 59, 771–781. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, Y.; Zhou, R.; Li, Y.; Gao, Y.; Tu, D.; Wilson, B.; Song, S.; Feng, J.; Hong, J.S.; et al. A novel role of NLRP3-generated IL-1β in the acute-chronic transition of peripheral lipopolysaccharide-elicited neuroinflammation: Implications for sepsis-associated neurodegeneration. J. Neuroinflamm. 2020, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Danielski, L.G.; Giustina, A.D.; Bonfante, S.; de Souza Goldim, M.P.; Joaquim, L.; Metzker, K.L.; Biehl, E.B.; Vieira, T.; de Medeiros, F.D.; da Rosa, N.; et al. NLRP3 Activation Contributes to Acute Brain Damage Leading to Memory Impairment in Sepsis-Surviving Rats. Mol. Neurobiol. 2020, 57, 5247–5262. [Google Scholar] [CrossRef]
- Chivero, E.T.; Guo, M.L.; Periyasamy, P.; Liao, K.; Callen, S.E.; Buch, S. HIV-1 Tat Primes and Activates Microglial NLRP3 Inflammasome-Mediated Neuroinflammation. J. Neurosci. 2017, 37, 3599–3609. [Google Scholar] [CrossRef] [Green Version]
- Katuri, A.; Bryant, J.; Heredia, A.; Makar, T.K. Role of the inflammasomes in HIV-associated neuroinflammation and neurocognitive disorders. Exp. Mol. Pathol. 2019, 108, 64–72. [Google Scholar] [CrossRef]
- He, X.; Yang, W.; Zeng, Z.; Wei, Y.; Gao, J.; Zhang, B.; Li, L.; Liu, L.; Wan, Y.; Zeng, Q.; et al. NLRP3-dependent pyroptosis is required for HIV-1 gp120-induced neuropathology. Cell. Mol. Immunol. 2020, 17, 283–299. [Google Scholar] [CrossRef]
- Hu, X.; Zeng, Q.; Xiao, J.; Qin, S.; Wang, Y.; Shan, T.; Hu, D.; Zhu, Y.; Liu, K.; Zheng, K.; et al. Herpes Simplex Virus 1 Induces Microglia Gasdermin D-Dependent Pyroptosis through Activating the NLR Family Pyrin Domain Containing 3 Inflammasome. Front. Microbiol. 2022, 13, 838808. [Google Scholar] [CrossRef]
- Chen, C.J.; Ou, Y.C.; Chang, C.Y.; Pan, H.C.; Lin, S.Y.; Liao, S.L.; Raung, S.L.; Chen, S.Y.; Chang, C.J. Src signaling involvement in Japanese encephalitis virus-induced cytokine production in microglia. Neurochem. Int. 2011, 58, 924–933. [Google Scholar] [CrossRef]
- He, Z.; Chen, J.; Zhu, X.; An, S.; Dong, X.; Yu, J.; Zhang, S.; Wu, Y.; Li, G.; Zhang, Y.; et al. NLRP3 Inflammasome Activation Mediates Zika Virus-Associated Inflammation. J. Infect. Dis. 2018, 217, 1942–1951. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef] [PubMed]
- de Rivero Vaccari, J.C.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. The inflammasome in times of COVID-19. Front. Immunol. 2020, 11, 583373. [Google Scholar] [CrossRef]
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y.; et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 2021, 12, 4664, Erratum in Nat. Commun. 2021, 12, 5306. [Google Scholar] [CrossRef]
- Yalcinkaya, M.; Liu, W.; Islam, M.N.; Kotini, A.G.; Gusarova, G.A.; Fidler, T.P.; Papapetrou, E.P.; Bhattacharya, J.; Wang, N.; Tall, A.R. Modulation of the NLRP3 inflammasome by SARS-CoV-2 Envelope protein. Sci. Rep. 2021, 11, 24432. [Google Scholar] [CrossRef]
- Olajide, O.A.; Iwuanyanwu, V.U.; Adegbola, O.D.; Al-Hindawi, A.A. SARS-CoV-2 Spike Glycoprotein S1 Induces Neuroinflammation in BV-2 Microglia. Mol. Neurobiol. 2022, 59, 445–458. [Google Scholar] [CrossRef]
- Ito, M.; Yanagi, Y.; Ichinohe, T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. 2012, 8, e1002857. [Google Scholar] [CrossRef] [Green Version]
- Moreira, J.D.; Iakhiaev, A.; Vankayalapati, R.; Jung, B.G.; Samten, B. Histone deacetylase-2 controls IL-1β production through the regulation of NLRP3 expression and activation in tuberculosis infection. iScience 2022, 25, 104799. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Litwiniuk, A.; Bik, W.; Kalisz, M.; Baranowska-Bik, A. Inflammasome NLRP3 Potentially Links Obesity-Associated Low-Grade Systemic Inflammation and Insulin Resistance with Alzheimer′s Disease. Int. J. Mol. Sci. 2021, 22, 5603. [Google Scholar] [CrossRef]
- Sobesky, J.L.; D′Angelo, H.M.; Weber, M.D.; Anderson, N.D.; Frank, M.G.; Watkins, L.R.; Maier, S.F.; Barrientos, R.M. Glucocorticoids Mediate Short-Term High-Fat Diet Induction of Neuroinflammatory Priming, the NLRP3 Inflammasome, and the Danger Signal HMGB1. eNeuro 2016, 3, ENEURO.0113-16.2016. [Google Scholar] [CrossRef] [Green Version]
- Keshk, W.A.; Ibrahim, M.A.; Shalaby, S.M.; Zalat, Z.A.; Elseady, W.S. Redox status, inflammation, necroptosis and inflammasome as indispensable contributors to high fat diet (HFD)-induced neurodegeneration; Effect of N-acetylcysteine (NAC). Arch. Biochem. Biophys. 2020, 680, 108227. [Google Scholar] [CrossRef]
- Wei, P.; Yang, F.; Zheng, Q.; Tang, W.; Li, J. The Potential Role of the NLRP3 Inflammasome Activation as a Link between Mitochondria ROS Generation and Neuroinflammation in Postoperative Cognitive Dysfunction. Front. Cell. Neurosci. 2019, 13, 73. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xiao, F.; Zhang, J.; Wang, X.; Ying, J.; Wei, G.; Chen, S.; Huang, X.; Yu, W.; Liu, X.; et al. Dexmedetomidine Mitigated NLRP3-Mediated Neuroinflammation via the Ubiquitin-Autophagy Pathway to Improve Perioperative Neurocognitive Disorder in Mice. Front. Pharmacol. 2021, 12, 646265. [Google Scholar] [CrossRef]
- Hirshman, N.A.; Hughes, F.M., Jr.; Jin, H.; Harrison, W.T.; White, S.W.; Doan, I.; Harper, S.N.; Leidig, P.D.; Purves, J.T. Cyclophosphamide-induced cystitis results in NLRP3-mediated inflammation in the hippocampus and symptoms of depression in rats. Am. J. Physiol. Renal Physiol. 2020, 318, F354–F362. [Google Scholar] [CrossRef]
- D′Espessailles, A.; Mora, Y.A.; Fuentes, C.; Cifuentes, M. Calcium-sensing receptor activates the NLRP3 inflammasome in LS14 preadipocytes mediated by ERK1/2 signaling. J. Cell. Physiol. 2018, 233, 6232–6240. [Google Scholar] [CrossRef]
- Wang, C.; Jia, Q.; Sun, C.; Jing, C. Calcium sensing receptor contribute to early brain injury through the CaMKII/NLRP3 pathway after subarachnoid hemorrhage in mice. Biochem. Biophys. Res. Commun. 2020, 530, 651–657. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, Y.; Wu, W.; Yin, Y.; Huang, D.; Wang, Y.; Li, W.; Li, W. Chronic glucocorticoids exposure enhances neurodegeneration in the frontal cortex and hippocampus via NLRP-1 inflammasome activation in male mice. Brain Behav. Immun. 2016, 52, 58–70. [Google Scholar] [CrossRef]
- Maturana, C.J.; Aguirre, A.; Sáez, J.C. High glucocorticoid levels during gestation activate the inflammasome in hippocampal oligodendrocytes of the offspring. Dev. Neurobiol. 2017, 77, 625–642. [Google Scholar] [CrossRef]
- Chivero, E.T.; Thangaraj, A.; Tripathi, A.; Periyasamy, P.; Guo, M.L.; Buch, S. NLRP3 Inflammasome Blockade Reduces Cocaine-Induced Microglial Activation and Neuroinflammation. Mol. Neurobiol. 2021, 58, 2215–2230. [Google Scholar] [CrossRef] [PubMed]
- Du, S.H.; Qiao, D.F.; Chen, C.X.; Chen, S.; Liu, C.; Lin, Z.; Wang, H.; Xie, W.B. Toll-Like Receptor 4 Mediates Methamphetamine-Induced Neuroinflammation through Caspase-11 Signaling Pathway in Astrocytes. Front. Mol. Neurosci. 2017, 10, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, E.; Liu, J.; Liu, H.; Wang, X.; Xiong, H. Inflammasome Activation by Methamphetamine Potentiates Lipopolysaccharide Stimulation of IL-1β Production in Microglia. J. Neuroimmune Pharmacol. 2018, 13, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.Y.; Koo, B.N.; Kim, S.Y.; Kam, E.H.; Nam, J.; Kim, E.J. Scopolamine promotes neuroinflammation and delirium-like neuropsychiatric disorder in mice. Sci. Rep. 2021, 11, 8376. [Google Scholar] [CrossRef]
- Lippai, D.; Bala, S.; Petrasek, J.; Csak, T.; Levin, I.; Kurt-Jones, E.A.; Szabo, G. Alcohol-induced IL-1β in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. J. Leukoc. Biol. 2013, 94, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Alfonso-Loeches, S.; Ureña-Peralta, J.; Morillo-Bargues, M.J.; Gómez-Pinedo, U.; Guerri, C. Ethanol-Induced TLR4/NLRP3 Neuroinflammatory Response in Microglial Cells Promotes Leukocyte Infiltration Across the BBB. Neurochem. Res. 2016, 41, 193–209. [Google Scholar] [CrossRef]
- Carranza-Aguilar, C.J.; Hernández-Mendoza, A.; Mejias-Aponte, C.; Rice, K.C.; Morales, M.; González-Espinosa, C.; Cruz, S.L. Morphine and Fentanyl Repeated Administration Induces Different Levels of NLRP3-Dependent Pyroptosis in the Dorsal Raphe Nucleus of Male Rats via Cell-Specific Activation of TLR4 and Opioid Receptors. Cell. Mol. Neurobiol. 2020, 42, 677–694. [Google Scholar] [CrossRef]
- Samir, P.; Kesavardhana, S.; Patmore, D.M.; Gingras, S.; Malireddi, R.K.S.; Karki, R.; Guy, C.S.; Briard, B.; Place, D.E.; Bhattacharya, A.; et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 2019, 573, 590–594. [Google Scholar] [CrossRef]
- Swaroop, S.; Mahadevan, A.; Shankar, S.K.; Adlakha, Y.K.; Basu, A. HSP60 critically regulates endogenous IL-1β production in activated microglia by stimulating NLRP3 inflammasome pathway. J. Neuroinflamm. 2018, 15, 177, Erratum in J. Neuroinflamm. 2018, 15, 317. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Yoon, J.H.; Ryu, J.H. Mitophagy: A balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016, 49, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Patra, S.; Bhol, C.S.; Panigrahi, D.P.; Praharaj, P.P.; Singh, A.; Patil, S.; Dhiman, R.; et al. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int. J. Biochem. Cell Biol. 2021, 136, 106013. [Google Scholar] [CrossRef]
- Leemans, J.C.; Cassel, S.L.; Sutterwala, F.S. Sensing damage by the NLRP3 inflammasome. Immunol. Rev. 2011, 243, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; He, Z.; Jacob, C.; Hu, X.; Liang, X.; Xiao, W.; Wan, L.; Xiao, P.; D′Ascenzo, N.; Ni, J.; et al. Effect of Increased IL-1β on Expression of HK in Alzheimer′s Disease. Int. J. Mol. Sci. 2021, 22, 1306. [Google Scholar] [CrossRef]
- Rivers-Auty, J.; Tapia, V.S.; White, C.S.; Daniels, M.J.D.; Drinkall, S.; Kennedy, P.T.; Spence, H.G.; Yu, S.; Green, J.P.; Hoyle, C.; et al. Zinc Status Alters Alzheimer′s Disease Progression through NLRP3-Dependent Inflammation. J. Neurosci. 2021, 41, 3025–3038. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, Z.M.; Wu, X.; Zhang, L.; Cao, Y.; Zhou, P. Distinct Molecular Mechanisms Underlying Potassium Efflux for NLRP3 Inflammasome Activation. Front. Immunol. 2020, 11, 609441. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Zhou, X.G.; Qiu, W.Q.; Yu, L.; Pan, R.; Teng, J.F.; Sang, Z.P.; Law, B.Y.; Zhao, Y.; Zhang, L.; Yan, L.; et al. Targeting microglial autophagic degradation of the NLRP3 inflammasome for identification of thonningianin A in Alzheimer′s disease. Inflamm. Regen. 2022, 42, 25. [Google Scholar] [CrossRef]
- Zhao, T.; Gao, J.; Van, J.; To, E.; Wang, A.; Cao, S.; Cui, J.Z.; Guo, J.P.; Lee, M.; McGeer, P.L.; et al. Age-related increases in amyloid beta and membrane attack complex: Evidence of inflammasome activation in the rodent eye. J. Neuroinflamm. 2015, 12, 121. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer′s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer′s Disease Pathogenesis: Role of Autophagy and Mitophagy Focusing in Microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Avila-Ferrufino, A.; Skuginna, V.; Susanti, H.E.; Anders, H.J. Quantitative expression of RIG-like helicase, NOD-like receptor and inflammasome-related mRNAs in humans and mice. Int. Immunol. 2010, 22, 717–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Activation and regulation of cellular inflammasomes: Gaps in our knowledge for central nervous system injury. J. Cereb. Blood Flow Metab. 2014, 34, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Nyúl-Tóth, Á.; Kozma, M.; Nagyőszi, P.; Nagy, K.; Fazakas, C.; Haskó, J.; Molnár, K.; Farkas, A.E.; Végh, A.G.; Váró, G.; et al. Expression of pattern recognition receptors and activation of the non-canonical inflammasome pathway in brain pericytes. Brain Behav. Immun. 2017, 64, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Johann, S.; Heitzer, M.; Kanagaratnam, M.; Goswami, A.; Rizo, T.; Weis, J.; Troost, D.; Beyer, C. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 2015, 63, 2260–2273. [Google Scholar] [CrossRef]
- Ebrahimi, T.; Rust, M.; Kaiser, S.N.; Slowik, A.; Beyer, C.; Koczulla, A.R.; Schulz, J.B.; Habib, P.; Bach, J.P. α1-antitrypsin mitigates NLRP3-inflammasome activation in amyloid β1–42-stimulated murine astrocytes. J. Neuroinflamm. 2018, 15, 282. [Google Scholar] [CrossRef]
- Sandhu, J.K.; Kulka, M. Decoding Mast Cell-Microglia Communication in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 1093. [Google Scholar] [CrossRef]
- Komleva, Y.K.; Lopatina, O.L.; Gorina, Y.V.; Chernykh, A.I.; Trufanova, L.V.; Vais, E.F.; Kharitonova, E.V.; Zhukov, E.L.; Vahtina, L.Y.; Medvedeva, N.N.; et al. Expression of NLRP3 Inflammasomes in Neurogenic Niche Contributes to the Effect of Spatial Learning in Physiological Conditions but Not in Alzheimer′s Type Neurodegeneration. Cell. Mol. Neurobiol. 2021, 42, 1355–1371. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer′s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Harte, M. Investigating markers of the NLRP3 inflammasome pathway in Alzheimer′s disease: A human post-mortem study. Genes 2021, 12, 1753. [Google Scholar] [CrossRef]
- Dal Prà, I.; Armato, U.; Chiarini, A. Family C G-Protein-Coupled Receptors in Alzheimer′s Disease and Therapeutic Implications. Front. Pharmacol. 2019, 10, 1282. [Google Scholar] [CrossRef]
- Dal Prà, I.; Armato, U.; Chioffi, F.; Pacchiana, R.; Whitfield, J.F.; Chakravarthy, B.; Gui, L.; Chiarini, A. The Aβ peptides-activated calcium-sensing receptor stimulates the production and secretion of vascular endothelial growth factor-A by normoxic adult human cortical astrocytes. Neuromol. Med. 2014, 16, 645–657. [Google Scholar] [CrossRef]
- Dal Prà, I.; Chiarini, A.; Pacchiana, R.; Gardenal, E.; Chakravarthy, B.; Whitfield, J.F.; Armato, U. Calcium-sensing receptors of human Astrocyte-Neuron Teams: Amyloid-β-driven mediators and therapeutic targets of Alzheimer′s Disease. Curr. Neuropharmacol. 2014, 12, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Dal Prà, I.; Armato, U.; Chiarini, A. Specific interactions of calcium-sensing receptors (CaSRs) with soluble amyloid-β peptides—A study using cultured normofunctioning adult human astrocytes. In Proceedings of the 2nd International Symposium on the Calcium-Sensing Receptor, San Diego, CA, USA, 3–4 March 2015; pp. 90–91. [Google Scholar]
- Hofer, A.M.; Brown, E.M. Extracellular calcium sensing and signaling. Nat. Rev. Mol. Cell Biol. 2003, 4, 530–538. [Google Scholar] [CrossRef]
- Gardenal, E.; Chiarini, A.; Armato, U.; Dal Prà, I.; Verkhratsky, A.; Rodríguez, J.J. Increased calcium-sensing receptor immunoreactivity in the hippocampus of a triple transgenic mouse model of Alzheimer′s Disease. Front. Neurosci. 2017, 11, 81. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-López, T.Y.; Orduña-Castillo, L.B.; Hernández-Vásquez, M.N.; Vázquez-Prado, J.; Reyes-Cruz, G. Calcium sensing receptor activates the NLRP3 inflammasome via a chaperone-assisted degradative pathway involving Hsp70 and LC3-II. Biochem. Biophys. Res. Commun. 2018, 505, 1121–1127. [Google Scholar] [CrossRef]
- Sokolowska, M.; Chen, L.Y.; Liu, Y.; Martinez-Anton, A.; Qi, H.Y.; Logun, C.; Alsaaty, S.; Park, Y.H.; Kastner, D.L.; Chae, J.J.; et al. Prostaglandin E2 Inhibits NLRP3 Inflammasome Activation through EP4 Receptor and Intracellular Cyclic AMP in Human Macrophages. J. Immunol. 2015, 194, 5472–5487. [Google Scholar] [CrossRef] [Green Version]
- Armato, U.; Chiarini, A.; Chakravarthy, B.; Chioffi, F.; Pacchiana, R.; Colarusso, E.; Whitfield, J.F.; Dal Prà, I. Calcium-sensing receptor antagonist (calcilytic) NPS 2143 specifically blocks the increased secretion of endogenous Aβ42 prompted by exogenous fibrillary or soluble Aβ25-35 in human cortical astrocytes and neurons-therapeutic relevance to Alzheimer′s disease. Biochim. Biophys. Acta 2013, 1832, 1634–1652. [Google Scholar] [CrossRef]
- Pi, M.; Faber, P.; Ekema, G.; Jackson, P.D.; Ting, A.; Wang, N.; Fontilla-Poole, M.; Mays, R.W.; Brunden, K.R.; Harrington, J.J.; et al. Identification of a novel extracellular cation-sensing G-protein-coupled receptor. J. Biol. Chem. 2005, 280, 40201–40209. [Google Scholar] [CrossRef] [Green Version]
- Pi, M.; Parrill, A.L.; Quarles, L.D. GPRC6A mediates the non-genomic effects of steroids. J. Biol. Chem. 2010, 285, 39953–39964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pi, M.; Wu, Y.; Quarles, L.D. GPRC6A mediates responses to osteocalcin in β-cells in vitro and pancreas in vivo. J. Bone Miner. Res. 2011, 26, 1680–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pi, M.; Quarles, L.D. GPRC6A regulates prostate cancer progression. Prostate 2011, 72, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Dutta, S.R.; Song, C.Y.; Oh, S.; Gonzalez, F.J.; Malik, K.U. Brain Testosterone-CYP1B1 (Cytochrome P450 1B1) Generated Metabolite 6β-Hydroxytestosterone Promotes Neurogenic Hypertension and Inflammation. Hypertension 2020, 76, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Bai, N.; Zhang, Q.; Zhang, W.; Liu, B.; Yang, F.; Brann, D.; Wang, R. G-protein-coupled estrogen receptor activation upregulates interleukin-1 receptor antagonist in the hippocampus after global cerebral ischemia: Implications for neuronal self-defense. J. Neuroinflamm. 2020, 17, 45. [Google Scholar] [CrossRef]
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell. 2013, 49, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk with NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer′s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef]
- McKee, C.M.; Coll, R.C. NLRP3 inflammasome priming: A riddle wrapped in a mystery inside an enigma. J. Leukoc. Biol. 2020, 108, 937–952. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Ye, X.J.; He, X.H.; Ouyang, D.Y. The Signaling Pathways Regulating NLRP3 Inflammasome Activation. Inflammation 2021, 44, 1229–1245. [Google Scholar] [CrossRef]
- Dierckx, T.; Haidar, M.; Grajchen, E.; Wouters, E.; Vanherle, S.; Loix, M.; Boeykens, A.; Bylemans, D.; Hardonnière, K.; Kerdine-Römer, S.; et al. Phloretin suppresses neuroinflammation by autophagy-mediated Nrf2 activation in macrophages. J. Neuroinflamm. 2021, 18, 148. [Google Scholar] [CrossRef]
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952. [Google Scholar] [CrossRef] [Green Version]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Tong, Z.; Jiang, S.; Zheng, W.; Zhao, J.; Zhou, X. The Roles of Endoplasmic Reticulum in NLRP3 Inflammasome Activation. Cells 2020, 9, 1219. [Google Scholar] [CrossRef]
- Jäger, E.; Murthy, S.; Schmidt, C.; Hahn, M.; Strobel, S.; Peters, A.; Stäubert, C.; Sungur, P.; Venus, T.; Geisler, M.; et al. Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat. Commun. 2020, 11, 4243. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Rossol, M.; Pierer, M.; Raulien, N.; Quandt, D.; Meusch, U.; Rothe, K.; Schubert, K.; Schöneberg, T.; Schaefer, M.; Krügel, U.; et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 2012, 3, 1329. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Kim, H.J.; Binas, B.; Kang, J.H.; Chung, I.Y. Inflammatory mediators ATP and S100A12 activate the NLRP3 inflammasome to induce MUC5AC production in airway epithelial cells. Biochem. Biophys. Res. Commun. 2018, 503, 657–664. [Google Scholar] [CrossRef]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer′s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef]
- Lim, J.C.; Lu, W.; Beckel, J.M.; Mitchell, C.H. Neuronal Release of Cytokine IL-3 Triggered by Mechanosensitive Autostimulation of the P2X7 Receptor Is Neuroprotective. Front. Cell. Neurosci. 2016, 10, 270. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Albalawi, F.; Beckel, J.M.; Lim, J.C.; Laties, A.M.; Mitchell, C.H. The P2X7 receptor links mechanical strain to cytokine IL-6 up-regulation and release in neurons and astrocytes. J. Neurochem. 2017, 141, 436–448. [Google Scholar] [CrossRef] [Green Version]
- Campagno, K.E.; Mitchell, C.H. The P2X7Receptor in Microglial Cells Modulates the Endolysosomal Axis, Autophagy, and Phagocytosis. Front. Cell. Neurosci. 2021, 15, 645244. [Google Scholar] [CrossRef] [PubMed]
- Shieh, C.H.; Heinrich, A.; Serchov, T.; van Calker, D.; Biber, K. P2X7-dependent, but differentially regulated release of IL-6, CCL2, and TNF-α in cultured mouse microglia. Glia 2014, 62, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Cieślak, M.; Wojtczak, A. Role of purinergic receptors in the Alzheimer′s disease. Purinergic Signal. 2018, 14, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erb, L.; Woods, L.T.; Khalafalla, M.G.; Weisman, G.A. Purinergic signaling in Alzheimer′s disease. Brain Res. Bull. 2019, 151, 25–37. [Google Scholar] [CrossRef]
- Duez, H.; Pourcet, B. Nuclear Receptors in the Control of the NLRP3 Inflammasome Pathway. Front. Endocrinol. 2021, 12, 630536. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Liang, Z.; Damianou, A.; Di Daniel, E.; Kessler, B.M. Inflammasome activation controlled by the interplay between post-translational modifications: Emerging drug target opportunities. Cell Commun. Signal. 2021, 19, 23. [Google Scholar] [CrossRef]
- Weber, A.N.R. Targeting the NLRP3 Inflammasome via BTK. Front. Cell Dev. Biol. 2021, 9, 630479. [Google Scholar] [CrossRef]
- Bezbradica, J.S.; Coll, R.C.; Schroder, K. Sterile signals generate weaker and delayed macrophage NLRP3 inflammasome responses relative to microbial signals. Cell. Mol. Immunol. 2017, 14, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Healy, L.M.; Yaqubi, M.; Ludwin, S.; Antel, J.P. Species differences in immune-mediated CNS tissue injury and repair: A (neuro)inflammatory topic. Glia 2020, 68, 811–829. [Google Scholar] [CrossRef]
- Zhang, C.J.; Jiang, M.; Zhou, H.; Liu, W.; Wang, C.; Kang, Z.; Han, B.; Zhang, Q.; Chen, X.; Xiao, J.; et al. TLR-stimulated IRAKM activates caspase-8 inflammasome in microglia and promotes neuroinflammation. J. Clin. Investing. 2018, 128, 5399–5412. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Elizagaray, M.L.; Gomes, M.T.R.; Guimaraes, E.S.; Rumbo, M.; Hozbor, D.F.; Oliveira, S.C.; Moreno, G. Canonical and Non-canonical Inflammasome Activation by Outer Membrane Vesicles Derived from Bordetella pertussis. Front. Immunol. 2020, 11, 1879. [Google Scholar] [CrossRef]
- Matikainen, S.; Nyman, T.A.; Cypryk, W. Function and Regulation of Noncanonical Caspase-4/5/11 Inflammasome. J. Immunol. 2020, 204, 3063–3069. [Google Scholar] [CrossRef]
- Yi, Y.S. Caspase-11 Noncanonical Inflammasome: A Novel Key Player in Murine Models of Neuroinflammation and Multiple Sclerosis. Neuroimmunomodulation 2021, 28, 195–203. [Google Scholar] [CrossRef]
- Zhang, D.; Qian, J.; Zhang, P.; Li, H.; Shen, H.; Li, X.; Chen, G. Gasdermin D serves as a key executioner of pyroptosis in experimental cerebral ischemia and reperfusion model both in vivo and in vitro. J. Neurosci. Res. 2019, 97, 645–660. [Google Scholar] [CrossRef]
- Wang, K.; Sun, Z.; Ru, J.; Wang, S.; Huang, L.; Ruan, L.; Lin, X.; Jin, K.; Zhuge, Q.; Yang, S. Ablation of GSDMD Improves Outcome of Ischemic Stroke Through Blocking Canonical and Non-canonical Inflammasomes Dependent Pyroptosis in Microglia. Front. Neurol. 2020, 11, 577927. [Google Scholar] [CrossRef]
- Carpenter, S.; Aiello, D.; Atianand, M.K.; Ricci, E.P.; Gandhi, P.; Hall, L.L.; Byron, M.; Monks, B.; Henry-Bezy, M.; Lawrence, J.B.; et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science 2013, 341, 789–792. [Google Scholar] [CrossRef] [Green Version]
- Heward, J.A.; Lindsay, M.A. Long non-coding RNAs in the regulation of the immune response. Trends Immunol. 2014, 35, 408–419. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [Green Version]
- Kiesel, P.; Gibson, T.J.; Ciesielczyk, B.; Bodemer, M.; Kaup, F.J.; Bodemer, W.; Zischler, H.; Zerr, I. Transcription of Alu DNA elements in blood cells of sporadic Creutzfeldt-Jakob disease (sCJD). Prion 2010, 4, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polesskaya, O.; Kananykhina, E.; Roy-Engel, A.M.; Nazarenko, O.; Kulemzina, I.; Baranova, A.; Vassetsky, Y.; Myakishev-Rempel, M. The role of Alu-derived RNAs in Alzheimer′s and other neurodegenerative conditions. Med. Hypotheses 2018, 115, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Saville, L.; Gollen, B.; Isaac, C.; Belay, A.; Mehla, J.; Patel, K.; Thakor, N.; Mohajerani, M.H.; Zovoilis, A. Increased processing of SINE B2 ncRNAs unveils a novel type of transcriptome deregulation in amyloid beta neuropathology. eLife 2020, 9, e61265. [Google Scholar] [CrossRef]
- Cheng, Y.; Saville, L.; Gollen, B.; Veronesi, A.A.; Mohajerani, M.; Joseph, J.T.; Zovoilis, A. Increased Alu RNA processing in Alzheimer brains is linked to gene expression changes. EMBO Rep. 2021, 22, e52255. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, Y.; Wang, Z.; Xu, C.; Qiao, S.; Liu, T.; Qi, K.; Tong, D.; Li, C. Bone Marrow Mesenchymal Stem Cell Exosome Attenuates Inflammasome-Related Pyroptosis via Delivering circ_003564 to Improve the Recovery of Spinal Cord Injury. Mol. Neurobiol. 2022, 59, 6771–6789. [Google Scholar] [CrossRef]
- Xue, Z.; Zhang, Z.; Liu, H.; Li, W.; Guo, X.; Zhang, Z.; Liu, Y.; Jia, L.; Li, Y.; Ren, Y.; et al. lincRNA-Cox2 regulates NLRP3 inflammasome and autophagy mediated neuroinflammation. Cell Death Differ. 2019, 26, 130–145. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Ding, T.; Chen, Y.; Long, T.; Xu, Q.; Lian, W.; Liu, W. LncRNA-Meg3 promotes Nlrp3-mediated microglial inflammation by targeting miR-7a-5p. Int. Immunopharmacol. 2021, 90, 107141. [Google Scholar] [CrossRef]
- Docrat, T.F.; Nagiah, S.; Chuturgoon, A.A. Metformin protects against neuroinflammation through integrated mechanisms of miR-141 and the NF-ĸB-mediated inflammasome pathway in a diabetic mouse model. Eur. J. Pharmacol. 2021, 903, 174146. [Google Scholar] [CrossRef]
- Cunha, C.; Gomes, C.; Vaz, A.R.; Brites, D. Exploring New Inflammatory Biomarkers and Pathways during LPS-Induced M1 Polarization. Mediat. Inflamm. 2016, 2016, 6986175. [Google Scholar] [CrossRef] [Green Version]
- Si, L.; Wang, H.; Wang, L. Suppression of miR-193a alleviates neuroinflammation and improves neurological function recovery after traumatic brain injury (TBI) in mice. Biochem. Biophys. Res. Commun. 2020, 523, 527–534. [Google Scholar] [CrossRef]
- Cao, Y.; Tan, X.; Lu, Q.; Huang, K.; Tang, X.; He, Z. miR-590-3 and SP1 Promote Neuronal Apoptosis in Patients with Alzheimer′s Disease via AMPK Signaling Pathway. Contrast Media Mol. Imaging 2021, 2021, 6010362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tao, J.; Zhang, S.; Lv, X. LncRNA MEG3 Reduces Hippocampal Neuron Apoptosis via the PI3K/AKT/mTOR Pathway in a Rat Model of Temporal Lobe Epilepsy. Neuropsychiatr. Dis. Treat. 2020, 16, 2519–2528. [Google Scholar] [CrossRef]
- Fan, Z.; Lu, M.; Qiao, C.; Zhou, Y.; Ding, J.H.; Hu, G. MicroRNA-7 Enhances Subventricular Zone Neurogenesis by Inhibiting NLRP3/Caspase-1 Axis in Adult Neural Stem Cells. Mol. Neurobiol. 2016, 53, 7057–7069. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson′s disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, G.H.; Wu, J.; Mou, F.F.; Xie, W.H.; Wang, F.B.; Wang, Q.L.; Fang, J.; Xu, Y.W.; Dong, Y.R.; Liu, J.R.; et al. Exosomes derived from hypoxia-preconditioned mesenchymal stromal cells ameliorate cognitive decline by rescuing synaptic dysfunction and regulating inflammatory responses in APP/PS1 mice. FASEB J. 2018, 32, 654–668. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Guo, L.; Yang, Y.; Guan, Q.; Shen, H.; Sheng, Y.; Jiao, Q. Mechanism of microRNA-22 in regulating neuroinflammation in Alzheimer′s disease. Brain Behav. 2020, 10, e01627. [Google Scholar] [CrossRef] [Green Version]
- Zhai, L.; Shen, H.; Sheng, Y.; Guan, Q. ADMSC Exo-MicroRNA-22 improve neurological function and neuroinflammation in mice with Alzheimer′s disease. J. Cell. Mol. Med. 2021, 25, 7513–7523, Erratum in J. Cell. Mol. Med. 2021, 25, 11037–11038. [Google Scholar] [CrossRef]
- Hu, L.T.; Wang, B.Y.; Fan, Y.H.; He, Z.Y.; Zheng, W.X. Exosomal miR-23b from bone marrow mesenchymal stem cells alleviates oxidative stress and pyroptosis after intracerebral hemorrhage. Neural Regen. Res. 2023, 18, 560–567. [Google Scholar] [CrossRef]
- Cao, Y.; Tan, X.; Lu, Q.; Huang, K.; Tang, X.; He, Z. MiR-29c-3p May Promote the Progression of Alzheimer′s Disease through BACE1. J. Healthc. Eng. 2021, 2021, 2031407. [Google Scholar] [CrossRef]
- Sha, S.; Shen, X.; Cao, Y.; Qu, L. Mesenchymal stem cells-derived extracellular vesicles ameliorate Alzheimer′s disease in rat models via the microRNA-29c-3p/BACE1 axis and the Wnt/β-catenin pathway. Aging 2021, 13, 15285–15306. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, H.; Wang, B.; Ao, Q.; He, Z. MicroRNA-152 attenuates neuroinflammation in intracerebral hemorrhage by inhibiting thioredoxin interacting protein (TXNIP)-mediated NLRP3 inflammasome activation. Int. Immunopharmacol. 2020, 80, 106141. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Z.; Xing, H.; Wang, Y.; Guo, Y. Exosomes derived from miR-188-3p-modified adipose-derived mesenchymal stem cells protect Parkinson′s disease. Mol. Ther. Nucleic Acids 2021, 23, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.Y.; Li, G.S.; Tu, C.; Chen, W.L.; Wang, X.W.; Wang, Y.N.; Peng, L.B.; Tan, F. MicroNAR-194-5p hinders the activation of NLRP3 inflammasomes and alleviates neuroinflammation during intracerebral hemorrhage by blocking the interaction between TRAF6 and NLRP3. Brain Res. 2021, 1752, 147228. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; Agostini, S.; Hernis, A.; Zanzottera, M.; Bianchi, A.; Clerici, M. Circulatory miR-223-3p Discriminates Between Parkinson′s and Alzheimer′s Patients. Sci. Rep. 2019, 9, 9393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Hu, Y.; Lu, R.; Ge, M.; Zhang, L. MicroRNA-374a-5p inhibits neuroinflammation in neonatal hypoxic-ischemic encephalopathy via regulating NLRP3 inflammasome targeted Smad6. Life Sci. 2020, 252, 117664. [Google Scholar] [CrossRef]
- Kaur, S.; Verma, H.; Dhiman, M.; Tell, G.; Gigli, G.L.; Janes, F.; Mantha, A.K. Brain Exosomes: Friend or Foe in Alzheimer′s Disease? Mol. Neurobiol. 2021, 58, 6610–6624. [Google Scholar] [CrossRef]
- Hu, Z.; Yuan, Y.; Zhang, X.; Lu, Y.; Dong, N.; Jiang, X.; Xu, J.; Zheng, D. Human Umbilical Cord Mesenchymal Stem Cell-Derived Exosomes Attenuate Oxygen-Glucose Deprivation/Reperfusion-Induced Microglial Pyroptosis by Promoting FOXO3a-Dependent Mitophagy. Oxid. Med. Cell. Longev. 2021, 2021, 6219715. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, M.; Liu, H.; Zhu, R.; He, H.; Zhou, Y.; Zhang, Y.; Li, C.; Liang, D.; Zeng, Q.; et al. Bone marrow mesenchymal stem cell-derived exosomes attenuate cerebral ischemia-reperfusion injury-induced neuroinflammation and pyroptosis by modulating microglia M1/M2 phenotypes. Exp. Neurol. 2021, 341, 113700. [Google Scholar] [CrossRef]
- Cui, G.H.; Guo, H.D.; Li, H.; Zhai, Y.; Gong, Z.B.; Wu, J.; Liu, J.S.; Dong, Y.R.; Hou, S.X.; Liu, J.R. RVG-modified exosomes derived from mesenchymal stem cells rescue memory deficits by regulating inflammatory responses in a mouse model of Alzheimer′s disease. Immun. Ageing 2019, 16, 10. [Google Scholar] [CrossRef] [Green Version]
- Chiarini, A.; Armato, U.; Gardenal, E.; Gui, L.; Dal Prà, I. Amyloid β-exposed human astrocytes overproduce phospho-Tau and overrelease it within exosomes, effects suppressed by calcilytic NPS 2143. Further implications for Alzheimer′s therapy. Front. Neurosci. 2017, 11, 217. [Google Scholar] [CrossRef] [Green Version]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer′s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Sun, Y.; He, Z.; Xu, Y.; Li, X.; Ding, J.; Lu, M.; Hu, G. Kynurenine regulates NLRP2 inflammasome in astrocytes and its implications in depression. Brain Behav. Immun. 2020, 88, 471–481. [Google Scholar] [CrossRef]
- Voet, S.; Mc Guire, C.; Hagemeyer, N.; Martens, A.; Schroeder, A.; Wieghofer, P.; Daems, C.; Staszewski, O.; Vande Walle, L.; Jordao, M.J.C.; et al. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat. Commun. 2018, 9, 2036. [Google Scholar] [CrossRef] [Green Version]
- Gaikwad, S.; Patel, D.; Agrawal-Rajput, R. CD40 Negatively Regulates ATP-TLR4-Activated Inflammasome in Microglia. Cell. Mol. Neurobiol. 2017, 37, 351–359. [Google Scholar] [CrossRef]
- Ma, S.; Wang, Y.; Zhou, X.; Li, Z.; Zhang, Z.; Wang, Y.; Huang, T.; Zhang, Y.; Shi, J.; Guan, F. MG53 Protects hUC-MSCs against Inflammatory Damage and Synergistically Enhances Their Efficacy in Neuroinflammation Injured Brain through Inhibiting NLRP3/Caspase-1/IL-1β Axis. ACS Chem. Neurosci. 2020, 11, 2590–2601. [Google Scholar] [CrossRef]
- Xiao, T.; Wan, J.; Qu, H.; Li, Y. Tripartite-motif protein 21 knockdown extenuates LPS-triggered neurotoxicity by inhibiting microglial M1 polarization via suppressing NF-κB-mediated NLRP3 inflammasome activation. Arch. Biochem. Biophys. 2021, 706, 108918. [Google Scholar] [CrossRef]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2019, 11, 480. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Franchi, L.; Núñez, G. The protein kinase PKR is critical for LPS-induced iNOS production but dispensable for inflammasome activation in macrophages. Eur. J. Immunol. 2013, 43, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.; Cooper, M.A.; O′Neill, L.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Heitzer, M.; Kaiser, S.; Kanagaratnam, M.; Zendedel, A.; Hartmann, P.; Beyer, C.; Johann, S. Administration of 17beta-Estradiol Improves Motoneuron Survival and Down-regulates Inflammasome Activation in Male SOD1 (G93A) ALS Mice. Mol. Neurobiol. 2017, 54, 8429–8443. [Google Scholar] [CrossRef] [PubMed]
- Aryanpour, R.; Zibara, K.; Pasbakhsh, P.; Jame′ei, S.B.; Namjoo, Z.; Ghanbari, A.; Mahmoudi, R.; Amani, S.; Kashani, I.R. 17β-Estradiol Reduces Demyelination in Cuprizone-fed Mice by Promoting M2 Microglia Polarity and Regulating NLRP3 Inflammasome. Neuroscience 2021, 463, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, R.; Wang, R.; Wang, J.; Vadlamudi, R.K.; Brann, D.W. 17β-Estradiol Regulates Microglia Activation and Polarization in the Hippocampus Following Global Cerebral Ischemia. Oxid. Med. Cell. Longev. 2018, 2018, 4248526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Huang, Y.; He, F.; Liu, J.; Li, M.; Sun, T.; Ren, W.; Hou, J.; Zhu, L. Dopamine D1 Receptor Agonist A-68930 Inhibits NLRP3 Inflammasome Activation, Controls Inflammation, and Alleviates Histopathology in a Rat Model of Spinal Cord Injury. Spine (Phila Pa 1976) 2016, 41, E330–E334. [Google Scholar] [CrossRef]
- Wang, S.; Yao, Q.; Wan, Y.; Wang, J.; Huang, C.; Li, D.; Yang, B. Adiponectin reduces brain injury after intracerebral hemorrhage by reducing NLRP3 inflammasome expression. Int. J. Neurosci. 2020, 130, 301–308. [Google Scholar] [CrossRef]
- Li, J.; Wu, D.M.; Yu, Y.; Deng, S.H.; Liu, T.; Zhang, T.; He, M.; Zhao, Y.Y.; Xu, Y. Amifostine ameliorates induction of experimental autoimmune encephalomyelitis: Effect on reactive oxygen species/NLRP3 pathway. Int. Immunopharmacol. 2020, 88, 106998. [Google Scholar] [CrossRef]
- Li, B.X.; Dai, X.; Xu, X.R.; Adili, R.; Neves, M.A.D.; Lei, X.; Shen, C.; Zhu, G.; Wang, Y.; Zhou, H.; et al. In vitro assessment and phase I randomized clinical trial of anfibatide a snake venom derived anti-thrombotic agent targeting human platelet GPIbα. Sci. Rep. 2021, 11, 11663. [Google Scholar] [CrossRef]
- Li, R.; Si, M.; Jia, H.Y.; Ma, Z.; Li, X.W.; Li, X.Y.; Dai, X.R.; Gong, P.; Luo, S.Y. Anfibatide alleviates inflammation and apoptosis via inhibiting NF-kappaB/NLRP3 axis in ischemic stroke. Eur. J. Pharmacol. 2022, 926, 175032. [Google Scholar] [CrossRef]
- Liu, P.; Gao, Q.; Guan, L.; Hu, Y.; Jiang, J.; Gao, T.; Sheng, W.; Xue, X.; Qiao, H.; Li, T. Atorvastatin attenuates surgery-induced BBB disruption and cognitive impairment partly by suppressing NF-κB pathway and NLRP3 inflammasome activation in aged mice. Acta Biochim. Biophys. Sin. 2021, 53, 528–537. [Google Scholar] [CrossRef]
- Jiang, W.; Li, M.; He, F.; Zhou, S.; Zhu, L. Targeting the NLRP3 inflammasome to attenuate spinal cord injury in mice. J. Neuroinflamm. 2017, 14, 207. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Zhang, L.; Shang, Y.; Zhu, Z.; Jin, S.; Guo, Z.; Wang, X. Concurrent suppression of Aβ aggregation and NLRP3 inflammasome activation for treating Alzheimer′s disease. Chem. Sci. 2022, 13, 2971–2980. [Google Scholar] [CrossRef]
- Wang, H.Q.; Song, K.Y.; Feng, J.Z.; Huang, S.Y.; Guo, X.M.; Zhang, L.; Zhang, G.; Huo, Y.C.; Zhang, R.R.; Ma, Y.; et al. Caffeine Inhibits Activation of the NLRP3 Inflammasome via Autophagy to Attenuate Microglia-Mediated Neuroinflammation in Experimental Autoimmune Encephalomyelitis. J. Mol. Neurosci. 2022, 72, 97–112. [Google Scholar] [CrossRef]
- de Oliveira, L.R.C.; Mimura, L.A.N.; Fraga-Silva, T.F.C.; Ishikawa, L.L.W.; Fernandes, A.A.H.; Zorzella-Pezavento, S.F.G.; Sartori, A. Calcitriol Prevents Neuroinflammation and Reduces Blood-Brain Barrier Disruption and Local Macrophage/Microglia Activation. Front. Pharmacol. 2020, 11, 161. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, X.; Chen, X.; Cai, Y.; Ma, Y.; Ma, J.; Zhang, Q.; Dai, L.; Fan, X.; Bai, Y. Choline Supplementation Ameliorates Behavioral Deficits and Alzheimer′s Disease-Like Pathology in Transgenic APP/PS1 Mice. Mol. Nutr. Food Res. 2019, 63, e1801407. [Google Scholar] [CrossRef]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D′Alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer′s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef]
- Bao, Y.; Zhu, Y.; He, G.; Ni, H.; Liu, C.; Ma, L.; Zhang, L.; Shi, D. Dexmedetomidine Attenuates Neuroinflammation in LPS-Stimulated BV2 Microglia Cells Through Upregulation Of miR-340. Drug Des. Devel. Ther. 2019, 13, 3465–3475. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Wang, J.X.; Du, Y.H.; Liu, Y.; Zhang, W.; Chen, J.F.; Liu, Y.J.; Zheng, M.; Wang, K.J.; He, G.Q. Dihydromyricetin inhibits microglial activation and neuroinflammation by suppressing NLRP3 inflammasome activation in APP/PS1 transgenic mice. CNS Neurosci. Ther. 2018, 24, 1207–1218. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Nizami, S.; Arunasalam, K.; Green, J.; Cook, J.; Lawrence, C.B.; Zarganes-Tzitzikas, T.; Davis, J.B.; Di Daniel, E.; Brough, D. Inhibition of the NLRP3 inflammasome by HSP90 inhibitors. Immunology 2021, 162, 84–91. [Google Scholar] [CrossRef]
- Gao, S.; Xu, T.; Guo, H.; Deng, Q.; Xun, C.; Liang, W.; Sheng, W. Ameliorative effects of echinacoside against spinal cord injury via inhibiting NLRP3 inflammasome signaling pathway. Life Sci. 2019, 237, 116978. [Google Scholar] [CrossRef]
- Kiasalari, Z.; Afshin-Majd, S.; Baluchnejadmojarad, T.; Azadi-Ahmadabadi, E.; Esmaeil-Jamaat, E.; Fahanik-Babaei, J.; Fakour, M.; Fereidouni, F.; Ghasemi-Tarie, R.; Jalalzade-Ogvar, S.; et al. Ellagic acid ameliorates neuroinflammation and demyelination in experimental autoimmune encephalomyelitis: Involvement of NLRP3 and pyroptosis. J. Chem. Neuroanat. 2021, 111, 101891. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Sun, J.; Kim, T.J.; Kim, Y.J.; Ko, S.B.; Kim, C.K.; Jia, X.; Yoon, B.W. Pretreatment with low-dose fimasartan ameliorates NLRP3 inflammasome-mediated neuroinflammation and brain injury after intracerebral hemorrhage. Exp. Neurol. 2018, 310, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Abu-Elfotuh, K.; Al-Najjar, A.H.; Mohammed, A.A.; Aboutaleb, A.S.; Badawi, G.A. Fluoxetine ameliorates Alzheimer′s disease progression and prevents the exacerbation of cardiovascular dysfunction of socially isolated depressed rats through activation of Nrf2/HO-1 and hindering TLR4/NLRP3 inflammasome signaling pathway. Int. Immunopharmacol. 2022, 104, 108488. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, Z.; He, X.; Yu, H.; Feng, J. Ghrelin Attenuates Neuroinflammation and Demyelination in Experimental Autoimmune Encephalomyelitis Involving NLRP3 Inflammasome Signaling Pathway and Pyroptosis. Front. Pharmacol. 2019, 10, 1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, J.; Tao, X.Y.; Teng, P.; Zhang, Y.; Guo, C.L.; Hu, L.; Qian, Y.N.; Jiang, C.Y.; Liu, W.T. Blocking ATP-sensitive potassium channel alleviates morphine tolerance by inhibiting HSP70-TLR4-NLRP3-mediated neuroinflammation. J. Neuroinflamm. 2017, 14, 228. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Yang, J.; Li, S.; Huang, R.; Zhang, D.; Zhao, J.; Wang, Q. Glibenclamide attenuates 2.5-hexanedione-induced neurotoxicity in the spinal cord of rats through mitigation of NLRP3 inflammasome activation, neuroinflammation and oxidative stress. Toxicol. Lett. 2020, 331, 152–158. [Google Scholar] [CrossRef]
- Shao, B.Z.; Wei, W.; Ke, P.; Xu, Z.Q.; Zhou, J.X.; Liu, C. Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS Neurosci. Ther. 2014, 20, 1021–1028. [Google Scholar] [CrossRef]
- Karkhah, A.; Saadi, M.; Pourabdolhossein, F.; Saleki, K.; Nouri, H.R. Indomethacin attenuates neuroinflammation and memory impairment in an STZ-induced model of Alzheimer′s like disease. Immunopharmacol. Immunotoxicol. 2021, 43, 758–766. [Google Scholar] [CrossRef]
- Cooper, M.A. Inzomelid is a CNS penetrant anti-inflammatory drug that blocks NLRP3 inflammasome activation targeted to prevent Synuclein Pathology and Dopaminergic Degeneration in Parkinson′s disease. In Proceedings of the 7th International Conference on Parkinson′s & Movement Disorders, London, UK, 11–12 November 2019. [Google Scholar]
- Kuwar, R.; Rolfe, A.; Di, L.; Xu, H.; He, L.; Jiang, Y.; Zhang, S.; Sun, D. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J. Neuroinflamm. 2019, 16, 81. [Google Scholar] [CrossRef] [Green Version]
- Lyu, D.; Wang, F.; Zhang, M.; Yang, W.; Huang, H.; Huang, Q.; Wu, C.; Qian, N.; Wang, M.; Zhang, H.; et al. Ketamine induces rapid antidepressant effects via the autophagy-NLRP3 inflammasome pathway. Psychopharmacology 2022, 239, 3201–3212. [Google Scholar] [CrossRef]
- Liu, S.; Wang, S.; Gu, R.; Che, N.; Wang, J.; Cheng, J.; Yuan, Z.; Cheng, Y.; Liao, Y. The XPO1 Inhibitor KPT-8602 Ameliorates Parkinson′s Disease by Inhibiting the NF-κB/NLRP3 Pathway. Front. Pharmacol. 2022, 13, 847605. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Feng, H.; Wang, H.; Wang, Y.; Mou, W.; Xu, G.; Zhang, P.; Li, R.; Shi, W.; Wang, Z.; et al. Licochalcone B specifically inhibits the NLRP3 inflammasome by disrupting NEK7-NLRP3 interaction. EMBO Rep. 2022, 23, e53499. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.R.; Tang, J.Y.; Wang, Y.Y.; Farooqi, A.A.; Yen, C.Y.; Yuan, S.F.; Huang, H.W.; Chang, H.W. Manoalide Preferentially Provides Antiproliferation of Oral Cancer Cells by Oxidative Stress-Mediated Apoptosis and DNA Damage. Cancers 2019, 11, 1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folmer, F.; Jaspars, M.; Schumacher, M.; Dicato, M.; Diederich, M. Marine Natural Products Targeting Phospholipases A2. Biochem. Pharmacol. 2010, 80, 1793–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salam, K.A.; Furuta, A.; Noda, N.; Tsuneda, S.; Sekiguchi, Y.; Yamashita, A.; Moriishi, K.; Nakakoshi, M.; Tsubuki, M.; Tani, H.; et al. Inhibition of Hepatitis C Virus NS3 Helicase by Manoalide. J. Nat. Prod. 2012, 75, 650–654. [Google Scholar] [CrossRef]
- Li, C.; Lin, H.; He, H.; Ma, M.; Jiang, W.; Zhou, R. Inhibition of the NLRP3 Inflammasome Activation by Manoalide Ameliorates Experimental Autoimmune Encephalomyelitis Pathogenesis. Front. Cell Dev. Biol. 2022, 10, 822236. [Google Scholar] [CrossRef]
- Fu, Q.; Li, J.; Qiu, L.; Ruan, J.; Mao, M.; Li, S.; Mao, Q. Inhibiting NLRP3 inflammasome with MCC950 ameliorates perioperative neurocognitive disorders, suppressing neuroinflammation in the hippocampus in aged mice. Int. Immunopharmacol. 2020, 82, 106317. [Google Scholar] [CrossRef]
- Swanton, T.; Beswick, J.A.; Hammadi, H.; Morris, L.; Williams, D.; de Cesco, S.; El-Sharkawy, L.; Yu, S.; Green, J.; Davis, J.B.; et al. Selective inhibition of the K+ efflux sensitive NLRP3 pathway by Cl- channel modulation. Chem. Sci. 2020, 11, 11720–11728. [Google Scholar] [CrossRef]
- Muñoz-Jurado, A.; Escribano, B.M.; Caballero-Villarraso, J.; Galván, A.; Agüera, E.; Santamaría, A.; Túnez, I. Melatonin and multiple sclerosis: Antioxidant, anti-inflammatory and immunomodulator mechanism of action. Inflammopharmacology 2022, 5, 1569–1596. [Google Scholar] [CrossRef]
- Madhu, L.N.; Kodali, M.; Attaluri, S.; Shuai, B.; Melissari, L.; Rao, X.; Shetty, A.K. Melatonin improves brain function in a model of chronic Gulf War Illness with modulation of oxidative stress, NLRP3 inflammasomes, and BDNF-ERK-CREB pathway in the hippocampus. Redox Biol. 2021, 43, 101973. [Google Scholar] [CrossRef]
- Fan, L.; Zhaohong, X.; Xiangxue, W.; Yingying, X.; Xiao, Z.; Xiaoyan, Z.; Jieke, Y.; Chao, L. Melatonin Ameliorates the Progression of Alzheimer′s Disease by Inducing TFEB Nuclear Translocation, Promoting Mitophagy, and Regulating NLRP3 Inflammasome Activity. Biomed. Res. Int. 2022, 2022, 8099459. [Google Scholar] [CrossRef]
- Farré-Alins, V.; Narros-Fernández, P.; Palomino-Antolín, A.; Decouty-Pérez, C.; Lopez-Rodriguez, A.B.; Parada, E.; Muñoz-Montero, A.; Gómez-Rangel, V.; López-Muñoz, F.; Ramos, E.; et al. Melatonin Reduces NLRP3 Inflammasome Activation by Increasing α7 nAChR-Mediated Autophagic Flux. Antioxidants 2020, 9, 1299. [Google Scholar] [CrossRef]
- Zheng, R.; Ruan, Y.; Yan, Y.; Lin, Z.; Xue, N.; Yan, Y.; Tian, J.; Yin, X.; Pu, J.; Zhang, B. Melatonin Attenuates Neuroinflammation by Down-Regulating NLRP3 Inflammasome via a SIRT1-Dependent Pathway in MPTP-Induced Models of Parkinson′s Disease. J. Inflamm. Res. 2021, 14, 3063–3075. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Li, S.; Huang, K.; Jiang, L.; Wang, Y. Metformin Alleviates LPS-Induced Acute Lung Injury by Regulating the SIRT1/NF-κB/NLRP3 Pathway and Inhibiting Endothelial Cell Pyroptosis. Front. Pharmacol. 2022, 13, 801337. [Google Scholar] [CrossRef]
- Chen, Q.; Yin, Y.; Li, L.; Zhang, Y.; He, W.; Shi, Y. Milrinone Ameliorates the Neuroinflammation and Memory Function of Alzheimer′s Disease in an APP/PS1 Mouse Model. Neuropsychiatr. Dis. Treat. 2021, 17, 2129–2139. [Google Scholar] [CrossRef]
- Garcez, M.L.; Mina, F.; Bellettini-Santos, T.; da Luz, A.P.; Schiavo, G.L.; Macieski, J.M.C.; Medeiros, E.B.; Marques, A.O.; Magnus, N.Q.; Budni, J. The Involvement of NLRP3 on the Effects of Minocycline in an AD-Like Pathology Induced by β-Amyloid Oligomers Administered to Mice. Mol. Neurobiol. 2019, 56, 2606–2617. [Google Scholar] [CrossRef]
- Cruz, S.L.; Armenta-Reséndiz, M.; Carranza-Aguilar, C.J.; Galván, E.J. Minocycline prevents neuronal hyperexcitability and neuroinflammation in medial prefrontal cortex, as well as memory impairment caused by repeated toluene inhalation in adolescent rats. Toxicol. Appl. Pharmacol. 2020, 395, 114980. [Google Scholar] [CrossRef]
- Chen, W.; Guo, C.; Huang, S.; Jia, Z.; Wang, J.; Zhong, J.; Ge, H.; Yuan, J.; Chen, T.; Liu, X.; et al. MitoQ attenuates brain damage by polarizing microglia towards the M2 phenotype through inhibition of the NLRP3 inflammasome after ICH. Pharmacol. Res. 2020, 161, 105122. [Google Scholar] [CrossRef]
- Chen, W.; Teng, X.; Ding, H.; Xie, Z.; Cheng, P.; Liu, Z.; Feng, T.; Zhang, X.; Huang, W.; Geng, D. Nrf2 protects against cerebral ischemia-reperfusion injury by suppressing programmed necrosis and inflammatory signaling pathways. Ann. Transl. Med. 2022, 10, 285. [Google Scholar] [CrossRef]
- Li, C.; Wang, J.; Fang, Y.; Liu, Y.; Chen, T.; Sun, H.; Zhou, X.F.; Liao, H. Nafamostat mesilate improves function recovery after stroke by inhibiting neuroinflammation in rats. Brain Behav. Immun. 2016, 56, 230–245. [Google Scholar] [CrossRef]
- Coll, R.C.; Schroder, K.; Pelegrín, P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol. Sci. 2022, 43, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, A.; Ran, Y.; Wei, C.; Xie, F.; Wu, J. Design, synthesis, and biological evaluation of phenyl vinyl sulfone based NLRP3 inflammasome inhibitors. Bioorg. Chem. 2022, 128, 106010. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zheng, B.; Yang, S.; Tang, X.; Wang, J.; Wei, D. The protective effects of phoenixin-14 against lipopolysaccharide-induced inflammation and inflammasome activation in astrocytes. Inflamm. Res. 2020, 69, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Dong, A.Q.; Yang, Y.P.; Jiang, S.M.; Yao, X.Y.; Qi, D.; Mao, C.J.; Cheng, X.Y.; Wang, F.; Hu, L.F.; Liu, C.F. Pramipexole inhibits astrocytic NLRP3 inflammasome activation via Drd3-dependent autophagy in a mouse model of Parkinson′s disease. Acta Pharmacol. Sin. 2022, 44, 32–43. [Google Scholar] [CrossRef]
- Yu, H.; Wu, M.; Lu, G.; Cao, T.; Chen, N.; Zhang, Y.; Jiang, Z.; Fan, H.; Yao, R. Prednisone alleviates demyelination through regulation of the NLRP3 inflammasome in a C57BL/6 mouse model of cuprizone-induced demyelination. Brain Res. 2018, 1678, 75–84. [Google Scholar] [CrossRef]
- Wei, C.; Guo, S.; Liu, W.; Jin, F.; Wei, B.; Fan, H.; Su, H.; Liu, J.; Zhang, N.; Fang, D.; et al. Resolvin D1 ameliorates Inflammation-Mediated Blood-Brain Barrier Disruption After Subarachnoid Hemorrhage in rats by Modulating A20 and NLRP3 Inflammasome. Front. Pharmacol. 2021, 11, 610734. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, J.; Zhao, X.; Chen, Z.; Wang, G.; Liu, A.; Wang, Q.; Zhou, W.; Xu, Y.; Wang, C. Phosphodiesterase-5 inhibitor sildenafil prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in APP/PS1 transgenic mice. Behav. Brain Res. 2013, 250, 230–237. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, Y.; Li, Q.; Chen, C.; Chen, H.; Song, Y.; Hua, F.; Zhang, Z. Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci. Lett. 2020, 736, 135279. [Google Scholar] [CrossRef]
- Liao, Y.J.; Pan, R.Y.; Kong, X.X.; Cheng, Y.; Du, L.; Wang, Z.C.; Yuan, C.; Cheng, J.B.; Yuan, Z.Q.; Zhang, H.Y. Correction: 1,2,4-Trimethoxybenzene selectively inhibits NLRP3 inflammasome activation and attenuates experimental autoimmune encephalomyelitis. Acta Pharmacol. Sin. 2022, 43, 504, Erratum in Acta Pharmacol. Sin. 2020, 42, 1769–1779. [Google Scholar] [CrossRef]
- Qiu, J.; Chen, Y.; Zhuo, J.; Zhang, L.; Liu, J.; Wang, B.; Sun, D.; Yu, S.; Lou, H. Urolithin A promotes mitophagy and suppresses NLRP3 inflammasome activation in lipopolysaccharide-induced BV2 microglial cells and MPTP-induced Parkinson′s disease model. Neuropharmacology 2022, 207, 108963. [Google Scholar] [CrossRef]
- Tian, D.; Xing, Y.; Gao, W.; Zhang, H.; Song, Y.; Tian, Y.; Dai, Z. Sevoflurane Aggravates the Progress of Alzheimer′s Disease Through NLRP3/Caspase-1/Gasdermin D Pathway. Front. Cell Dev. Biol. 2022, 9, 801422. [Google Scholar] [CrossRef]
- Das, S.; Mishra, K.P.; Ganju, L.; Singh, S.B. Andrographolide - A promising therapeutic agent, negatively regulates glial cell derived neurodegeneration of prefrontal cortex, hippocampus and working memory impairment. J. Neuroimmunol. 2017, 313, 161–175. [Google Scholar] [CrossRef]
- Gugliandolo, E.; D′Amico, R.; Cordaro, M.; Fusco, R.; Siracusa, R.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. Neuroprotective Effect of Artesunate in Experimental Model of Traumatic Brain Injury. Front. Neurol. 2018, 9, 590. [Google Scholar] [CrossRef] [Green Version]
- Ju, I.G.; Huh, E.; Kim, N.; Lee, S.; Choi, J.G.; Hong, J.; Oh, M.S. Artemisiae Iwayomogii Herba inhibits lipopolysaccharide-induced neuroinflammation by regulating NF-κB and MAPK signaling pathways. Phytomedicine 2021, 84, 153501. [Google Scholar] [CrossRef]
- Li, M.; Li, H.; Fang, F.; Deng, X.; Ma, S. Astragaloside IV attenuates cognitive impairments induced by transient cerebral ischemia and reperfusion in mice via anti-inflammatory mechanisms. Neurosci. Lett. 2017, 639, 114–119. [Google Scholar] [CrossRef]
- Jin, X.; Liu, M.Y.; Zhang, D.F.; Zhong, X.; Du, K.; Qian, P.; Yao, W.F.; Gao, H.; Wei, M.J. Baicalin mitigates cognitive impairment and protects neurons from microglia-mediated neuroinflammation via suppressing NLRP3 inflammasomes and TLR4/NF-κB signaling pathway. CNS Neurosci. Ther. 2019, 25, 575–590. [Google Scholar] [CrossRef]
- Lee, C.M.; Lee, D.S.; Jung, W.K.; Yoo, J.S.; Yim, M.J.; Choi, Y.H.; Park, S.; Seo, S.K.; Choi, J.S.; Lee, Y.M.; et al. Benzyl isothiocyanate inhibits inflammasome activation in E. coli LPS-stimulated BV2 cells. Int. J. Mol. Med. 2016, 38, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Wu, D.M.; Li, J.; Deng, S.H.; Liu, T.; Zhang, T.; He, M.; Zhao, Y.Y.; Xu, Y. Bixin Attenuates Experimental Autoimmune Encephalomyelitis by Suppressing TXNIP/NLRP3 Inflammasome Activity and Activating NRF2 Signaling. Front. Immunol. 2020, 11, 593368. [Google Scholar] [CrossRef]
- Satoh, T.; Trudler, D.; Oh, C.K.; Lipton, S.A. Potential Therapeutic Use of the Rosemary Diterpene Carnosic Acid for Alzheimer′s Disease, Parkinson′s Disease, and Long-COVID through NRF2 Activation to Counteract the NLRP3 Inflammasome. Antioxidants 2022, 11, 124. [Google Scholar] [CrossRef]
- Shi, W.; Xu, G.; Zhan, X.; Gao, Y.; Wang, Z.; Fu, S.; Qin, N.; Hou, X.; Ai, Y.; Wang, C.; et al. Carnosol inhibits inflammasome activation by directly targeting HSP90 to treat inflammasome-mediated diseases. Cell Death Dis. 2020, 11, 252. [Google Scholar] [CrossRef] [Green Version]
- Chu, X.; Zhang, L.; Zhou, Y.; Fang, Q. Cucurbitacin B alleviates cerebral ischemia/reperfusion injury by inhibiting NLRP3 inflammasome-mediated inflammation and reducing oxidative stress. Biosci. Biotechnol. Biochem. 2022, 11, zbac065. [Google Scholar] [CrossRef] [PubMed]
- González-Cofrade, L.; Cuadrado, I.; Amesty, Á.; Estévez-Braun, A.; de Las Heras, B.; Hortelano, S. Dehydroisohispanolone as a Promising NLRP3 Inhibitor Agent: Bioevaluation and Molecular Docking. Pharmaceuticals 2022, 15, 825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, D.; Yao, X.; Wen, J.; Wang, Y.; Zhang, Y. DMTHB ameliorates memory impairment in Alzheimer′s disease mice through regulation of neuroinflammation. Neurosci. Lett. 2022, 785, 136770. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, Y.; Yu, L.; Xu, Y. Esculentoside A exerts anti-inflammatory activity in microglial cells. Int. Immunopharmacol. 2017, 51, 148–157. [Google Scholar] [CrossRef]
- Zheng, X.; Gong, T.; Tang, C.; Zhong, Y.; Shi, L.; Fang, X.; Chen, D.; Zhu, Z. Gastrodin improves neuroinflammation-induced cognitive dysfunction in rats by regulating NLRP3 inflammasome. BMC Anesthesiol. 2022, 22, 371. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Zhang, J.; Gao, Y.; Li, S.; Chang, C.; Yu, D.; Yang, G. Ginkgolide B inhibits NLRP3 inflammasome activation and promotes microglial M2 polarization in Aβ1-42-induced microglia cells. Neurosci. Lett. 2021, 764, 136206. [Google Scholar] [CrossRef]
- Shao, L.; Dong, C.; Geng, D.; He, Q.; Shi, Y. Ginkgolide B inactivates the NLRP3 inflammasome by promoting autophagic degradation to improve learning and memory impairment in Alzheimer′s disease. Metab. Brain Dis. 2022, 37, 329–341. [Google Scholar] [CrossRef]
- Jiang, J.; Sun, X.; Akther, M.; Lian, M.L.; Quan, L.H.; Koppula, S.; Han, J.H.; Kopalli, S.R.; Kang, T.B.; Lee, K.H. Ginsenoside metabolite 20(S)-protopanaxatriol from Panax ginseng attenuates inflammation-mediated NLRP3 inflammasome activation. J. Ethnopharmacol. 2020, 251, 112564. [Google Scholar] [CrossRef]
- Gao, Y.; Li, J.; Wang, J.; Li, X.; Li, J.; Chu, S.; Li, L.; Chen, N.; Zhang, L. Ginsenoside Rg1 prevent and treat inflammatory diseases: A review. Int. Immunopharmacol. 2020, 87, 106805. [Google Scholar] [CrossRef]
- Wang, J.; Wang, D.; Zhou, Z.; Zhang, X.; Zhang, C.; He, Y.; Liu, C.; Yuan, C.; Yuan, D.; Wang, T. Saponins from Panax japonicus alleviate HFD-induced impaired behaviors through inhibiting NLRP3 inflammasome to upregulate AMPA receptors. Neurochem. Int. 2021, 148, 105098. [Google Scholar] [CrossRef]
- Yi, Y.S. Roles of ginsenosides in inflammasome activation. J. Ginseng Res. 2019, 43, 172–178. [Google Scholar] [CrossRef]
- Yi, Y.S. New mechanisms of ginseng saponin-mediated anti-inflammatory action via targeting canonical inflammasome signaling pathways. J. Ethnopharmacol. 2021, 278, 114292. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Tiwari, V.; Gangadhar, N.M.; Rashid, M.; Sultana, N.; Singh, S.K.; Shukla, S.; Wahajuddin, M. Isoformononetin, a dietary isoflavone protects against streptozotocin induced rat model of neuroinflammation through inhibition of NLRP3/ASC/IL-1 axis activation. Life Sci. 2021, 286, 119989. [Google Scholar] [CrossRef]
- Zeng, J.; Chen, Y.; Ding, R. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J. Neuroinflamm. 2017, 14, 119. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.H.; Lv, H.N.; Cui, Q.H.; Tu, P.F.; Jiang, Y.; Zeng, K.W. Isosibiricin inhibits microglial activation by targeting the dopamine D1/D2 receptor-dependent NLRP3/caspase-1 inflammasome pathway. Acta Pharmacol. Sin. 2020, 41, 173–180. [Google Scholar] [CrossRef]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, S.F.; Sureda, A.; Dehpour, A.R.; Shirooie, S.; Silva, A.S.; Devi, K.P.; Ahmed, T.; Ishaq, N.; Hashim, R.; Sobarzo-Sánchez, E.; et al. Regulation of autophagy by polyphenols: Paving the road for treatment of neurodegeneration. Biotechnol. Adv. 2018, 36, 1768–1778. [Google Scholar] [CrossRef]
- Han, X.; Sun, S.; Sun, Y.; Song, Q.; Zhu, J.; Song, N.; Chen, M.; Sun, T.; Xia, M.; Ding, J.; et al. Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: Implications for Parkinson disease. Autophagy 2019, 15, 1860–1881. [Google Scholar] [CrossRef]
- Mokarizadeh, N.; Karimi, P.; Erfani, M.; Sadigh-Eteghad, S.; Fathi Maroufi, N.; Rashtchizadeh, N. β-Lapachone attenuates cognitive impairment and neuroinflammation in beta-amyloid induced mouse model of Alzheimer′s disease. Int. Immunopharmacol. 2020, 81, 106300. [Google Scholar] [CrossRef]
- Xiong, R.; Zhou, X.G.; Tang, Y.; Wu, J.M.; Sun, Y.S.; Teng, J.F.; Pan, R.; Law, B.Y.; Zhao, Y.; Qiu, W.Q.; et al. Lychee seed polyphenol protects the blood-brain barrier through inhibiting Aβ(25-35)-induced NLRP3 inflammasome activation via the AMPK/mTOR/ULK1-mediated autophagy in bEnd.3 cells and APP/PS1 mice. Phytother. Res. 2021, 35, 954–973. [Google Scholar] [CrossRef]
- Qiu, W.Q.; Pan, R.; Tang, Y.; Zhou, X.G.; Wu, J.M.; Yu, L.; Law, B.Y.; Ai, W.; Yu, C.L.; Qin, D.L.; et al. Lychee seed polyphenol inhibits Aβ-induced activation of NLRP3 inflammasome via the LRP1/AMPK mediated autophagy induction. Biomed. Pharmacother. 2020, 130, 110575. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.Y.; Wang, R.C.; Pan, Y.L.; Yue, Z.G.; Zhou, R.; Xie, P.; Tang, Z.S. Mangiferin inhibited neuroinflammation through regulating microglial polarization and suppressing NF-κB, NLRP3 pathway. Chin. J. Nat. Med. 2021, 19, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Luo, S.; Zhao, S.; Yin, S.; Li, X.; Mou, T. Myricitrin attenuates memory impairment in a rat model of sepsis-associated encephalopathy via the NLRP3/Bax/Bcl pathway. Folia Neuropathol. 2019, 57, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, T.; Lu, M. Myricitrin attenuates hypoxic-ischemia-induced brain injury in neonatal rats by mitigating oxidative stress and nuclear factor erythroid 2-related factor 2/hemeoxygenase-1/antioxidant response element signaling pathway. Phcog. Mag. 2021, 17, 828–835. [Google Scholar] [CrossRef]
- Wang, H.; Guo, Y.; Qiao, Y.; Zhang, J.; Jiang, P. Nobiletin Ameliorates NLRP3 Inflammasome-Mediated Inflammation Through Promoting Autophagy via the AMPK Pathway. Mol. Neurobiol. 2020, 57, 5056–5068. [Google Scholar] [CrossRef]
- Al Rihani, S.B.; Darakjian, L.I.; Kaddoumi, A. Oleocanthal-Rich Extra-Virgin Olive Oil Restores the Blood-Brain Barrier Function through NLRP3 Inflammasome Inhibition Simultaneously with Autophagy Induction in TgSwDI Mice. ACS Chem. Neurosci. 2019, 10, 3543–3554. [Google Scholar] [CrossRef]
- Wang, S.; Yang, H.; Yu, L.; Jin, J.; Qian, L.; Zhao, H.; Xu, Y.; Zhu, X. Oridonin attenuates Aβ1-42-induced neuroinflammation and inhibits NF-κB pathway. PloS ONE 2014, 9, e104745. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, X.; Gong, Q.; Shi, J.; Li, F. Osthole Improves Cognitive Function of Vascular Dementia Rats: Reducing Aβ Deposition via Inhibition NLRP3 Inflammasome. Biol. Pharm. Bull. 2020, 43, 1315–1323. [Google Scholar] [CrossRef]
- Chen, D.B.; Gao, H.W.; Peng, C.; Pei, S.Q.; Dai, A.R.; Yu, X.T.; Zhou, P.; Wang, Y.; Cai, B. Quinones as preventive agents in Alzheimer′s diseases: Focus on NLRP3 inflammasomes. J. Pharm. Pharmacol. 2020, 72, 1481–1490. [Google Scholar] [CrossRef]
- Han, X.; Xu, T.; Fang, Q.; Zhang, H.; Yue, L.; Hu, G.; Sun, L. Quercetin hinders microglial activation to alleviate neurotoxicity via the interplay between NLRP3 inflammasome and mitophagy. Redox Biol. 2021, 44, 102010. [Google Scholar] [CrossRef]
- Li, H.; Chen, F.J.; Yang, W.L.; Qiao, H.Z.; Zhang, S.J. Quercetin improves cognitive disorder in aging mice by inhibiting NLRP3 inflammasome activation. Food Funct. 2021, 12, 717–725. [Google Scholar] [CrossRef]
- Kiasalari, Z.; Afshin-Majd, S.; Baluchnejadmojarad, T.; Azadi-Ahmadabadi, E.; Fakour, M.; Ghasemi-Tarie, R.; Jalalzade-Ogvar, S.; Khodashenas, V.; Tashakori-Miyanroudi, M.; Roghani, M. Sinomenine Alleviates Murine Experimental Autoimmune Encephalomyelitis Model of Multiple Sclerosis through Inhibiting NLRP3 Inflammasome. J. Mol. Neurosci. 2021, 71, 215–224. [Google Scholar] [CrossRef]
- Atluri, V.S.R.; Tiwari, S.; Rodriguez, M.; Kaushik, A.; Yndart, A.; Kolishetti, N.; Yatham, M.; Nair, M. Inhibition of Amyloid-Beta Production, Associated Neuroinflammation, and Histone Deacetylase 2-Mediated Epigenetic Modifications Prevent Neuropathology in Alzheimer′s Disease in vitro Model. Front. Aging Neurosci. 2020, 11, 342. [Google Scholar] [CrossRef] [Green Version]
- Cadoná, F.C.; de Souza, D.V.; Fontana, T.; Bodenstein, D.F.; Ramos, A.P.; Sagrillo, M.R.; Salvador, M.; Mota, K.; Davidson, C.B.; Ribeiro, E.E.; et al. Açaí (Euterpe oleracea Mart.) as a Potential Anti-neuroinflammatory Agent: NLRP3 Priming and Activating Signal Pathway Modulation. Mol. Neurobiol. 2021, 58, 4460–4476. [Google Scholar] [CrossRef]
- Yu, S.H.; Sun, X.; Kim, M.K.; Akther, M.; Han, J.H.; Kim, T.Y.; Jiang, J.; Kang, T.B.; Lee, K.H. Chrysanthemum indicum extract inhibits NLRP3 and AIM2 inflammasome activation via regulating ASC phosphorylation. J. Ethnopharmacol. 2019, 239, 111917. [Google Scholar] [CrossRef]
- Jeong, Y.H.; Kim, T.I.; Oh, Y.C.; Ma, J.Y. Chrysanthemum indicum Prevents Hydrogen Peroxide-Induced Neurotoxicity by Activating the TrkB/Akt Signaling Pathway in Hippocampal Neuronal Cells. Nutrients 2021, 13, 3690. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, G.; Li, Z.; Xiao, X.; Tang, J.; Bai, Z. NLRP3 Inflammasome Pharmacological Inhibitors in Glycyrrhiza for NLRP3-Driven Diseases Treatment: Extinguishing the Fire of Inflammation. J. Inflamm. Res. 2022, 15, 409–422. [Google Scholar] [CrossRef]
- Kim, N.; Do, J.; Ju, I.G.; Jeon, S.H.; Lee, J.K.; Oh, M.S. Picrorhiza kurroa Prevents Memory Deficits by Inhibiting NLRP3 Inflammasome Activation and BACE1 Expression in 5xFAD Mice. Neurotherapeutics 2020, 17, 189–199. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, X.; Zhang, X.; Huang, L.; Sun, Y.; Cheng, Z.; Xu, W.; Li, C.G.; Zheng, Y.; Huang, M. Pien-Tze-Huang, a Chinese patent formula, attenuates NLRP3 inflammasome-related neuroinflammation by enhancing autophagy via the AMPK/mTOR/ULK1 signaling pathway. Biomed. Pharmacother. 2021, 141, 111814. [Google Scholar] [CrossRef]
- Yin, X.L.; Wu, H.M.; Zhang, B.H.; Zhu, N.W.; Chen, T.; Ma, X.X.; Zhang, L.Y.; Lv, L.; Zhang, M.; Wang, F.Y.; et al. Tojapride prevents CaSR-mediated NLRP3 inflammasome activation in oesophageal epithelium irritated by acidic bile salts. J. Cell. Mol. Med. 2020, 24, 1208–1219. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Fang, B.Y.; Meng, X.B.; Zhang, S.X.; Wang, H.; Gao, G.; Liu, F.; Wu, Y.; Hu, J.; Sun, G.B.; et al. Folium Ginkgo extract and tetramethylpyrazine sodium chloride injection (Xingxiong injection) protects against focal cerebral ischaemia/reperfusion injury via activating the Akt/Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Pharm. Biol. 2022, 60, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Hong, J.Y.; Jeon, W.J.; Lee, J.; Baek, S.H.; Ha, I.H. Lycopus lucidus Turcz Exerts Neuroprotective Effects Against H2O2-Induced Neuroinflammation by Inhibiting NLRP3 Inflammasome Activation in Cortical Neurons. J. Inflamm. Res. 2021, 14, 1759–1773. [Google Scholar] [CrossRef]
- Denes, A.; Coutts, G.; Lénárt, N.; Cruickshank, S.M.; Pelegrin, P.; Skinner, J.; Rothwell, N.; Allan, S.M.; Brough, D. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl. Acad. Sci. USA 2015, 112, 4050–4055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.L.; Wang, X.; Shao, L.; Jiang, G.T.; Min, J.W.; Mei, X.Y.; He, X.H.; Liu, W.H.; Huang, W.X.; Peng, B.W. TRPV1 mediates astrocyte activation and interleukin-1β release induced by hypoxic ischemia (HI). J. Neuroinflamm. 2019, 16, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schölwer, I.; Habib, P.; Voelz, C.; Rolfes, L.; Beyer, C.; Slowik, A. NLRP3 Depletion Fails to Mitigate Inflammation but Restores Diminished Phagocytosis in BV-2 Cells After In Vitro Hypoxia. Mol. Neurobiol. 2020, 57, 2588–2599. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, J.; Li, D.; Li, P.; Zhou, X.; Li, Y.; He, Z.; Qin, L.; Liang, L.; Luo, X. Interleukin-10 inhibits interleukin-1β production and inflammasome activation of microglia in epileptic seizures. J. Neuroinflamm. 2019, 16, 66. [Google Scholar] [CrossRef] [Green Version]
- Liew, F.; Girard, J.P.; Turnquist, H. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Jiao, M.; Li, X.; Chen, L.; Wang, X.; Yuan, B.; Liu, T.; Dong, Q.; Mei, H.; Yin, H. Neuroprotective effect of astrocyte-derived IL-33 in neonatal hypoxic-ischemic brain injury. J. Neuroinflamm. 2020, 17, 251. [Google Scholar] [CrossRef]
- Strangward, P.; Haley, M.J.; Albornoz, M.G.; Barrington, J.; Shaw, T.; Dookie, R.; Zeef, L.; Baker, S.M.; Winter, E.; Tzeng, T.C.; et al. Targeting the IL33-NLRP3 axis improves therapy for experimental cerebral malaria. Proc. Natl. Acad. Sci. USA 2018, 115, 7404–7409. [Google Scholar] [CrossRef] [Green Version]
- Bellut, M.; Raimondi, A.T.; Haarmann, A.; Zimmermann, L.; Stoll, G.; Schuhmann, M.K. NLRP3 Inhibition Reduces rt-PA Induced Endothelial Dysfunction under Ischemic Conditions. Biomedicines 2022, 10, 762. [Google Scholar] [CrossRef]
- Xu, Q.; Ye, Y.; Wang, Z.; Zhu, H.; Li, Y.; Wang, J.; Gao, W.; Gu, L. NLRP3 Knockout Protects against Lung Injury Induced by Cerebral Ischemia-Reperfusion. Oxid. Med. Cell. Longev. 2022, 2022, 6260102. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, M.; Ji, M.; Zhang, D.; Chen, B.; Gong, W.; Li, X.; Zhou, Y.; Dong, C.; Wen, G.; et al. The neuroprotective mechanism of lithium after ischaemic stroke. Commun. Biol. 2022, 5, 105. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Zhao, W.; Li, L.; Li, L.; Sun, Y.; Shao, J.; Ren, X.; Zang, W.; Cao, J. Role of spinal RIP3 in inflammatory pain and electroacupuncture-mediated analgesic effect in mice. Life Sci. 2022, 306, 120839. [Google Scholar] [CrossRef]
- Zhong, X.; Chen, B.; Li, Z.; Lin, R.; Ruan, S.; Wang, F.; Liang, H.; Tao, J. Electroacupuncture Ameliorates Cognitive Impairment Through the Inhibition of NLRP3 Inflammasome Activation by Regulating Melatonin-Mediated Mitophagy in Stroke Rats. Neurochem. Res. 2022, 47, 1917–1930, Erratum in Neurochem. Res. 2022, 47, 1931–1933. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer′s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [Green Version]
- Lučiūnaitė, A.; McManus, R.M.; Jankunec, M.; Rácz, I.; Dansokho, C.; Dalgėdienė, I.; Schwartz, S.; Brosseron, F.; Heneka, M.T. Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J. Neurochem. 2020, 155, 650–661. [Google Scholar] [CrossRef] [Green Version]
- Couturier, J.; Stancu, I.C.; Schakman, O.; Pierrot, N.; Huaux, F.; Kienlen-Campard, P.; Dewachter, I.; Octave, J.N. Activation of phagocytic activity in astrocytes by reduced expression of the inflammasome component ASC and its implication in a mouse model of Alzheimer disease. J. Neuroinflamm. 2016, 13, 20. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, S.B.; Bhagavan, S.M.; Kaur, H.; Giler, G.E.; Kempuraj, D.; Thangavel, R.; Ahmed, M.E.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.; et al. Glia Maturation Factor in the Pathogenesis of Alzheimer′s disease. Open Access J. Neurol. Neurosurg. 2019, 12, 79–82. [Google Scholar]
- Friker, L.L.; Scheiblich, H.; Hochheiser, I.V.; Brinkschulte, R.; Riedel, D.; Latz, E.; Geyer, M.; Heneka, M.T. β-Amyloid Clustering around ASC Fibrils Boosts Its Toxicity in Microglia. Cell Rep. 2020, 30, 3743–3754.e6. [Google Scholar] [CrossRef]
- Murphy, N.; Grehan, B.; Lynch, M.A. Glial uptake of amyloid beta induces NLRP3 inflammasome formation via cathepsin-dependent degradation of NLRP10. Neuromolecular Med. 2014, 16, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Slowik, A.; Lammerding, L.; Zendedel, A.; Habib, P.; Beyer, C. Impact of steroid hormones E2 and P on the NLRP3/ASC/Casp1 axis in primary mouse astroglia and BV-2 cells after in vitro hypoxia. J. Steroid Biochem. Mol. Biol. 2018, 183, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Liu, Y.; Yu, D.; Wang, M.; Hou, Y. The neuroprotection of progesterone against Aβ-induced NLRP3-Caspase-1 inflammasome activation via enhancing autophagy in astrocytes. Int. Immunopharmacol. 2019, 74, 105669. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Dixon, B.J.; Doycheva, D.M.; Li, B.; Zhang, Y.; Hu, Q.; He, Y.; Guo, Z.; Nowrangi, D.; Flores, J.; et al. IRE1α inhibition decreased TXNIP/NLRP3 inflammasome activation through miR-17-5p after neonatal hypoxic-ischemic brain injury in rats. J. Neuroinflamm. 2018, 15, 32. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Xu, Y.; Wang, X.; Guo, C.; Wang, T.; Wang, Z.Y. Dl-3-n-Butylphthalide Inhibits NLRP3 Inflammasome and Mitigates Alzheimer′s-Like Pathology via Nrf2-TXNIP-TrX Axis. Antioxid. Redox. Signal. 2019, 30, 1411–1431. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, J.; Chen, Y.; Shang, H.; Zhang, W.; Yu, J.; He, L.; Xing, C.; Zhuang, C. Direct inhibition of Keap1-Nrf2 Protein-Protein interaction as a potential therapeutic strategy for Alzheimer′s disease. Bioorg. Chem. 2020, 103, 104172. [Google Scholar] [CrossRef]
- Saad El-Din, S.; Rashed, L.; Medhat, E.; Emad Aboulhoda, B.; Desoky Badawy, A.; Mohammed ShamsEldeen, A.; Abdelgwad, M. Active form of vitamin D analogue mitigates neurodegenerative changes in Alzheimer′s disease in rats by targeting Keap1/Nrf2 and MAPK-38p/ERK signaling pathways. Steroids 2020, 156, 108586. [Google Scholar] [CrossRef]
- Yang, X.; Ji, J.; Liu, C.; Zhou, M.; Li, H.; Ye, S.; Hu, Q. HJ22, a Novel derivative of piperine, attenuates ibotenic acid-induced cognitive impairment, oxidative stress, apoptosis and inflammation via inhibiting the protein-protein interaction of Keap1-Nrf2. Int. Immunopharmacol. 2020, 83, 106383. [Google Scholar] [CrossRef]
- Yang, X.; Zhi, J.; Leng, H.; Chen, Y.; Gao, H.; Ma, J.; Ji, J.; Hu, Q. The piperine derivative HJ105 inhibits Aβ1-42-induced neuroinflammation and oxidative damage via the Keap1-Nrf2-TXNIP axis. Phytomedicine 2021, 87, 153571. [Google Scholar] [CrossRef]
- Bharti, V.; Tan, H.; Zhou, H.; Wang, J.F. Txnip mediates glucocorticoid-activated NLRP3 inflammatory signaling in mouse microglia. Neurochem. Int. 2019, 131, 104564. [Google Scholar] [CrossRef]
- Gussago, C.; Casati, M.; Ferri, E.; Arosio, B. The Triggering Receptor Expressed on Myeloid Cells-2 (TREM-2) as Expression of the Relationship between Microglia and Alzheimer′s Disease: A Novel Marker for a Promising Pathway to Explore. J. Frailty Aging 2019, 8, 54–56. [Google Scholar] [CrossRef]
- Wang, S.Y.; Gong, P.Y.; Yan, E.; Zhang, Y.D.; Jiang, T. The role of TREML2 in Alzheimer′s disease. J. Alzheimers Dis. 2020, 76, 799–806. [Google Scholar] [CrossRef]
- Sierksma, A.; Lu, A.; Mancuso, R.; Fattorelli, N.; Thrupp, N.; Salta, E.; Zoco, J.; Blum, D.; Buee, L.; De Strooper, B.; et al. Novel Alzheimer risk genes determine the microglia response to amyloid-beta but not to TAU pathology. EMBO Mol. Med. 2020, 12, e10606. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, C.C.; Atagi, Y.; Chen, X.F.; Jia, L.; Yang, L.; He, W.; Zhang, X.; Kang, S.S.; Rosenberry, T.L.; et al. Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiol. Aging 2016, 42, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.Y.; Fu, X.X.; Duan, R.; Wei, B.; Cao, H.M.; Yan, E.; Chen, S.Y.; Zhang, Y.D.; Jiang, T. The Alzheimer′s disease-associated gene TREML2 modulates inflammation by regulating microglia polarization and NLRP3 inflammasome activation. Neural Regen. Res. 2023, 18, 434–438. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, A.B.; Hennessy, E.; Murray, C.L.; Nazmi, A.; Delaney, H.J.; Healy, D.; Fagan, S.G.; Rooney, M.; Stewart, E.; Lewis, A.; et al. Acute systemic inflammation exacerbates neuroinflammation in Alzheimer′s disease: IL-1β drives amplified responses in primed astrocytes and neuronal network dysfunction. Alzheimers Dement. 2021, 17, 1735–1755. [Google Scholar] [CrossRef]
- Saresella, M.; Piancone, F.; Marventano, I.; Zoppis, M.; Hernis, A.; Zanette, M.; Trabattoni, D.; Chiappedi, M.; Ghezzo, A.; Canevini, M.P.; et al. Multiple inflammasome complexes are activated in autistic spectrum disorders. Brain Behav. Immun. 2016, 57, 125–133. [Google Scholar] [CrossRef]
- Ahmed, M.E.; Iyer, S.; Thangavel, R.; Kempuraj, D.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.; Zaheer, A. Co-Localization of Glia Maturation Factor with NLRP3 Inflammasome and Autophagosome Markers in Human Alzheimer′s Disease Brain. J. Alzheimers Dis. 2017, 60, 1143–1160. [Google Scholar] [CrossRef] [Green Version]
- Stancu, I.C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.N.; et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Li, X.; Wang, J.; Wang, H. The Role of the Effects of Autophagy on NLRP3 Inflammasome in Inflammatory Nervous System Diseases. Front. Cell Dev. Biol. 2021, 9, 657478. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Xiao, D.; Zhao, Y.; Tan, B.; Long, Z.; Yu, L.; He, G. Enhanced Autolysosomal Function Ameliorates the Inflammatory Response Mediated by the NLRP3 Inflammasome in Alzheimer′s Disease. Front. Aging Neurosci. 2021, 13, 629891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Shao, X.Y.; Qi, G.J.; Chen, Q.; Bu, L.L.; Chen, L.J.; Shi, J.; Ming, J.; Tian, B. Cdk5-Dependent Activation of Neuronal Inflammasomes in Parkinson′s Disease. Mov. Disord. 2016, 31, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Deora, V.; Albornoz, E.A.; Zhu, K.; Woodruff, T.M.; Gordon, R. The Ketone Body β-Hydroxybutyrate Does Not Inhibit Synuclein Mediated Inflammasome Activation in Microglia. J. Neuroimmune Pharmacol. 2017, 12, 568–574. [Google Scholar] [CrossRef]
- Shippy, D.C.; Wilhelm, C.; Viharkumar, P.A.; Raife, T.J.; Ulland, T.K. β-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer′s disease pathology. J. Neuroinflamm. 2020, 17, 280. [Google Scholar] [CrossRef]
- Sarkar, S.; Malovic, E.; Harishchandra, D.S.; Ghaisas, S.; Panicker, N.; Charli, A.; Palanisamy, B.N.; Rokad, D.; Jin, H.; Anantharam, V.; et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson′s disease. NPJ Park. Dis. 2017, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O′Neill, L.A.; et al. Inflammasome Inhibition Prevents a-Synuclein Pathology and Dopaminergic Neurodegeneration in Mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xia, Y.; Yin, S.; Wan, F.; Hu, J.; Kou, L.; Sun, Y.; Wu, J.; Zhou, Q.; Huang, J.; et al. Targeting Microglial α-Synuclein/TLRs/NF-kappaB/NLRP3 Inflammasome Axis in Parkinson′s Disease. Front. Immunol. 2021, 12, 719807. [Google Scholar] [CrossRef]
- Scheiblich, H.; Bousset, L.; Schwartz, S.; Griep, A.; Latz, E.; Melki, R.; Heneka, M.T. Microglial NLRP3 Inflammasome Activation upon TLR2 and TLR5 Ligation by Distinct α-Synuclein Assemblies. J. Immunol. 2021, 207, 2143–2154. [Google Scholar] [CrossRef]
- von Herrmann, K.M.; Salas, L.A.; Martinez, E.M.; Young, A.L.; Howard, J.M.; Feldman, M.S.; Christensen, B.C.; Wilkins, O.M.; Lee, S.L.; Hickey, W.F.; et al. NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson′s disease. NPJ Park. Dis. 2018, 4, 24. [Google Scholar] [CrossRef]
- Fan, Z.; Pan, Y.T.; Zhang, Z.Y.; Yang, H.; Yu, S.Y.; Zheng, Y.; Ma, J.H.; Wang, X.M. Systemic Activation of NLRP3 Inflammasome and Plasma a-Synuclein Levels Are Correlated with Motor Severity and Progression in Parkinson′s Disease. J. Neuroinflamm. 2020, 17, 11. [Google Scholar] [CrossRef] [Green Version]
- Anderson, F.L.; von Herrmann, K.M.; Andrew, A.S.; Kuras, Y.I.; Young, A.L.; Scherzer, C.R.; Hickey, W.F.; Lee, S.L.; Havrda, M.C. Plasma-borne indicators of inflammasome activity in Parkinson′s disease patients. NPJ Park. Dis. 2021, 7, 2. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, X.N.; Fang, J.N.; Hua, F.F.; Han, J.Y.; Yuan, Z.Q.; Xie, A.M. The mechanism behind activation of the Nod-like receptor family protein 3 inflammasome in Parkinson′s disease. Neural. Regen. Res. 2022, 17, 898–904. [Google Scholar] [CrossRef]
- Simola, N.; Morelli, M.; Carta, A.R. The 6-hydroxydopamine model of Parkinson′s disease. Neurotox. Res. 2007, 11, 151–167. [Google Scholar] [CrossRef]
- Si, X.L.; Fang, Y.J.; Li, L.F.; Gu, L.Y.; Yin, X.Z.; Tian, J.; Yan, Y.P.; Pu, J.L.; Zhang, B.R. From inflammasome to Parkinson′s disease: Does the NLRP3 inflammasome facilitate exosome secretion and exosomal alpha-synuclein transmission in Parkinson′s disease? Exp. Neurol. 2021, 336, 113525. [Google Scholar] [CrossRef]
- Chen, J.; Mao, K.; Yu, H.; Wen, Y.; She, H.; Zhang, H.; Liu, L.; Li, M.; Li, W.; Zou, F. p38-TFEB pathways promote microglia activation through inhibiting CMA-mediated NLRP3 degradation in Parkinson′s disease. J. Neuroinflamm. 2021, 18, 295. [Google Scholar] [CrossRef]
- Panicker, N.; Kam, T.I.; Wang, H.; Neifert, S.; Chou, S.C.; Kumar, M.; Brahmachari, S.; Jhaldiyal, A.; Hinkle, J.T.; Akkentli, F.; et al. Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson′s disease. Neuron 2022, 110, 2422–2437.e9. [Google Scholar] [CrossRef]
- Gris, D.; Ye, Z.; Iocca, H.A.; Wen, H.; Craven, R.R.; Gris, P.; Huang, M.; Schneider, M.; Miller, S.D.; Ting, J.P. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J. Immunol. 2010, 185, 974–981. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, S.; Río, J.; Urcelay, E.; Nurtdinov, R.; Bustamante, M.F.; Fernández, O.; Oliver, B.; Zettl, U.; Brassat, D.; Killestein, J.; et al. NLRP3 inflammasome is associated with the response to IFN-β in patients with multiple sclerosis. Brain 2015, 138, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, S.; Costa, C.; Eixarch, H.; Keller, C.W.; Amman, L.; Martínez-Banaclocha, H.; Midaglia, L.; Sarró, E.; Machín-Díaz, I.; Villar, L.M.; et al. NLRP3 inflammasome as prognostic factor and therapeutic target in primary progressive multiple sclerosis patients. Brain 2020, 143, 1414–1430. [Google Scholar] [CrossRef]
- Olcum, M.; Tastan, B.; Kiser, C.; Genc, S.; Genc, K. Microglial NLRP3 inflammasome activation in multiple sclerosis. Adv. Protein Chem. Struct. Biol. 2020, 119, 247–308. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yu, H.; Bu, Z.; Wen, L.; Yan, L.; Feng, J. Focus on the Role of the NLRP3 Inflammasome in Multiple Sclerosis: Pathogenesis, Diagnosis, and Therapeutics. Front. Mol. Neurosci. 2022, 15, 894298. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.L.; Oliveira, E.M.; Pontillo, A. Variants in NLRP3 and NLRC4 inflammasome associate with susceptibility and severity of multiple sclerosis. Mult. Scler. Relat. Disord. 2019, 29, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Vidmar, L.; Maver, A.; Drulović, J.; Sepčić, J.; Novaković, I.; Ristič, S.; Šega, S.; Peterlin, B. Multiple Sclerosis patients carry an increased burden of exceedingly rare genetic variants in the inflammasome regulatory genes. Sci. Rep. 2019, 9, 9171. [Google Scholar] [CrossRef] [Green Version]
- Keane, R.W.; Dietrich, W.D.; de Rivero Vaccari, J.P. Inflammasome Proteins as Biomarkers of Multiple Sclerosis. Front. Neurol. 2018, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, A.; Quintana, F.J. The NLRP3 inflammasome in progressive multiple sclerosis. Brain 2020, 143, 1286–1288. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, N.; Gran, B.; Constantinescu, C.S. Are current disease-modifying therapeutics in multiple sclerosis justified based on studies in experimental autoimmune encephalomyelitis? J. Neurochem. 2010, 115, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Williams, K.L.; Gunn, M.D.; Shinohara, M.L. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2012, 109, 10480–10485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Kuo, A.; Brockman, D.A.; Cooper, M.A.; Smith, M.T. Pharmacological inhibition of the NLRP3 inflammasome as a potential target for multiple sclerosis induced central neuropathic pain. Inflammopharmacology 2018, 26, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Bakhshi, S.; Shamsi, S. MCC950 in the treatment of NLRP3-mediated inflammatory diseases: Latest evidence and therapeutic outcomes. Int. Immunopharmacol. 2022, 106, 108595. [Google Scholar] [CrossRef]
- Hou, B.; Zhang, Y.; Liang, P.; He, Y.; Peng, B.; Liu, W.; Han, S.; Yin, J.; He, X. Inhibition of the NLRP3-inflammasome prevents cognitive deficits in experimental autoimmune encephalomyelitis mice via the alteration of astrocyte phenotype. Cell Death Dis. 2020, 11, 377. [Google Scholar] [CrossRef]
- Inoue, M.; Shinohara, M.L. The role of interferon-β in the treatment of multiple sclerosis and experimental autoimmune encephalomyelitis—In the perspective of inflammasomes. Immunology 2013, 139, 11–18. [Google Scholar] [CrossRef]
- Gros-Louis, F.; Gaspar, C.; Rouleau, G.A. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 956–972. [Google Scholar] [CrossRef] [Green Version]
- Jaarsma, D.; Teuling, E.; Haasdijk, E.D.; De Zeeuw, C.I.; Hoogenraad, C.C. Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J. Neurosci. 2008, 28, 2075–2088. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Boillee, S.; Roberts, E.A.; Garcia, M.L.; McAlonis-Downes, M.; Mikse, O.R.; Cleveland, D.W.; Goldstein, L.S. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA 2008, 105, 7594–7599. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Ezzi, S.A.; Urushitani, M.; Julien, J.P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007, 102, 170–178. [Google Scholar] [CrossRef]
- Roberts, K.; Zeineddine, R.; Corcoran, L.; Li, W.; Campbell, I.L.; Yerbury, J.J. Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a cytotoxic phenotype. Glia 2013, 61, 409–419. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Elliott, E.; Rifai, O.M.; O′Shaughnessy, J.; McDade, K.; Abrahams, S.; Chandran, S.; Smith, C.; Gregory, J.M. NLRP3 inflammasome as a key molecular target underlying cognitive resilience in amyotrophic lateral sclerosis. J. Pathol. 2022, 256, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Kadhim, H.; Deltenre, P.; Martin, J.J.; Sébire, G. In-situ expression of Interleukin-18 and associated mediators in the human brain of sALS patients: Hypothesis for a role for immune-inflammatory mechanisms. Med. Hypotheses 2016, 86, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Gugliandolo, A.; Giacoppo, S.; Bramanti, P.; Mazzon, E. NLRP3 Inflammasome Activation in a Transgenic Amyotrophic Lateral Sclerosis Model. Inflammation 2018, 41, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Debye, B.; Schmülling, L.; Zhou, L.; Rune, G.; Beyer, C.; Johann, S. Neurodegeneration and NLRP3 inflammasome expression in the anterior thalamus of SOD1(G93A) ALS mice. Brain Pathol. 2018, 28, 14–27. [Google Scholar] [CrossRef]
- Michaelson, N.; Facciponte, D.; Bradley, W.; Stommel, E. Cytokine expression levels in ALS: A potential link between inflammation and BMAA-triggered protein misfolding. Cytokine Growth Factor Rev. 2017, 37, 81–88. [Google Scholar] [CrossRef]
- Van Schoor, E.; Ospitalieri, S.; Moonen, S.; Tomé, S.O.; Ronisz, A.; Ok, O.; Weishaupt, J.; Ludolph, A.C.; Van Damme, P.; Van Den Bosch, L.; et al. Increased pyroptosis activation in white matter microglia is associated with neuronal loss in ALS motor cortex. Acta Neuropathol. 2022, 144, 393–411. [Google Scholar] [CrossRef]
- Stallings, N.R.; Puttaparthi, K.; Luther, C.M.; Burns, D.K.; Elliott, J.L. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol. Dis. 2010, 40, 404–414. [Google Scholar] [CrossRef]
- Quek, H.; Cuní-López, C.; Stewart, R.; Colletti, T.; Notaro, A.; Nguyen, T.H.; Sun, Y.; Guo, C.C.; Lupton, M.K.; Roberts, T.L.; et al. ALS monocyte-derived microglia-like cells reveal cytoplasmic TDP-43 accumulation, DNA damage, and cell-specific impairment of phagocytosis associated with disease progression. J. Neuroinflamm. 2022, 19, 58. [Google Scholar] [CrossRef]
- Kenney, C.; Powell, S.; Jankovic, J. Autopsy-proven Huntington′s disease with 29 trinucleotide repeats. Mov. Disord. 2007, 22, 127–130. [Google Scholar] [CrossRef]
- Fusco, F.R.; Paldino, E. Role of Phosphodiesterases in Huntington′s Disease. Adv. Neurobiol. 2017, 17, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Paldino, E.; Fusco, F.R. Emerging Role of NLRP3 Inflammasome/Pyroptosis in Huntington′s Disease. Int. J. Mol. Sci. 2022, 23, 8363. [Google Scholar] [CrossRef] [PubMed]
- Menalled, L.B.; Chesselet, M.F. Mouse models of Huntington′s disease. Trends Pharmacol. Sci. 2002, 23, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Paldino, E.; D′Angelo, V.; Sancesario, G.; Fusco, F.R. Pyroptotic cell death in the R6/2 mouse model of Huntington′s disease: New insight on the inflammasome. Cell Death Discov. 2020, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Paldino, E.; D′Angelo, V.; Laurenti, D.; Angeloni, C.; Sancesario, G.; Fusco, F.R. Modulation of Inflammasome and Pyroptosis by Olaparib, a PARP-1 Inhibitor.; in the R6/2 Mouse Model of Huntington′s Disease. Cells 2020, 9, 2286. [Google Scholar] [CrossRef]
- Chen, K.P.; Hua, K.F.; Tsai, F.T.; Lin, T.Y.; Cheng, C.Y.; Yang, D.I.; Hsu, H.T.; Ju, T.C. A selective inhibitor of the NLRP3 inflammasome as a potential therapeutic approach for neuroprotection in a transgenic mouse model of Huntington′s disease. J. Neuroinflamm. 2022, 19, 56. [Google Scholar] [CrossRef]
- Siew, J.J.; Chen, H.M.; Chen, H.Y.; Chen, H.L.; Chen, C.M.; Soong, B.W.; Wu, Y.R.; Chang, C.P.; Chan, Y.C.; Lin, C.H.; et al. Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington′s disease. Nat. Commun. 2019, 10, 3473. [Google Scholar] [CrossRef] [Green Version]
- Barake, F.; Soza, A.; González, A. Galectins in the brain: Advances in neuroinflammation, neuroprotection and therapeutic opportunities. Curr. Opin. Neurol. 2020, 33, 381–390. [Google Scholar] [CrossRef]
- Tricarico, P.M.; Caracciolo, I.; Crovella, S.; D′Agaro, P. Zika virus induces inflammasome activation in the glial cell line U87-MG. Biochem. Biophys. Res. Commun. 2017, 492, 597–602. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, Q.; Wu, Y.; Ma, L.; Zhang, Z.; Liu, T.; Jin, S.; She, Y.; Li, Y.P.; Cui, J. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. 2018, 37, e99347. [Google Scholar] [CrossRef]
- Wang, W.; Li, G.; De, W.; Luo, Z.; Pan, P.; Tian, M.; Wang, Y.; Xiao, F.; Li, A.; Wu, K.; et al. Zika virus infection induces host inflammatory responses by facilitating NLRP3 inflammasome assembly and interleukin-1β secretion. Nat. Commun. 2018, 9, 106. [Google Scholar] [CrossRef] [Green Version]
- Ramos, H.J.; Lanteri, M.C.; Blahnik, G.; Negash, A.; Suthar, M.S.; Brassil, M.M.; Sodhi, K.; Treuting, P.M.; Busch, M.P.; Norris, P.J.; et al. IL-1β signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. 2012, 8, e1003039. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.M.; Lin, C.C.; Lee, I.T.; Lin, Y.H.; Yang, C.M.; Chen, W.J.; Jou, M.J.; Hsiao, L.D. Japanese encephalitis virus induces matrix metalloproteinase-9 expression via a ROS/c-Src/PDGFR/PI3K/Akt/MAPKs-dependent AP-1 pathway in rat brain astrocytes. J. Neuroinflamm. 2012, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Zhao, Z.; Anees, A.; Li, Y.; Ashraf, U.; Chen, Z.; Song, Y.; Chen, H.; Cao, S.; Ye, J. p21-activated kinase 4 signaling promotes Japanese encephalitis virus-mediated inflammation in astrocytes. Front. Cell. Infect. Microbiol. 2017, 7, 271. [Google Scholar] [CrossRef]
- Ashraf, U.; Ding, Z.; Deng, S.; Ye, J.; Cao, S.; Chen, Z. Pathogenicity and virulence of Japanese encephalitis virus: Neuroinflammation and neuronal cell damage. Virulence 2021, 12, 968–980. [Google Scholar] [CrossRef]
- Burdo, T.H.; Lackner, A.; Williams, K.C. Monocyte/macrophages, and their role in HIV neuropathogenesis. Immunol. Rev. 2013, 254, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.G.; Reinke, S.N.; Mamik, M.K.; McKenzie, B.A.; Maingat, F.; Branton, W.G.; Broadhurst, D.I.; Power, C. Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS. Retrovirology 2014, 11, 35. [Google Scholar] [CrossRef] [Green Version]
- Breitinger, U.; Farag, N.S.; Sticht, H.; Breitinger, H.G. Viroporins: Structure, function, and their role in the life cycle of SARS-CoV-2. Int. J. Biochem. Cell Biol. 2022, 145, 106185. [Google Scholar] [CrossRef]
- Poeck, H.; Bscheider, M.; Gross, O.; Finger, K.; Roth, S.; Rebsamen, M.; Hannesschläger, N.; Schlee, M.; Rothenfusser, S.; Barchet, W.; et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 2010, 11, 63–69, Addendum in Nat. Immunol. 2014, 15, 109. [Google Scholar] [CrossRef]
- Rajan, J.V.; Rodriguez, D.; Miao, E.A.; Aderem, A. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J. Virol. 2011, 85, 4167–4172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, M.P.; Iba, M.; Nath, A.; Masliah, E.; Kim, C. Does SARS-CoV-2 affect neurodegenerative disorders? TLR2, a potential receptor for SARS-CoV-2 in the CNS. Exp. Mol. Med. 2022, 54, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Hung, E.C.; Chim, S.S.; Chan, P.K.; Tong, Y.K.; Ng, E.K.; Chiu, R.W.; Leung, C.B.; Sung, J.J.; Tam, J.S.; Lo, Y.M. Detection of SARS coronavirus RNA in the cerebrospinal fluid of a patient with severe acute respiratory syndrome. Clin. Chem. 2003, 49, 2108–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepehrinezhad, A.; Rezaeitalab, F.; Shahbazi, A.; Sahab-Negah, S. A Computational-Based Drug Repurposing Method Targeting SARS-CoV-2 and its Neurological Manifestations Genes and Signaling Pathways. Bioinform. Biol. Insights. 2021, 15, 11779322211026728. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front. Neurol. 2020, 11, 573095. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, D.E.; Oliveira-Giacomelli, A.; Glaser, T.; Arnaud-Sampaio, V.F.; Andrejew, R.; Dieckmann, L.; Baranova, J.; Lameu, C.; Ratajczak, M.Z.; Ulrich, H. Hyperactivation of P2X7 receptors as a culprit of COVID-19 neuropathology. Mol. Psychiatry 2021, 26, 1044–1059. [Google Scholar] [CrossRef]
- Sepehrinezhad, A.; Gorji, A.; Sahab Negah, S. SARS-CoV-2 may trigger inflammasome and pyroptosis in the central nervous system: A mechanistic view of neurotropism. Inflammopharmacology 2021, 29, 1049–1059. [Google Scholar] [CrossRef]
- Helms, J.; Kremer, S.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Kummerlen, C.; Collange, O.; Boulay, C.; Fafi-Kremer, S.; Ohana, M.; et al. Neurologic features in severe SARS-CoV-2 infection. N. Engl. J. Med. 2020, 382, 2268–2270. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, W.; Lukiw, W. Ubiquity of the SARS-CoV-2 receptor ACE2 and upregulation in limbic regions of Alzheimer′s disease brain. Folia Neuropathol. 2021, 59, 232–238. [Google Scholar] [CrossRef]
- Ding, Q.; Shults, N.V.; Gychka, S.G.; Harris, B.T.; Suzuki, Y.J. Protein Expression of Angiotensin-Converting Enzyme 2 (ACE2) is Upregulated in Brains with Alzheimer′s Disease. Int. J. Mol. Sci. 2021, 22, 1687. [Google Scholar] [CrossRef]
- Theobald, S.J.; Simonis, A.; Georgomanolis, T.; Kreer, C.; Zehner, M.; Eisfeld, H.S.; Albert, M.C.; Chhen, J.; Motameny, S.; Erger, F.; et al. Long-lived macrophage reprogramming drives spike protein-mediated inflammasome activation in COVID-19. EMBO Mol. Med. 2021, 13, e14150. [Google Scholar] [CrossRef]
- Xu, H.; Akinyemi, I.A.; Chitre, S.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology 2022, 568, 13–22. [Google Scholar] [CrossRef]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef]
- Fatima, S.; Zaidi, S.S.; Alsharidah, A.S.; Aljaser, F.S.; Banu, N. Possible Prophylactic Approach for SARS-CoV-2 Infection by Combination of Melatonin, Vitamin C and Zinc in Animals. Front. Vet. Sci. 2020, 7, 585789. [Google Scholar] [CrossRef]
- Ding, H.G.; Deng, Y.Y.; Yang, R.Q.; Wang, Q.S.; Jiang, W.Q.; Han, Y.L.; Huang, L.Q.; Wen, M.Y.; Zhong, W.H.; Li, X.S.; et al. Hypercapnia induces IL-1β overproduction via activation of NLRP3 inflammasome: Implication in cognitive impairment in hypoxemic adult rats. J. Neuroinflamm. 2018, 15, 4. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Golenbock, D.; Latz, E.; Morgan, D.; Brown, R. Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimers Res. Ther. 2020, 12, 69. [Google Scholar] [CrossRef]
- Farheen, S.; Agrawal, S.; Zubair, S.; Agrawal, A.; Jamal, F.; Altaf, I.; Kashif Anwar, A.; Umair, S.M.; Owais, M. Pathophysiology of aging and immune-senescence: Possible correlates with comorbidity and mortality in middle-aged and old COVID-19 patients. Front. Aging 2021, 2, 748591. [Google Scholar] [CrossRef]
- Flud, V.V.; Shcherbuk, Y.A.; Shcherbuk, A.Y.; Leonov, V.I.; Al-Sahli, O.A. Neurological complications and consequences of new coronavirus COVID-19 infection in elderly and old patients (literature review). Adv. Gerontol. 2022, 35, 231–242. [Google Scholar]
- Fu, Y.W.; Xu, H.S.; Liu, S.J. COVID-19 and neurodegenerative diseases. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 4535–4544. [Google Scholar] [CrossRef]
- Baazaoui, N.; Iqbal, K. COVID-19 and Neurodegenerative Diseases: Prion-Like Spread and Long-Term Consequences. J. Alzheimers Dis. 2022, 88, 399–416. [Google Scholar] [CrossRef]
- Bernardini, A.; Gigli, G.L.; Janes, F.; Pellitteri, G.; Ciardi, C.; Fabris, M.; Valente, M. Creutzfeldt-Jakob disease after COVID-19: Infection-induced prion protein misfolding? A case report. Prion 2022, 16, 78–83. [Google Scholar] [CrossRef]
- Tetz, G.; Tetz, V. Prion-like Domains in Spike Protein of SARS-CoV-2 Differ across Its Variants and Enable Changes in Affinity to ACE2. Microorganisms 2022, 10, 280. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.; Stewart, W.; Dams-O′Connor, K.; Diaz-Arrastia, R.; Horton, L.; Menon, D.K.; Polinder, S. The chronic and evolving neurological consequences of traumatic brain injury. Lancet Neurol. 2017, 16, 813–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Kim, H.J.; Kim, J.U.; Yook, T.H.; Kim, K.H.; Lee, J.Y.; Yang, G. A Novel Treatment Strategy by Natural Products in NLRP3 Inflammasome-Mediated Neuroinflammation in Alzheimer′s and Parkinson′s Disease. Int. J. Mol. Sci. 2021, 22, 1324. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.G.; Zhou, X.G.; Qiao, G.; Yu, L.; Tang, Y.; Yan, L.; Qiu, W.Q.; Pan, R.; Yu, C.L.; Law, B.Y.; et al. Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res. Rev. 2021, 65, 101202. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, B.A.; Mamik, M.K.; Saito, L.B.; Boghozian, R.; Monaco, M.C.; Major, E.O.; Lu, J.Q.; Branton, W.G.; Power, C. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, E6065–E6074. [Google Scholar] [CrossRef] [Green Version]
- Yap, J.K.Y.; Pickard, B.S.; Chan, E.W.L.; Gan, S.Y. The Role of Neuronal NLRP1 Inflammasome in Alzheimer′s Disease: Bringing Neurons into the Neuroinflammation Game. Mol. Neurobiol. 2019, 56, 7741–7753. [Google Scholar] [CrossRef]
- Nagyőszi, P.; Nyúl-Tóth, Á.; Fazakas, C.; Wilhelm, I.; Kozma, M.; Molnár, J.; Haskó, J.; Krizbai, I.A. Regulation of NOD-like receptors and inflammasome activation in cerebral endothelial cells. J. Neurochem. 2015, 135, 551–564. [Google Scholar] [CrossRef]
- Feng, Y.S.; Tan, Z.X.; Wu, L.Y.; Dong, F.; Zhang, F. The involvement of NLRP3 inflammasome in the treatment of neurodegenerative diseases. Biomed. Pharmacother. 2021, 38, 111428. [Google Scholar] [CrossRef]
- Lahooti, B.; Chhibber, T.; Bagchi, S.; Varahachalam, S.P.; Jayant, R.D. Therapeutic role of inflammasome inhibitors in neurodegenerative disorders. Brain Behav. Immun. 2021, 91, 771–783. [Google Scholar] [CrossRef]
- Corcoran, S.E.; Halai, R.; Cooper, M.A. Pharmacological Inhibition of the Nod-Like Receptor Family Pyrin Domain Containing 3 Inflammasome with MCC950. Pharmacol. Rev. 2021, 73, 968–1000. [Google Scholar] [CrossRef]
- Soriano-Teruel, P.M.; García-Laínez, G.; Marco-Salvador, M.; Pardo, J.; Arias, M.; DeFord, C.; Merfort, I.; Vicent, M.J.; Pelegrín, P.; Sancho, M.; et al. Identification of an ASC oligomerization inhibitor for the treatment of inflammatory diseases. Cell Death Dis. 2021, 12, 1155. [Google Scholar] [CrossRef]
- De Souza, N. Model organisms: Mouse models challenged. Nat. Methods 2013, 10, 288. [Google Scholar] [CrossRef]
- Pound, P.; Ritskes-Hoitinga, M. Is it possible to overcome issues of external validity in preclinical animal research? Why most animal models are bound to fail. J. Transl. Med. 2018, 16, 304. [Google Scholar] [CrossRef] [Green Version]
- Gharib, W.H.; Robinson-Rechavi, M. When orthologs diverge between human and mouse. Brief. Bioinform. 2011, 12, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; Mcdonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [Green Version]
- Greek, R.; Menache, A. Systematic reviews of animal models: Methodology versus epistemology. Int. J. Med. Sci. 2013, 10, 206–221. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, A. The flaws and human harms of animal experimentation. Camb. Q. Health Ethics. 2015, 24, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Balls, M. It′s Time to Include Harm to Humans in Harm-Benefit Analysis—But How to Do It, that is the Question. Altern. Lab. Anim. 2021, 49, 182–196. [Google Scholar] [CrossRef]
- Chiarini, A.; Gardenal, E.; Whitfield, J.F.; Chakravarthy, B.; Armato, U.; Dal Pra, I. Preventing the spread of Alzheimer′s disease neuropathology: A role for calcilytics? Curr. Pharm. Biotechnol. 2015, 16, 696–706. [Google Scholar] [CrossRef]
- Chiarini, A.; Armato, U.; Liu, D.; Dal Prà, I. Calcium-Sensing Receptor Antagonist NPS 2143 Restores Amyloid Precursor Protein Physiological Non-Amyloidogenic Processing in Aβ-Exposed Adult Human Astrocytes. Sci. Rep. 2017, 7, 1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer′s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]





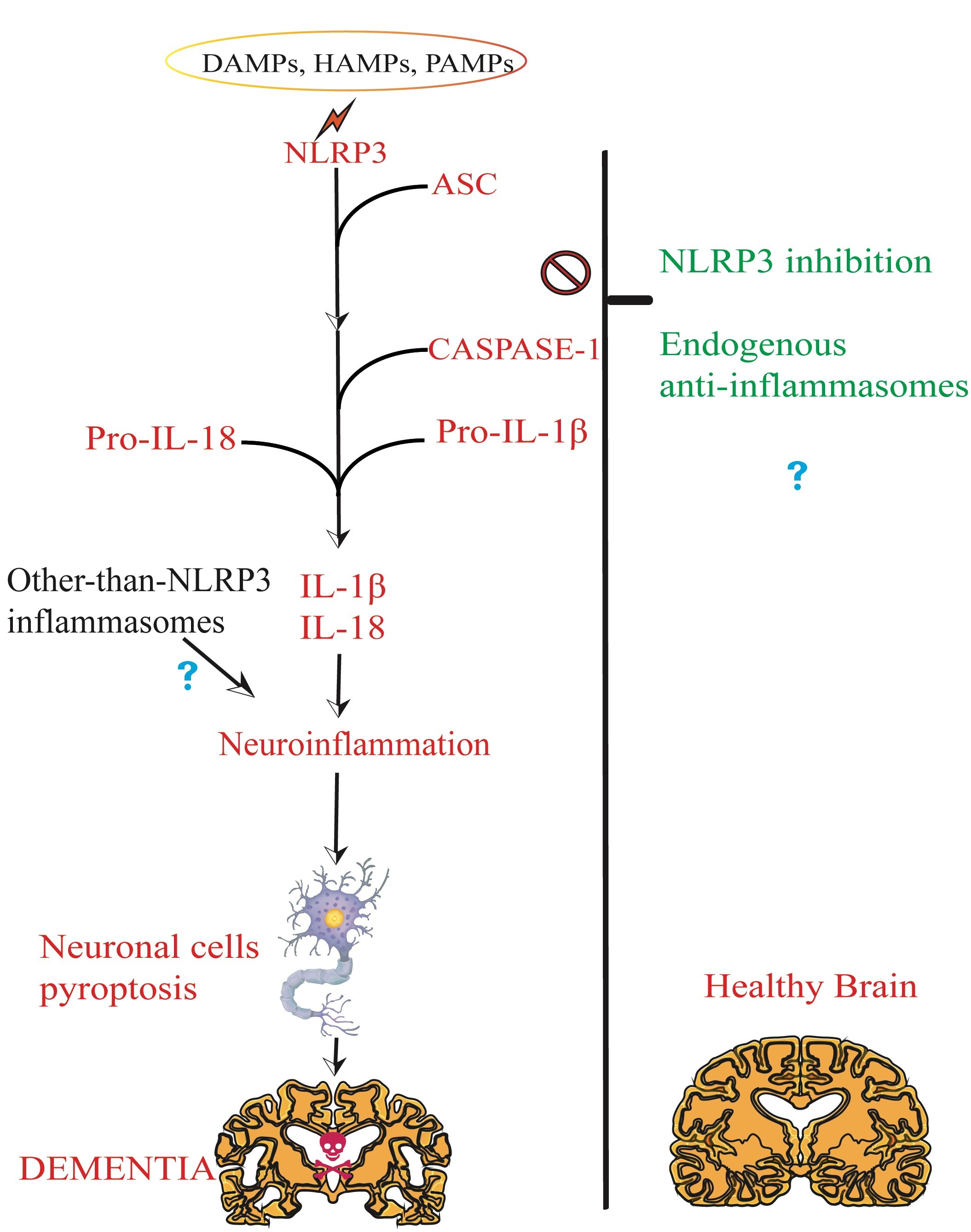{kind=link}
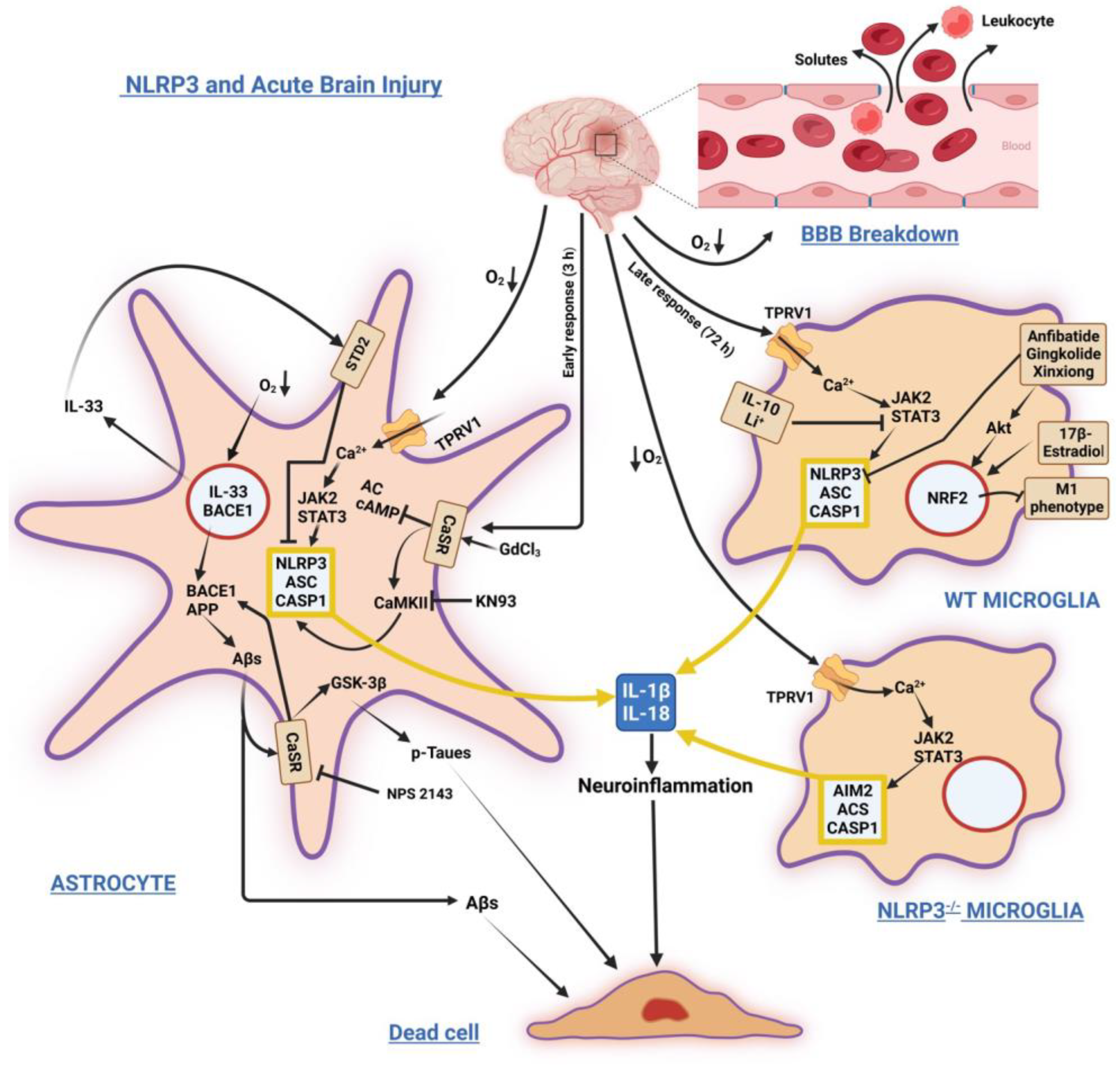{kind=link}
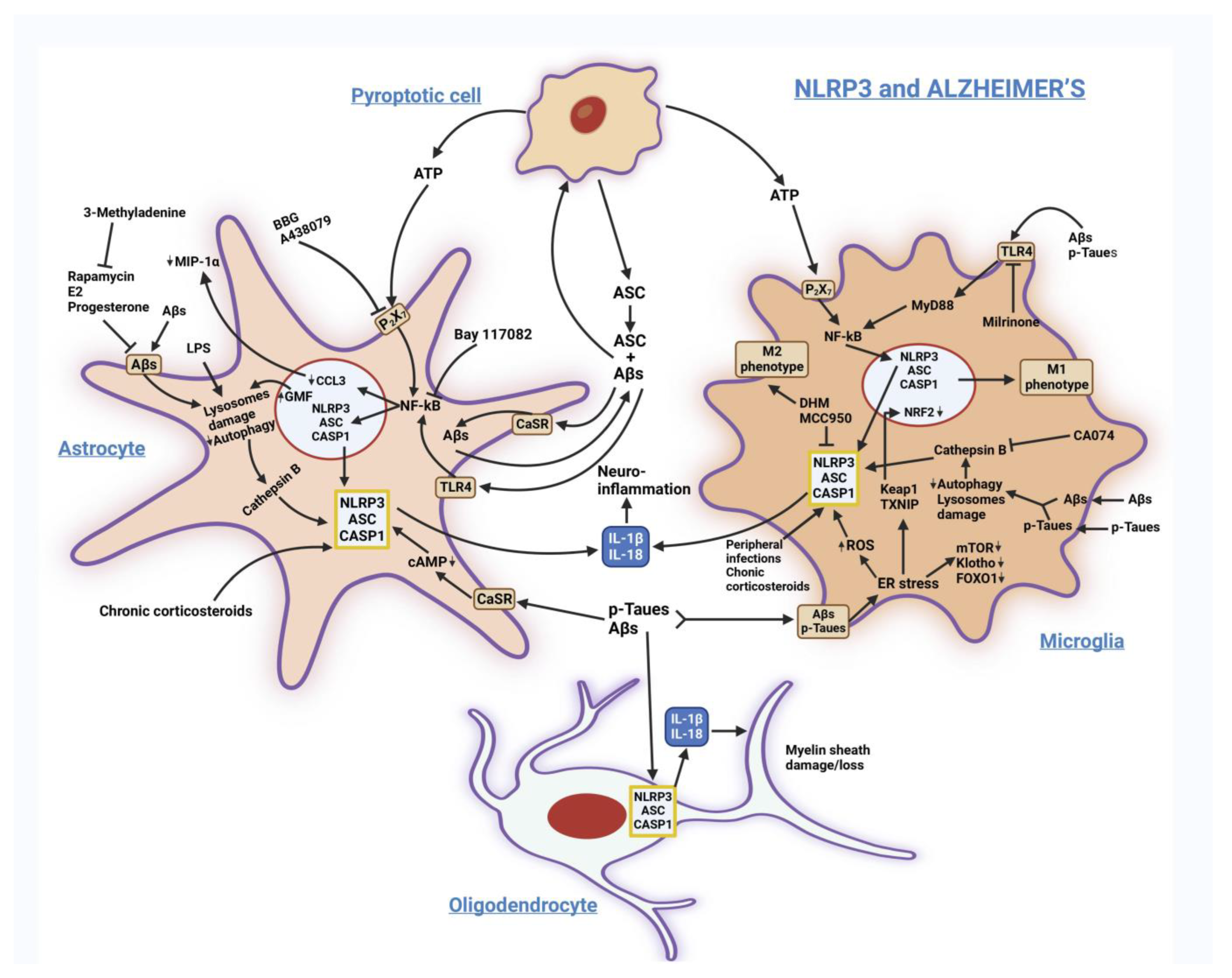{kind=link}
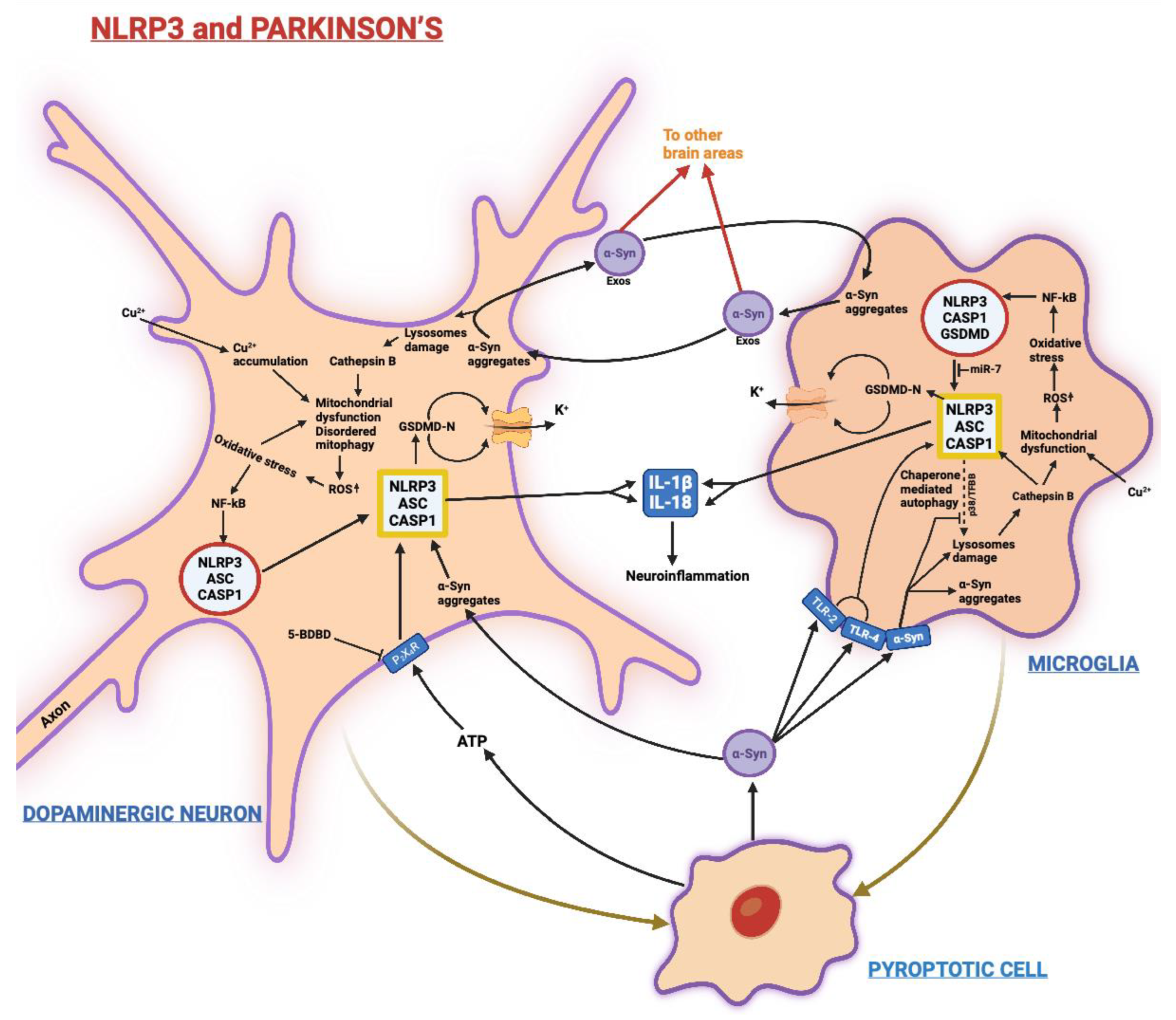{kind=link}
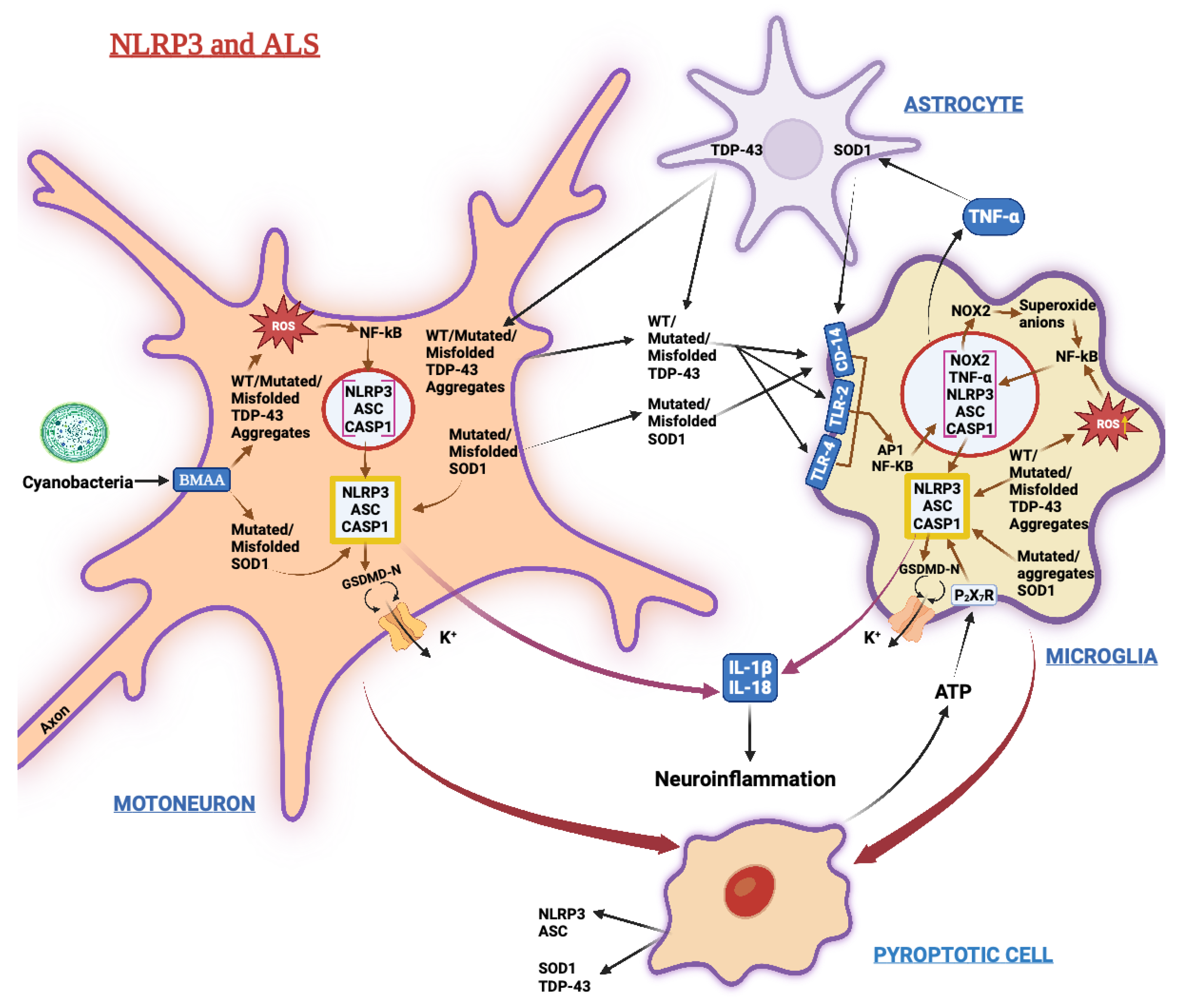{kind=link}
| Condition/Factor | Mechanisms | References |
|---|---|---|
| Vascular ailments Stroke Intracerebral hemorrhage Hemorrhagic stroke | Mitochondrial disfunction after hypoxic ischemia/reperfusion (HI/R) Chronic hypoxia | [32,33,34,35,36,37,38,39,40] |
| Seizures Mesial lobe temporal epilepsy Soman or A255 (nerve agent) exposure | Acetyl- and butyryl-cholinesterase inhibition | [41,42] |
| Metal accumulation Manganese (Mn), Lead (Pb) Copper (Cu) Cadmium (Cd) Aluminium/alum | Metal-induced neurotoxicity ↑§ ROS & NF-κB-p65 pathway CaSR and GCP6RA signaling | [43,44,45,46,47,48,49,50,51] See also Box 1 |
| Mechanical stresses and strains Skull trauma Optic nerve trauma Elevated intracranial pressure Glaucoma | Osteopontin NIMA-related kinase 7 (or NEK7) P2X7 receptor activation HMGB1/caspase-8 pathway | [52,53,54,55,56,57,58,59] See also Box 2 |
| Neurodegenerative diseases Alzheimer’s disease (AD) Tauopathies Parkinson’s disease (PD) Amyotrophic lateral sclerosis (ALS) Huntington’s disease (HD) Prion disease (PrPSc) | Aβs, autophagy block, NEK7 p-Taues paired helical filaments ER stress, ↑ ROS α-Synuclein aggregates Mutated SOD1, TDP-43 Expanded CAG repeats in HTT/OT15 gene Prion protein seeding | [60,61,62,63,64,65,66,67,68,69,70,71] |
| Environmental pollution PM2.5 | Increased ROS production by microglia | [72,73] |
| Infectious diseases Sepsis (bacteria, fungi) West Nile Virus (WNV) HIV-1 Herpes Virus 1 Japanese Encephalitis Virus (JEV) Zika Virus (ZIKV) SARS-CoV-2 Encephalomyocarditis Virus (EMCV) Tuberculosis | Bacterial and fungal toxins Intensified IL-1β signaling Tat and gp120 proteins Gasdermin D-dependent pyroptosis ROS-dependent activation of Src/Ras/Raf/ERK/NF-κB signaling axis NS5 protein and ↑ ROS S1 spike glycoprotein, viroporin ORF3a/8 viroporin ORF2b Early secreted antigenic target protein of 6 kDa (ESAT-6) | [74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90] |
| Metabolic disorders Atherosclerosis Gout Obesity/high-fat diet Nonalcoholic hepatic steatosis Type-2 diabetes mellitus (T2DM) | Hypercholesterolemia Urates→NEK7 Glucocorticoids and fatty acids surpluses→TNFR and Toll-like receptors ROS, NO, hydroperoxides, scavenger receptors, mTOR | [91,92,93,94] |
| Iatrogenic factors Postoperative cognitive dysfunction Cyclophosphamide cystitis GdCl3, cinacalcet Glucocorticoids (elevated levels) | Drugs, infection, electrolyte imbalance TNF-α Calcimimetic•CaSR/ERK1/2/CaMKII NLRP1 and NLRP3 inflammasomes | [95,96,97,98,99,100,101] See also Box 1 |
| Psychotropic drugs Cocaine Methamphetamine Scopolamine Ethanol Morphine Fentanyl | σ-1 receptor TLR-4 ↑ Dhx58, S100a, Lrm4 genes TLR-4 μ-3 and κ opioid receptors μ-opioid receptor | [102,103,104,105,106,107,108] |
| Cellular stress and injury ATP Pore-inducing agents Phagocytosed protein polymers ROS Cardiolipin Raised IL-1β levels Reduced cyclic AMP (cAMP) levels Zn2+ deficiency K+ efflux Ca2+ and Cl− influx | Purinergic receptor signaling DDX3X protein/NLRP3 complexes Heat shock protein 60 (HSP60) and TLR-4-p38 MAPKs axis Oxidized mtDNA and proteins Lysosome-released cathepsin B Mitochondria-released hexokinase, ROS NLRP3 activation Ionic imbalances | [25,109,110,111,112,113,114,115,116,117,118,119] |
| Aging Inflammaging | ↑ Membrane attack complexes (MAC) Reduced mitochondrial fission and fusion Declined mitophagy Mitochondrial damage Selective autophagy-mediated mitochondrial homeostasis (in microglia) | [33,112,120,121,122] |
| (A) Activation. | |||
| RNAs | Model | Mechanisms | References |
| LncRNA-Cox2 | Murine microglia | ↑ § Transcription of NLRP3 and ASC TLR-mediated signaling pathways Autophagy block Microglia activation | [180,181,188] |
| LncRNA-Meg3 | Murine microglia | miR-7a-5 downregulation | [189] |
| miR-141 | Brain tissue of diabetic mice | NF-ĸB-mediated NLRP3 expression | [190] |
| Exo-miR-124 Exo-miR-146a Exo-miR-155 | LPS-primed N9 microglia cells | ↑ TLR4/TLR2/NF-ĸB axis | [191] |
| miR-193 | Murine brain cortex Murine microglia | ↑ Expression of NLRP3, ASC, cleaved caspase-1 and mature IL-1β | [192] |
| miR-590-3 | In silico AD patients’ data | Promoted neurons’ death via AMPK signaling | [193] |
| P3Alu-RNAs | Primary human retinal pigment cells | ERK1/2 and NLRP3 activation, neurons’ death | [184] |
| (B) Inhibition. | |||
| RNAs | Model | Mechanisms | References |
| circRNA_003564 | Spinal cord injury (rat model) | ↓ § NLRP3, caspase-1, mature IL-1β, Il-18, GsdmD ↓ Pyroptosis | [187] |
| LncRNA-Meg3 | Rat hippocampal neuronal model of temporal epilepsy | PI3K/AKT/mTOR pathway activation | [194] |
| miR-7 | Murine neural stem cells | NLRP3/caspase-1 suppressor | [195,196] |
| Exo-miR-21 | APP/PS1 2xTg AD-model mouse | Improved memory | [197] |
| miR-22, Exo-miR-22 | APP/PS1 2xTg AD-model mouse PC12 cells | Downregulated NLRP3 | [198,199] |
| Exo-miR-23b | Rat model of intracerebral hemorrhage | Antioxidant effects via PTEN/NRF2 inhibition | [200] |
| miR-29c-3p Exo-miR-29c-3p | PC12 cells AD-model rat | Suppression of BACE1, p-Tau, and pyroptosis via Wnt/β-catenin pathway | [201,202] |
| miR-152 | Microglial BV2 cell Hippocampal neuronal HT22 cell line Rat model of intracerebral hemorrhage | TXNIP-mediated block of NLRP3 activation | [203] |
| Exo-miR-188-3p | PD-model mouse MN9D dopaminergic neuronal cells | Suppression of NLR3/pyroptosis | [204] |
| miR-194-5p | Rat model of intra- cerebral hemorrhage | Blocked NLRP3/TRAF6 interaction | [205] |
| miR-223-3p | Serum samples from PD, AD, and MCI patients, and healthy controls | Negative NLRP3 regulation | [206] |
| miR-374a-5p | Rat model of hypoxic-ischemia encephalopathy | Suppressor of SMAD6/NLRP3 in microglia | [207] |
| Compound [References] | IUPAC Name | Main Molecular Activity | Main Biological Activity | Experimental Model |
|---|---|---|---|---|
| 17β-Estradiol (E2) [223,224,225] See also Box 1 | (8R,9S,13S,14S,17S)-13-methyl-6,7,8,9,11,12,14,15,16,17-decahydrocyclopenta[a]phenanthrene-3,17-diol | Ligand for estrogen receptor-α (ER-α) and -β (ER-β), and for G-protein coupled receptor 1 (GPER1) | ↓§ NLRP3, ASC, cleaved caspase-1, IL-1β ↓§ M1 microglia ↑ M2 microglia | Male SOD1(G93A) ALS-model mice Global brain ischemia-model rodents |
| A43879 [226] See also Box 2 | 3-[[5-(2,3-dichlorophenyl)-tetrazol-1-yl]methyl]pyridine hydrochloride | P2X7 purinergic receptor antagonist | ↓ P2X7 receptor signaling ↓ NLRP3 | Spinal cord injury-model animal |
| Adiponectin [227] | Protein | Ligand for Adipo-R1 and Adipo-R2 receptors | ↓ NLRP3, IL-1β, IL-18 ↑ Autophagy via AMPK pathway | Intracerebral hemorrhage-model rat |
| Amifostine [228] | 2-(3-aminopropylamino)ethyl-sulfanylphosphonic acid | Protects against the DNA-damaging effects of ionizing radiations and chemotherapy drug-induced ROS | ↓ ROS, pyroptosis | Experimental autoimmune encephalomyelitis (EAE)-model rat |
| α1-Antitrypsin (A1AT) [128] | Protein | Protease inhibitor | ↓ Aβ1–42-driven NLRP3 activation | Mouse primary cortical astrocytes |
| Anfibatide [229,230] | Dimeric protein | Antagonist of the glycoprotein Ib IX-V (GPIb) complex | ↓ NLRP3/NF-κB axis, cleaved caspase-1 and -3, and Bax ↑ Bcl2 | Cerebral HI/R injury-model rat |
| Atorvastatin [231] | (3R,5R)-7-[2-(4-fluorophenyl)-3-phenyl-4-(phenylcarbamoyl)-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoic acid | Inhibitor of hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase | ↓ NLRP3/NF-κB signaling axis | Surgery-induced BBB disruption in aged mice |
| Bay117082 [232] | (E)-3-(4-methylphenyl)-sulfonylprop-2-enenitrile | Calcium channel blocker | ↓ ATPase activity of NLRP3 | Spinal cord injury-model animal |
| BPBA [233] | (2-[2-(benzo[d]thiazol-2-yl) phenyl-amino] benzoic acid) | Inhibitor of self- and Cu2+- or Zn2+-induced Aβs aggregation | ↓ Aβs aggregation and neurotoxicity ↓ NLRP3 and IL-1β | Aβ-induced paralysis in transgenic Caenorabditis elegans |
| Caffeine [234] | 1,3,7-trimethypurine-2,6-dione | Antagonist of all adenosine receptor subtypes (A1, A2a, A2b, A3) in the CNS PDE inhibitor | ↓ Rapamycin (mTOR) axis and Bax ↑ Autophagy | EAE-model C57BL/6 mice Mouse microglia BV2 microglial cells |
| Calcitriol [235] | (1R,3S,5Z)-5-[(2E)-2-[(1R,3aS,7aR)-1-[(2R)-6-hydroxy-6-methylheptan-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1H-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol | Ligand for vitamin D receptors | ↓ ROS, NLRP3, caspase-1, IL-1β, CX3CR1, CCL17, Tbx21 ↓ Spinal cord demyelination | EAE-model C57BL/6 mice |
| Choline [236] | 2-hydroxyethyl-(trimethyl)azanium | Methyl donor Ligand for choline transporters, CTL1 included | ↓ NLRP3, Aβs deposition, and microgliosis | APP/PS1 AD-model mice |
| Dapansutrile (i.e., OLT1177) [237] | 3-methylsulfonyl propanenitrile | Direct NLRP3 ATPase inhibitor | ↓ Microglia activation and Aβs plaque numbers in the cerebral cortex ↓ IL-1β and IL-6 ↑ Dendritic spine density Successful Phase I clinical trial | APP/PS1 AD-model mice |
| Dexmedetomidine (Dexm) [96,238] | 5-[(1S)-1-(2,3-dimethylphenyl)ethyl]-1H-imidazole | Specific and selective α-2 adrenoceptor agonist | ↓ NF-κB and proinflammatory cytokines via miR-340 upregulation ↑ Autophagy | LPS-stimulated BV2 microglia cells |
| Dihydromyricetin [239] | (2R,3R)-3,5,7-trihydroxy-2-(3,4,5-trihydroxyphenyl)-2,3-dihydrochromen-4-one | Antioxidant, anti-binge hangover, and anti-cancer activity | ↓ NLRP3 ↑ Aβ clearance ↑ Expression of neprilysin ↑ M2 microglial phenotype | APP/PS1 AD-model mice |
| A-68930 [240] | 1-(aminomethyl)-3-phenyl-3,4-dihydro-1H-isochromene-5,6-diol;hydrochloride | Potent and selective Dopamine D1-like receptor agonist | ↓ NLRP3 activation | LPS-induced systemic inflammation mouse model |
| Bromocriptine | (6aR,9R)-5-bromo-N-[(1S,2S,4R,7S)-2-hydroxy-7-(2-methylpropyl)-5,8-dioxo-4-propan-2-yl-3-oxa-6,9-diazatricyclo[7.3.0.02,6]dodecan-4-yl]-7-methyl-6,6a,8,9-tetrahydro-4H-indolo[4,3-fg]quinoline-9-carboxamide | Dopamine D2 receptor agonist | ↑ NLRP3 ubiquitination via cAMP | Neurotoxin MPTP-treated mice |
| Dopamine [226] | 4-(2-aminoethyl)benzene- 1,2-diol | Agonist for the five Dopamine receptor subtypes (D1, D2, D3, D4, D5) | ↓ IL-1β and IL-18 secretion | Spinal cord injury-model rat |
| LY171555 | (4aR,8aR)-5-propyl-1,4,4a,6,7,8,8a,9-octahydropyrazolo[3,4-g]quinoline;hydrochloride | Specific dopamine D2 receptor agonist | ||
| Quinerolane | (5aR,9aR)-6-propyl-5a,7,8,9,9a,10-hexahydro-5H-pyrido[2,3-g]quinazolin-2-amine | Dopamine D2 and D3 receptors agonist | ||
| EC144 [241] | 5-[2-amino-4-chloro-7-[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]pyrrolo[2,3-d]pyrimidin-5-yl]-2-methylpent-4-yn-2-ol | Selective inhibitor of heat shock protein 90 (HSP90) | ↓ IL-1β and IL-18 | Peritonitis-model animal |
| Echinacoside [242] | [(2R,3R,4R,5R,6R)-6-[2-(3,4-dihydroxyphenyl)ethoxy]-5-hydroxy-2-[[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]-4-[(2S,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxyoxan-3-yl] (E)-3-(3,4-dihydroxyphenyl)prop-2-enoate | Neuroprotective effects via undefined upstream mechanisms | ↓ NLRP3, NF-κB-p65, and ROS | Spinal cord injury-model animal LPS-treated BV2 microglial cells |
| Ellagic acid [243] | 6,7,13,14-tetrahydroxy-2,9-dioxatetracyclo[6.6.2.04,16.011,15]hexadeca-1(15),4,6,8(16),11,13-hexaene-3,10-dione | ATP-competitive inhibitor of constitutively active CK2 Ser/Thr protein kinase | ↓ caspase-1, IL-6, IL-10, IL-17A, TNF-α, GFAP, and Iba1 | EAE-model mouse |
| Fimasartan [244] | 2-[2-butyl-4-methyl-6-oxo-1-[[4-[2-(2H-tetrazol-5-yl) phenyl]phenyl]methyl]pyrimidin-5-yl]-N,N-dimethylethanethioamide | Angiotensin II receptor antagonist | ↓ NLRP3/ASC/caspase-1 and NF-κB pathways | Intracerebral hemorrhage-model rat Hemolysate-treated BV2 microglia |
| Fluoxetine [245] | N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]propan-1-amine | Serotonin reuptake inhibitor | ↓ NF-κB, TLR-4, NLRP3, caspase-1, TNF-α, IL-1β ↓ AChE activity, Aβ, Tau protein, MDA | Depression- and AD-model animals |
| Ghrelin [246] | (4S)-4-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[(2-aminoacetyl)amino]-3-hydroxypropanoyl]amino]-3-hydroxypropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]-1-oxopropan-2-…………………………………. yl]amino]-5-oxopentanoic acid | Ligand for GHS-R1a receptor | ↓ NF-κB/NLRP3 axis, IL-6, COX2, TNF-α, NOS-2, and pyroptosis | EAE-model animal |
| Glibenclamide [247,248] | 5-chloro-N-[2-[4-(cyclohexylcarbamoylsulfamoyl)phenyl]ethyl]-2-methoxybenzamide | Classic KATP channel blocker | ATP-sensitive K+ channel inhibitor ↓ NLRP3 ↓ Release of HSP70 ↓ NLRP3, GsdmD-cleavage, ↓ Oxidative stress, demyelination, axon degeneration | Morphine-induced neuroinflammation animal and cellular models Hexanendione-induced neurotoxicity-model animal |
| HU-308 [249] | [(1R,4R,5R)-4-[2,6-dimethoxy-4-(2-methyloctan-2-yl)phenyl]-6,6-dimethyl-2-bicyclo[3.1.1]hept-2-enyl]methanol | Activator of cannabinoid receptor 2 | ↑ Autophagy | BV2 microglia cells EAE-model animals |
| Indomethacin [250] | 2-[1-(4-chlorobenzoyl)-5-methoxy-2-methylindol-3-yl]acetic acid | Prostaglandin G/H synthase 2 or cyclo-oxygenase (COX) enzyme inhibitor | ↓ NLRC4 and NLRP3 genes ↓ IL-1β, caspase-1, and p-Taues | Streptozotocin (STZ)-induced AD-like model |
| Inzomelid [251] | 1-(1,2,3,5,6,7-hexahydro-s-indacen-4-yl)-3-(1-propan-2-ylpyrazol-3-yl)sulfonylurea | Nonspecific and reversible inhibitor of the cyclo-oxygenase (COX) enzyme or prostaglandin G/H synthase | ↓ NLRP3 | ClinicalTrial.gov NCT04015076 |
| JC124 [252] | 5-chloro-2-methoxy-N-[2-[4-(methylsulfamoyl)phenyl]ethyl]benzamide) | Specific inhibitor of expression of NLRP3 and its adaptor protein ASC | ↓ NLRP3, ASC, IL-1β, TNFα, NOS-2, caspase-1, and pyroptosis | Traumatic brain injury in male rats |
| Ketamine [253] | 2-(2-chlorophenyl)-2-(methylamino)cyclohexan-1-one | NMDA receptors antagonist | ↓ NF-κB, NLRP3, ASC, caspase-1, IL-1β ↑ Autophagy | Depressive-like-model rat |
| KPT-8602 [254] | (E)-3-[3-[3,5-bis(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]-2-pyrimidin-5-ylprop-2-enamide | Exportin 1 (XPO1) nuclear transport inhibitor | ↓ Exportin 1 ↓ NLRP3/NF-κB signaling axis | LPS-treated macrophages LPS-induced inflammation mouse model MPTP mouse model of PD |
| Licochalcone B [255] | (E)-3-(3,4-dihydroxy-2-methoxyphenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one | Specific inhibitor of NEK7-NLRP3 interaction | ↓ Canonical and non-canonical NLRP3 inflammasome activation | Murine macrophages Mouse models of LPS-induced septic shock, peritonitis, and non-alcoholic steatohepatitis |
| Manoalide [256,257,258,259] | (2R)-2-hydroxy-3-[(2R,6R)-6-hydroxy-5-[(E)-4-methyl-6-(2,6,6-trimethylcyclohexen-1-yl)hex-3-enyl]-3,6-dihydro-2H-pyran-2-yl]-2H-furan-5-one | Inhibitor of NEK7-NLRP3 activating interaction | ↓ Canonical and non-canonical NLRP3 inflammasome activation | EAE-model animal |
| MCC950 (i.e., CRID3) [222,260] | 1,2,3,5,6,7-hexahydro-s-indacen-4-ylcarbamoyl-[4-(2-hydroxypropan-2-yl)furan-2-yl]sulfonylazanide | Selectively and specifically binds NLRP3 NATCH domain hindering Walker B motif function thereby inhibiting NLRP3 conformational modifications and oligomerization | ↓ NLRP3 ↑ Aβ-phagocytic capability of microglia ↓ IL-1β, IL-18, TNF-α, NLRP3, ASC, cleaved caspase-1, Iba1-, and GFAP-positive cells ↑ BDNF and PSD95 expression | APP/PS1 transgenic AD-model mouse LPS + ATP-induced microglia Perioperative neurocognitive disorders-model mice |
| Mefenamic, Tolfenamic, Flufenamic, Meclofenamic acids [261] | 2-(2,3-dimethylanilino)benzoic acid 2-(3-chloro-2-methylanilino)benzoic acid 2-[3-(trifluoromethyl)anilino]benzoic acid 2-(2,6-dichloro-3-methylanilino)benzoic acid | Cyclooxygenase (COX) inhibitors Cl- channel inhibitors | ↓ NLRP3 and IL-1β processing and release | LPS-primed primary bone marrow-derived macrophages |
| Melatonin [262,263,264,265,266] | N-[2-(5-methoxy-1H-indol-3-yl)ethyl]acetamide | Natural hormone of the pineal gland acting through its receptors | ↑ TFEB nuclear translocation ↑ mitophagy ↓ NLRP3, IL-18, IL-6, and IL-1β ↓ ROS ↑ Sirtuin 1 ↑ α7-nAChR-mediated “autophagic flux” | Aβ 25–35-treated SH-SY5Y cells APP/PS1 AD-model mice Chronic Gulf War syndrome |
| Metformin (MET) [267] | 3-(diaminomethylidene)-1,1-dimethylguanidine | AMP-activated protein kinase (AMPK) agonist | ↓ NF-κB signaling pathway ↑ Sirtuin 1 ↓ NLRP3-mediated ECs pyroptosis | LPS-stimulated lung tissues and pulmonary endothelial cells |
| Milrinone [268] | 6-methyl-2-oxo-5-pyridin-4-yl-1H-pyridine-3-carbonitrile | Inhibitor of phosphodiesterase III | ↑ cAMP ↓ TLR4/MyD88/NF-κB axis ↓ IL-1β, IL-6, TNF-α ↓ Aβ, p-Tau, ROS | LPS/Aβ-treated BV2 microglial cells APP/PS1 AD-model mouse |
| Minocycline [269,270] | (4S,4aS,5aR,12aR)-4,7-bis(dimethylamino)-1,10,11,12a-tetrahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4H-tetracene-2-carboxamide | Caspase-1 negative modulator | ↓ TLR-2, MyD88, NLRP3/NF-κB axis, IL-1β | AD-like dementia-model mouse |
| Mitoquinone (MitoQ) [271] | 10-(4,5-dimethoxy-2-methyl-3,6-dioxocyclohexa-1,4-dien-1-yl)decyl-triphenylphosphanium | Selectively accumulates inside mitochondria with anti-oxidant action | ↓ Mitochondrial ROS, NLRP3 activation, IL-1β, and IL-18 ↑ M2 phenotype microglia | Intracerebral hemorrhage-model mouse FeCl2-treated microglia |
| N-acetylcysteine [272] | (2R)-2-acetamido-3-sulfanylpropanoic acid | Stimulator of glutathione synthetase | ↓ ROS ↑ NRF2-induced NAD(P)H quinone dehydrogenase 1 (NQO1) | Ischemic stroke-model rat |
| Nafamostat mesylate [273] | (6-carbamimidoyl naphthalen-2-yl) 4-(diaminomethyl-ideneamino)benzoate | Synthetic inhibitor of serine proteases with a wide spectrum of activity | ↓ NLRP3/NF-κB signaling ↓ TNF-α, IL-1β, NOS-2, COX-2, IL-18 | Stroke-model animal |
| NT-0796 [274] | unknown | Orally available brain- penetrant NLRP3 inhibitor | ↓ NLRP3 | ANZCTR.org.au ACTRN126210010828-97 |
| Phenyl vinyl sulfone [275] | ethenylsulfonylbenzene | Cysteine protease inhibitor | ↓ NLRP3-mediated IL-1β release | LPS+ATP-treated J774A.1 cells LPS intraperitoneally injected C57BL/6 mouse |
| Phoenixin-14 [276] See also Box 1 | protein | Ligand for the multiple function G protein-coupled receptor GPR173 | ↓ HMGB1-mediated NLRP3 activation ↓ IL-1β and IL-18 | LPS-treated mouse primary astrocytes |
| Pramipexole [277] | 6S)-6-N-propyl-4,5,6,7-tetrahydro-1,3-benzothiazole-2,6-diamine | Dopamine-D3 receptors agonist | ↑ Autophagy ↓ NLRP3, ASC, cleaved caspase-1 IL-1β, IL-18 | LPS+ATP-stimulated primary mouse astrocytes PD-model mouse |
| Prednisone (PDN) [278] | (8S,9S,10R,13S,14S,17R)-17-hydroxy-17-(2-hydroxyacetyl)-10,13-dimethyl-6,7,8,9,12,14,15,16-octahydrocyclopenta[a]phenanthrene-3,11-dione | Glucocorticoid receptor agonist | ↓ NLRP3 activation ↓ TNF-α, CCL8, CXCL10, CXCL16 ↓ astrocytes and microglia activation | Cuprizone (CPZ)- induced demyelination-model mouse |
| Resolvin D1 [279] See also Box 1 | (4Z,7S,8R,9E,11E,13Z,15E,17S,19Z)-7,8,17-trihydroxydocosa-4,9,11,13,15,19-hexaenoic acid | Ligand for N-formyl peptide receptor-2 and GPR-32 | ↑ A20 expression ↓ NLRP3/NF-κB axis | Subarachnoid hemorrhage-model rat |
| Sildenafil [280] | 5-[2-ethoxy-5-(4-methylpiperazin-1-yl)sulfonylphenyl]-1-methyl-3-propyl-6H-pyrazolo[4,3-d]pyrimidin-7-one | 3′,5′-cyclic GMP (cGMP)-specific phosphodiesterase inhibitor | ↓ NLRP3 ↓ Hippocampal Aβ1–40 and Aβ1–42 ↑ Brain cGMP levels | APP/PS1 AD-model mouse |
| TAK-242 (CLI-095) [103,281] | (R)-Ethyl 6-(N-(2-chloro-4-fluorophenyl)sulfamoyl)cyclohex-1-enecarboxylate | TLR-4 signal transduction inhibitors | ↓ TLR-4-NF-κB-caspase-11 axis ↓ NLRP3, IL-1β, and IL-18 | Methamphetamine-treated mouse and primary astrocytes Aβ1–42-treated BV2 microglia and HT-22 neurons |
| 1,2,4-TTB [282] | 1,2,4-Trimethoxybenzene | Inhibitor of NLRP3 oligomer formation | ↓ Nigericin- or ATP-mediated NLRP3 activation | Murine bone marrow-derived macrophages (BMDMs) Primary mouse microglia EAE-model mice |
| Urolithin A [283] | 3,8-dihydroxybenzo[c]chromen-6-one | Gut microflora processed derivative of ellagic acid | ↓ NLRP3 activation via mitophagy promotion in microglia | LPS- or MPTP-treated BV2 microglial cells MPTP PD-model mouse |
| VX-765 [284] | (2S)-1-[(2S)-2-[(4-amino-3-chlorobenzoyl)amino]-3,3-dimethylbutanoyl]-N-[(2R,3S)-2-ethoxy-5-oxooxolan-3-yl]pyrrolidine-2-carboxamide | Competitive inhibitor of ICE/caspase-1 (active metabolite: VRT-043198) | ↓ NLRP3/caspase-1/GsdmD pathway | APP/PS1 AD-model mice BV2 microglial cells |
| (A) Agents. | ||||
| Natural Compounds and Sources | Chemical Class | Biological Activities | Experimental Models | References |
| Andrographolide from the roots and leaves of the plant Creat or Green chireta (Andrographis paniculata Wall. ex Nees) | labdane diterpenoid | ↓§ P2X7 receptor signaling ↓ HMGB1-induced TLR-4-NFκB signaling | LPS-activated mixed glial cells LPS-treated mouse | [285] see also Box 2 |
| Artesunate/Artemisinin from Artemisiae Iwayomogii Herba | sesquiterpene lactone | ↓ Inflammatory response and neuron death ↑§ Expression of BDNF, GDNF, and NT-3 neurotrophins | Traumatic brain injury-model mouse LPS-stimulated BV-2 microglial cells LPS-treated mouse | [286,287] |
| Astragaloside IV from Astragalus membranaceus (i.e., Huangqi) | pentacyclic triterpenoid | Antioxidant activity | Transient cerebral ischemia/reperfusion (I/R)-model mice | [288] |
| Baicalin from the root of Scutellaria baicalensis Georgi | flavonoid | ↓ TLR-4/NF-κB/NLRP3 axis | APP/PS1 AD-model mice LPS/Aβ-stimulated BV2 microglial cells | [289] |
| Benzyl isothiocyanate from cruciferous vegetables | benzene | ↓ NLRP3 activation via mitochondria- generated ROS inhibition ↓ NF-κB signaling | LPS-induced BV2 microglial cells | [290] |
| Bixin from the seeds of the Achiote tree (i.e., Bixa orellana) | apocarotenoid | Suppression of thioredoxin-interacting protein (TXNIP)-NLRP3 activity | EAE-model mouse | [291] |
| Carnosic acid (CA) Carnosol (CS) from Rosmarinus officinalis | abietane-type tricyclic diterpenes | ↑ KEAP1 (Kelch-like ECH-associated protein 1)/NRF2 (erythroid 2–related factor 2) transcriptional pathway activation ↓ HSP 90 inhibition | APP/PS1 AD-model mice Primary mouse bone marrow-derived macrophages | [292,293] |
| Cucurbitacin B from Cucurbitaceae | tetracyclic triterpene | ↓ NLRP3, caspase-1 self-activation, and IL-1β release | Ischemia/reperfusion injury-model rat | [294] |
| Dehydroisohispanolone diterpene (DT1) from Ballota hispanica (Labiatae) | labdane (bicyclic diterpene) | ↓ NF-κB and NLRP3 signaling | Nigericin-activated murine bone marrow-derived macrophages | [295] |
| Demethylene-tetrahydroberberine (DMTHB) from Berberis vulgaris, Berberis aristata | alkaloid | ↓ NLRP3 inflammasome’s activation ↓ IL-6 signaling | AD-model mice | [296] |
| Esculentoside A from the roots of Indian pokeweed (i.e., Phytolacca esculenta Van Houtte) | triterpene saponin | ↓ NF-κB, MAPKs and NLRP3 pathways | LPS-activated murine primary microglia cells and BV2 microglia cells | [297] |
| Gastrodin from rhizome of Gastrodia elata Blume | phenolic glycoside | ↓ TLR4-NF-κB-NLRP3 axis and microglia-mediated neuroinflammation | LPS-treated rats | [298] |
| Ginkgolide B (BN-52021) from Ginkgo biloba and Machilus wangchiana | diterpenoid esters | ↓ NLRP3 and microglia-mediated neuroinflammation ↑ NLRP3 autophagic degradation | Aβ1–42-induced BV2 cells LPS-primed BV2 cells senescence-accelerated male mouse prone 8 (SAMP8) | [299,300] |
| Ginsenosides (Rb1, Rg1, Rg3, Rg5, Rh1, Compound K, Chikusetsusaponin IVa, Gintonin, and 20(S)-Protopanaxatriol) from Panax ginseng C.A. Meyer; Panax quinquefolius L. (i.e., American Ginseng); and Panax japonicus T. Nees | saponins | ↓ NLRP3, NLRP1, AIM-2, and caspase-1 self-activation ↓ brain load of Aβs ↑ soluble (s)APP-α | AD in rodent models Depression-like behavior in rat model Post-traumatic stress disorder-like behavior in rodent model Stroke model High fat diet-model mouse | [301,302,303,304,305] |
| Isoformononetin from Cicer arietinum L. (chickpea) | methoxyisoflavone | ↓ NLRP3, NLRP2, ASC, NFκB-p65, IL-1β, caspase-1 proteins, and ROS | Streptozotocin-treated rat | [306] |
| Isoliquiritigenin from the Chinese herbal medicine Glycyrrhiza (Guo Lao) | isoflavone | ↓ NLRP3 ↑ NRF2-induced antioxidant activity | Hippocampal organotypic slice cultures after oxygen/glucose deprivation (OGD) | [307] |
| Isosibiricin from orange jasmine (i.e., Murraya exotica or paniculata) | coumarin | NLRP3-inhibition mediated by Dopamine D1/2 receptors | LPS-primed mouse BV-2 microglial cells | [308] |
| Kaempferol from several herbs in TCM | polyphenol flavonoid | ↑ NLRP3 autophagic degradation | PD-model mouse LPS-primed BV-2 microglial cells | [309,310,311] |
| β-Lapachone from the Lapacho tree or Jacaranda (i.e., Tabebuia Avellaneda Lorentz) | benzochromenone | Antioxidant activity | Multiple sclerosis and AD-model animals | [312] |
| Lychee seed polyphenols (LSPs) from the Litchi chinensis tree | polyphenols | ↑ Autophagy via the AMPK/mTOR/ULK1 axis ↑ Tight junctions’ expression ↑ LRP1 (i.e., low-density lipoprotein receptor-related protein 1), Beclin 1, and LC-3II proteins | Aβ-induced BV2 microglia cells APP/PS1 AD-model mouse | [313,314] |
| Mangiferin from the rhizome of Anemarrhena asphodeloides Bunge | C-glucoside xanthone | ↓ NF-κB and NLRP3 signaling ↓ Microglial M1 polarization | LPS-induced BV2 cells | [315] |
| Myricitrin from the root bark of the tallow shrub (i.e., Myrica cerifera L.) | polyphenol hydroxy flavonoid | ↓ NLRP3/Bax/Bcl2 axis NF-κB inactivation Antioxidant activity | Rat model of sepsis-linked encephalopathy Brain HI-model rat | [316,317] |
| Neferine from the green seed embryos of the lotus plant ( i.e., Nelumbo nucifera Gaertn) | bisbenzylisoquinoline alkaloid | ↓ NLRP3-mediated neuronal pyroptosis | Neonatal HI brain damage model rat PC12 cells | [35] |
| Nobiletin from Citrus L. fruits | polymethoxylated flavonoid | ↓ NLRP3 ↑ Autophagy via AMPK/mTOR/ULK1 axis | LPS-treated rat brain and BV2 cells | [318] |
| Oleocanthal from extra-virgin olive oil | phenylethanoid | ↓ NLRP3 ↑ Autophagy via AMPK/mTOR/ULK1 axis | AD-model TgSwDI Mouse | [319] |
| Oridonin from Isodon Rubescens (Hemsl.) H. Hara | (1S,2S,5S,8R,9S,10S,11R,15S,18R)-9,10,15,18-tetrahydroxy-12,12-dimethyl-6-methylidene-17-oxapentacyclo[7.6.2.15,8.01,11.02,8]octadecan-7-one | Binds NLRP3’s NACHT domain blocking NEK-7-NLRP3 activating interaction ↓ NF-κB pathway, Aβ1–42-elicited neuroinflammation, and pyroptosis | Aβ1–42-induced AD mice | [320] |
| Osthole from the roots of various medicinal plants, including Cnidium monnieri L. and Angelica pubescens (Japan’s Shishiudo). | 7-methoxy-8-(3-methylpent-2-enyl) coumarin | ↓ NLRP3 ↓ brain load of Aβs | Rat model of chronic cerebral ischemic hypoperfusion | [321] |
| Purpurin from Rubia tinctorum L. Rhein from Rheum rhabarbarum | anthraquinones | ↓ NLRP3, caspase-1 self-activation, and IL-1β release | AD-model animals Perirhinal cortex high-fat-diet-induced animal model | [322] |
| Quercetin (plant pigment) | flavonoid | Antioxidant activity ↓ NLRP3-pyroptosis-mediated IL-1β release ↑ Sirtuin | LPS-induced primary microglial cells and BV2 cells LPS-induced PD model mouse Depression-model mouse SAMP8 mice | [323,324] |
| Sinomenine from the roots of the climbing plant Sinomenium acutum (Thumb.) | alkaloid | Antioxidant and anti-inflammatory activity | EAE-model mouse | [325] |
| Thonningianin A from Penthorum chinense | ellagitannin polyphenol | ↑ NLRP3 autophagic degradation via AMPK/ULK1 and Raf/MEK/ERK axis | In vitro and in vivo AD models, including, C. elegans, APP/PS1 mice, BV-2 cells, and PC-12 cells | [119] |
| Withaferin from Indian ginseng (i.e., Withania somnifera) | steroidal lactone | ↓ Gene expression of NF-κB and associated neuroinflammatory molecules | SH-SY5Y cells transfected with APP plasmid (SH-APP) | [326] |
| (B) Herbal Extracts. | ||||
| Herbal/Fruit Extract | Source | Biological Activity | Experimental Model | References |
| Açaí extract | Berries of the Euterpe oleracea Mart. palm tree | Antioxidant activity | LPS- or nigericin-activated microglia (EOC 13.31) cells | [327] |
| Crysanthemum indicum extract (CIE) | TCM (main components: chlorogenic acid, luteoloside, and 3,5-dicaffeoylquinic acid) | Antioxidant activity ↑ TrkB/Akt/CREB/BDNF and Akt/Nrf-2/ARE axes | H2O2-induced oxidative toxicity in hippocampal HT22 neuronal cell line | [328,329] |
| Glycyrrhiza (Guo Lao) | TCM (main components: licochalcone, isochalcone A, echinatin, isoliquiritigenin, and glycyrrhizin) | ↓ NLRP3, TNF-α, IL-1β, and IL-18 ↑ AMPK/NRF2/antioxidant response element (ARE) signaling | LPS-induced chondrocyte pyroptosis LPS-induced macrophage cells Ischemic brain damage-model animal | [307,330] |
| Kutki | Ayurvedic medicine from rhizomes and roots of Picrorhiza kurroa | ↓ NLRP3 and BACE-1 expression | 5xFAD-model mice | [331] |
| Pien-Tze-Huang | TCM, including Radix et Rhizoma Notoginseng, Moschus, Calculus Bovis, and Snake Gall | ↓ NLRP3 ↑ Autophagy via AMPK/mTOR/ULK1 axis | LPS-induced BV2 microglial cells cerebral ischemia/reperfusion impaired rats | [332] |
| Tojapride | TCM (main components: Cyperus rotundus L. (i.e., Nagar motha in India), Perilla frutescens L. (i.e., Basionym), and Aurantii Fructus Immaturus L., the natural flavanone glycosides Naringin and Neohesperidin. | ↓ CaSR-mediated NLRP3 inflammasome’s activation | Esophageal epithelial cells (reflux esophagitis) | [333] see also Box 1 |
| Xingxiong | Extract from Ginkgo biloba L. or Ginkgo folium L. and tetramethylpyrazine sodium chloride | ↓ NLRP3 ↑Akt/NRF2 axis | Focal cerebral I/R damage | [334] |
| Ze Lan | Rhizomes or rootstalks of Lycopus lucidus | ↓ NLRP3 | H2O2-induced oxidative injury in rat embryo cortical neurons | [335] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiarini, A.; Gui, L.; Viviani, C.; Armato, U.; Dal Prà, I. NLRP3 Inflammasome’s Activation in Acute and Chronic Brain Diseases—An Update on Pathogenetic Mechanisms and Therapeutic Perspectives with Respect to Other Inflammasomes. Biomedicines 2023, 11, 999. https://doi.org/10.3390/biomedicines11040999
Chiarini A, Gui L, Viviani C, Armato U, Dal Prà I. NLRP3 Inflammasome’s Activation in Acute and Chronic Brain Diseases—An Update on Pathogenetic Mechanisms and Therapeutic Perspectives with Respect to Other Inflammasomes. Biomedicines. 2023; 11(4):999. https://doi.org/10.3390/biomedicines11040999
Chicago/Turabian StyleChiarini, Anna, Li Gui, Chiara Viviani, Ubaldo Armato, and Ilaria Dal Prà. 2023. "NLRP3 Inflammasome’s Activation in Acute and Chronic Brain Diseases—An Update on Pathogenetic Mechanisms and Therapeutic Perspectives with Respect to Other Inflammasomes" Biomedicines 11, no. 4: 999. https://doi.org/10.3390/biomedicines11040999