
The Y831C Mutation of the POLG Gene in Dementia
 , , , ,
, , , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
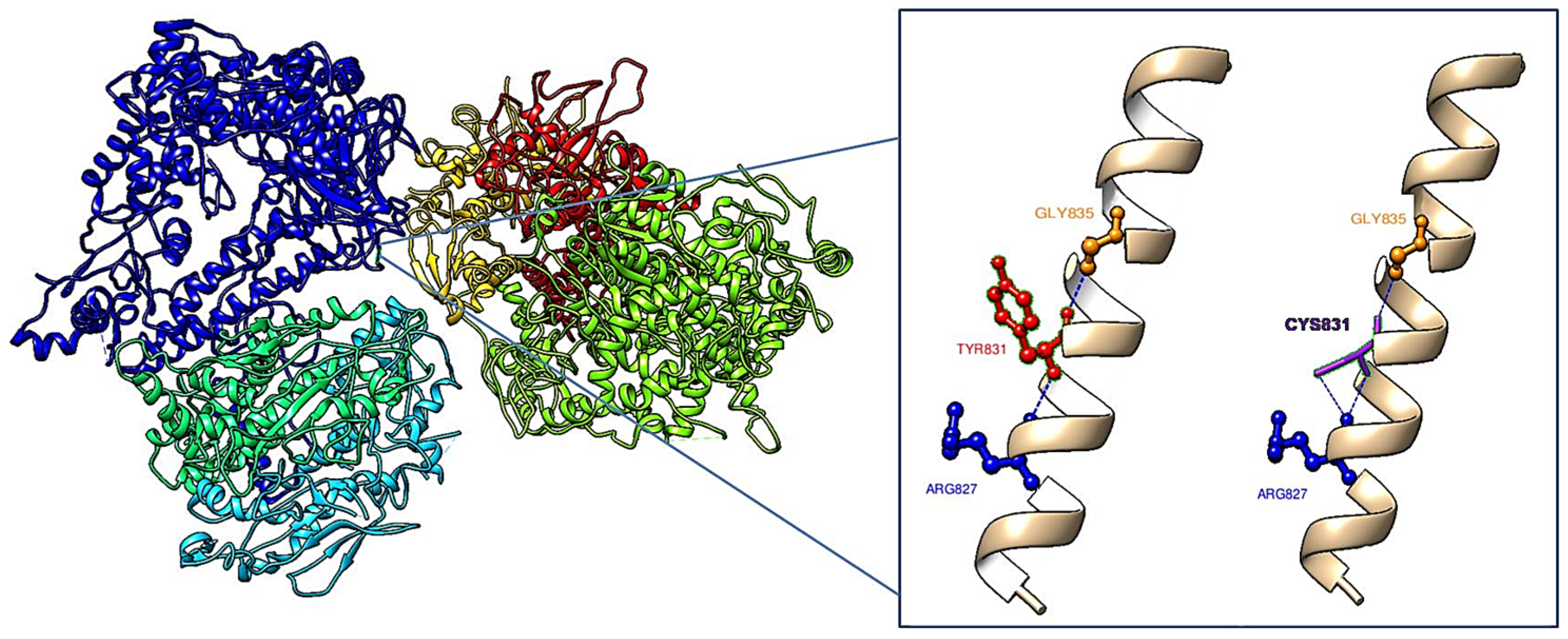
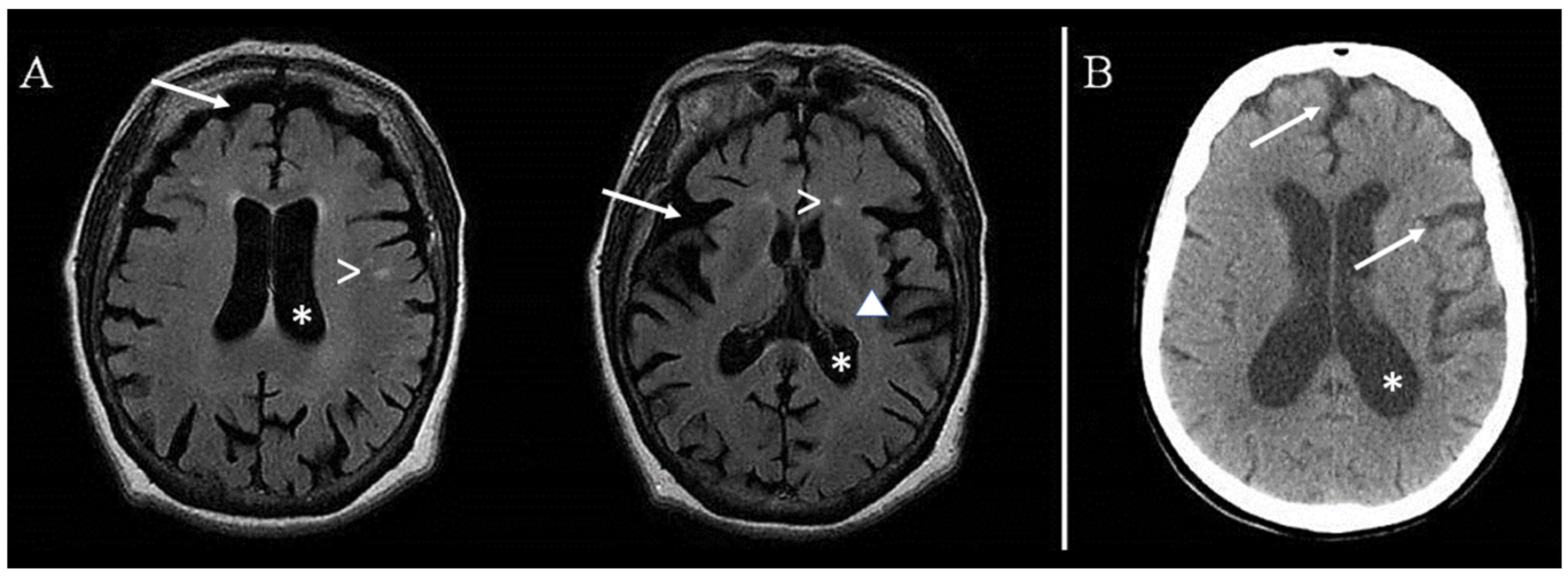
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, J.; Gennatas, E.D.; Kramer, J.H.; Miller, B.L.; Seeley, W.W. Predicting Regional Neurodegeneration from the Healthy Brain Functional Connectome. Neuron 2012, 73, 1216–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salemi, M.; Lanza, G.; Mogavero, M.P.; Cosentino, F.I.I.; Borgione, E.; Iorio, R.; Ventola, G.M.; Marchese, G.; Salluzzo, M.G.; Ravo, M.; et al. A Transcriptome Analysis of MRNAs and Long Non-Coding RNAs in Patients with Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 1535. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Gitler, A.D. Defects in Trafficking Bridge Parkinson’s Disease Pathology and Genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Canter, R.G.; Penney, J.; Tsai, L.-H. The Road to Restoring Neural Circuits for the Treatment of Alzheimer’s Disease. Nature 2016, 539, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss-Coray, T. Ageing, Neurodegeneration and Brain Rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [Green Version]
- Lanza, G.; Centonze, S.S.; Destro, G.; Vella, V.; Bellomo, M.; Pennisi, M.; Bella, R.; Ciavardelli, D. Shiatsu as an Adjuvant Therapy for Depression in Patients with Alzheimer’s Disease: A Pilot Study. Complement. Ther. Med. 2018, 38, 74–78. [Google Scholar] [CrossRef]
- Cantone, M.; Lanza, G.; Ranieri, F.; Opie, G.M.; Terranova, C. Editorial: Non-Invasive Brain Stimulation in the Study and Modulation of Metaplasticity in Neurological Disorders. Front. Neurol. 2021, 12, 721906. [Google Scholar] [CrossRef]
- Lanza, G.; Casabona, J.A.; Bellomo, M.; Cantone, M.; Fisicaro, F.; Bella, R.; Pennisi, G.; Bramanti, P.; Pennisi, M.; Bramanti, A. Update on Intensive Motor Training in Spinocerebellar Ataxia: Time to Move a Step Forward? J. Int. Med. Res. 2020, 48, 300060519854626. [Google Scholar] [CrossRef] [Green Version]
- Caruso, G.; Godos, J.; Privitera, A.; Lanza, G.; Castellano, S.; Chillemi, A.; Bruni, O.; Ferri, R.; Caraci, F.; Grosso, G. Phenolic Acids and Prevention of Cognitive Decline: Polyphenols with a Neuroprotective Role in Cognitive Disorders and Alzheimer’s Disease. Nutrients 2022, 14, 819. [Google Scholar] [CrossRef]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-Cell Autonomous Toxicity in Neurodegenerative Disorders: ALS and Beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marambaud, P.; Dreses-Werringloer, U.; Vingtdeux, V. Calcium Signaling in Neurodegeneration. Mol. Neurodegener. 2009, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Kiaei, M. New Hopes and Challenges for Treatment of Neurodegenerative Disorders: Great Opportunities for Young Neuroscientists. Basic Clin. Neurosci. 2013, 4, 3–4. [Google Scholar] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Briston, T.; Hicks, A.R. Mitochondrial Dysfunction and Neurodegenerative Proteinopathies: Mechanisms and Prospects for Therapeutic Intervention. Biochem. Soc. Trans. 2018, 46, 829–842. [Google Scholar] [CrossRef] [Green Version]
- Muddapu, V.R.; Dharshini, S.A.P.; Chakravarthy, V.S.; Gromiha, M.M. Neurodegenerative Diseases—Is Metabolic Deficiency the Root Cause? Front. Neurosci. 2020, 14, 213. [Google Scholar] [CrossRef] [Green Version]
- Lanza, G.; Cantone, M.; Musso, S.; Borgione, E.; Scuderi, C.; Ferri, R. Early-Onset Subcortical Ischemic Vascular Dementia in an Adult with MtDNA Mutation 3316G > A. J. Neurol. 2018, 265, 968–969. [Google Scholar] [CrossRef]
- Yakubovskaya, E.; Chen, Z.; Carrodeguas, J.A.; Kisker, C.; Bogenhagen, D.F. Functional Human Mitochondrial DNA Polymerase Gamma Forms a Heterotrimer. J. Biol. Chem. 2006, 281, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Nicholas Russo, S.; Shah, E.G.; Copeland, W.C.; Koenig, M.K. A New Pathogenic POLG Variant. Mol. Genet. Metab. Rep. 2022, 32, 100890. [Google Scholar] [CrossRef]
- Hsieh, P.-C.; Wang, C.-C.; Tsai, C.-L.; Yeh, Y.-M.; Lee, Y.S.; Wu, Y.-R. POLG R964C and GBA L444P Mutations in Familial Parkinson’s Disease: Case Report and Literature Review. Brain Behav. 2019, 9, e01281. [Google Scholar] [CrossRef] [Green Version]
- Eerola, J.; Luoma, P.T.; Peuralinna, T.; Scholz, S.; Paisan-Ruiz, C.; Suomalainen, A.; Singleton, A.B.; Tienari, P.J. POLG1 Polyglutamine Tract Variants Associated with Parkinson’s Disease. Neurosci. Lett. 2010, 477, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anvret, A.; Westerlund, M.; Sydow, O.; Willows, T.; Lind, C.; Galter, D.; Belin, A.C. Variations of the CAG Trinucleotide Repeat in DNA Polymerase γ (POLG1) Is Associated with Parkinson’s Disease in Sweden. Neurosci. Lett. 2010, 485, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Davidzon, G.; Greene, P.; Mancuso, M.; Klos, K.J.; Ahlskog, J.E.; Hirano, M.; DiMauro, S. Early-Onset Familial Parkinsonism Due to POLG Mutations. Ann. Neurol. 2006, 59, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Mao, W.; Xu, E.; Cai, Y.; Wang, C.; Chhetri, J.K.; Chan, P. Novel POLG Mutation in a Patient with Early-Onset Parkinsonism, Progressive External Ophthalmoplegia and Optic Atrophy. Int. J. Neurosci. 2020, 130, 319–321. [Google Scholar] [CrossRef]
- Mancuso, M.; Filosto, M.; Bellan, M.; Liguori, R.; Montagna, P.; Baruzzi, A.; DiMauro, S.; Carelli, V. POLG Mutations Causing Ophthalmoplegia, Sensorimotor Polyneuropathy, Ataxia, and Deafness. Neurology 2004, 62, 316–318. [Google Scholar] [CrossRef]
- Barthélémy, C.; de Baulny, H.O.; Lombès, A. D-Loop Mutations in Mitochondrial DNA: Link with Mitochondrial DNA Depletion? Hum. Genet. 2002, 110, 479–487. [Google Scholar] [CrossRef]
- Stopińska, K.; Grzybowski, T.; Malyarchuk, B.A.; Derenko, M.V.; Miścicka-Sliwka, D. Optimization of the Y831C Mutation Detection in Human DNA Polymerase Gamma by Allelic Discrimination Assay. Acta Biochim. Pol. 2006, 53, 591–595. [Google Scholar] [CrossRef]
- Scuderi, C.; Borgione, E.; Castello, F.; Lo Giudice, M.; Santa Paola, S.; Giambirtone, M.; Di Blasi, F.D.; Elia, M.; Amato, C.; Città, S.; et al. The in Cis T251I and P587L POLG1 Base Changes: Description of a New Family and Literature Review. Neuromuscul. Disord. 2015, 25, 333–339. [Google Scholar] [CrossRef]
- Wong, L.-J.C.; Naviaux, R.K.; Brunetti-Pierri, N.; Zhang, Q.; Schmitt, E.S.; Truong, C.; Milone, M.; Cohen, B.H.; Wical, B.; Ganesh, J.; et al. Molecular and Clinical Genetics of Mitochondrial Diseases Due to POLG Mutations. Hum. Mutat. 2008, 29, E150–E172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodbridge, P.; Liang, C.; Davis, R.L.; Vandebona, H.; Sue, C.M. POLG Mutations in Australian Patients with Mitochondrial Disease. Intern. Med. J. 2013, 43, 150–156. [Google Scholar] [CrossRef]
- Min, J.; Farooq, M.U.; Glisson, C. Adult Phenotypic Spectrum of Headache, Myopathy and Ischemic Stroke Associated with Mitochondrial POLG Mutation. Austin J. Cerebrovasc. Dis. Stroke 2014, 1, 1021. [Google Scholar]
- Da Pozzo, P.; Cardaioli, E.; Rubegni, A.; Gallus, G.N.; Malandrini, A.; Rufa, A.; Battisti, C.; Carluccio, M.A.; Rocchi, R.; Giannini, F.; et al. Novel POLG Mutations and Variable Clinical Phenotypes in 13 Italian Patients. Neurol. Sci. 2017, 38, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Spracklen, T.F.; Kasher, P.R.; Kraus, S.; Botha, T.L.; Page, D.J.; Kamuli, S.; Booi, Z.; Chin, A.; Laing, N.; Keavney, B.D.; et al. Identification of a POLG Variant in a Family with Arrhythmogenic Cardiomyopathy and Left Ventricular Fibrosis. Circ. Genom. Precis. Med. 2021, 14, e003138. [Google Scholar] [CrossRef] [PubMed]
- Luoma, P.T.; Eerola, J.; Ahola, S.; Hakonen, A.H.; Hellström, O.; Kivistö, K.T.; Tienari, P.J.; Suomalainen, A. Mitochondrial DNA Polymerase Gamma Variants in Idiopathic Sporadic Parkinson Disease. Neurology 2007, 69, 1152–1159. [Google Scholar] [CrossRef]
- Taanman, J.-W.; Rahman, S.; Pagnamenta, A.T.; Morris, A.A.M.; Bitner-Glindzicz, M.; Wolf, N.I.; Leonard, J.V.; Clayton, P.T.; Schapira, A.H.V. Analysis of Mutant DNA Polymerase Gamma in Patients with Mitochondrial DNA Depletion. Hum. Mutat. 2009, 30, 248–254. [Google Scholar] [CrossRef]
- Ylönen, S.; Ylikotila, P.; Siitonen, A.; Finnilä, S.; Autere, J.; Majamaa, K. Variations of Mitochondrial DNA Polymerase γ in Patients with Parkinson’s Disease. J. Neurol. 2013, 260, 3144–3149. [Google Scholar] [CrossRef]
- Hikmat, O.; Tzoulis, C.; Chong, W.K.; Chentouf, L.; Klingenberg, C.; Fratter, C.; Carr, L.J.; Prabhakar, P.; Kumaraguru, N.; Gissen, P.; et al. The Clinical Spectrum and Natural History of Early-Onset Diseases Due to DNA Polymerase Gamma Mutations. Genet. Med. 2017, 19, 1217–1225. [Google Scholar] [CrossRef]
- Ortega-Moreno, L.; Giráldez, B.G.; Soto-Insuga, V.; Losada-Del Pozo, R.; Rodrigo-Moreno, M.; Alarcón-Morcillo, C.; Sánchez-Martín, G.; Díaz-Gómez, E.; Guerrero-López, R.; Serratosa, J.M.; et al. Molecular Diagnosis of Patients with Epilepsy and Developmental Delay Using a Customized Panel of Epilepsy Genes. PLoS ONE 2017, 12, e0188978. [Google Scholar] [CrossRef]
- Tiangyou, W.; Hudson, G.; Ghezzi, D.; Ferrari, G.; Zeviani, M.; Burn, D.J.; Chinnery, P.F. POLG1 in Idiopathic Parkinson Disease. Neurology 2006, 67, 1698–1700. [Google Scholar] [CrossRef]
- Gui, Y.; Xu, Z.; Lv, W.; Liu, H.; Zhao, J.-J.; Hu, X.-Y. Association of Mitochondrial DNA Polymerase γ Gene POLG1 Polymorphisms with Parkinsonism in Chinese Populations. PLoS ONE 2012, 7, e50086. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, T.; Ishiwata, M.; Kakiuchi, C.; Fuke, S.; Iwata, N.; Ozaki, N.; Kunugi, H.; Minabe, Y.; Nakamura, K.; Iwata, Y.; et al. Enrichment of Deleterious Variants of Mitochondrial DNA Polymerase Gene(POLG1) in Bipolar Disorder. Psychiatry Clin. Neurosci. 2017, 71, 518–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| N. | Sex | Age Years | Age at Onset Years | Diagnosis | Family History | Neurological and/or Neuropsychiatric Comorbidities |
|---|---|---|---|---|---|---|
| 1 | F | 60 | 54 | FTD | Yes (mother) | Parkinsonism, lacunar CVD |
| 2 | M | 76 | 67 | PD | No | Mild dementia, RBD, OSAS, chronic diffuse CVD |
| 3 | M | 70 | 60 | AD | Yes (mother) | Chronic diffuse CVD |
| 4 | M | 69 | 63 | DLB | No | None |
| 5 | F | 65 | 60 | CBD | No | Mild dementia, lacunar CVD |
| 6 | M | 60 | 58 | FTD | Yes (NS) | Parkinsonism, lacunar CVD |
| 7 | M | 73 | 69 | PD | No | RBD, OSAS, lacunar CVD |
| 8 | F | 83 | 77 | AD | No | Parkinsonism, chronic diffuse CVD |
| 9 | M | 67 | 59 | DLB | No | None |
| 10 | M | 68 | 64 | PD | No | MCI, depression, lacunar CVD |
| 11 | M | 68 | 65 | PD | No | MCI, depression |
| 12 | F | 82 | 52 | DLB | Yes (NS) | Parkinsonism, chronic diffuse CVD, schizophrenia |
| 13 | M | 80 | 69 | PD | No | MCI, obsessive-compulsive disorder, chronic diffuse CVD |
| 14 | M | 92 | 85 | DLB | No | Lacunar CVD |
| 15 | M | 68 | 62 | PSP | Yes (NS) | MCI, depression |
| 16 | F | 71 | 68 | PD | No | Dementia, OSAS, chronic diffuse CVD |
| 17 | M | 62 | 61 | PD | Yes (NS) | Lacunar CVD |
| 18 | M | 80 | 77 | PD | Yes (NS) | MCI, lacunar CVD |
| 19 | M | 71 | 78 | PD | No | RBD, chronic diffuse CVD |
| 20 | M | 74 | 70 | DLB | No | Parkinsonism, RBD, OSAS |
| 21 | M | 52 | 49 | PD | No | RBD, lacunar CVD |
| 22 | M | 77 | 72 | PD | Yes (NS) | RBD, OSAS, narcolepsy, chronic diffuse CVD |
| 23 | M | 69 | 66 | FTD | No | Previous traumatic brain injury |
| 24 | M | 76 | 72 | DLB | Yes (NS) | Parkinsonism, previous stroke |
| 25 | M | 76 | 75 | PD | No | Lacunar CVD |
| 26 | F | 70 | 66 | DLB | Yes (NS) | Parkinsonism, RBD, chronic diffuse CVD |
| 27 | F | 69 | 67 | AD | No | Lacunar CVD |
| 28 | F | 63 | 58 | PD | Yes (NS) | Lacunar CVD, distal motor-sensory polyneuropathy |
| 29 | M | 57 | 51 | AD | No | Depression |
| 30 | M | 66 | 56 | PD | No | RBD, chronic diffuse CVD, anxiety disorder |
| 31 | M | 60 | 57 | PD | No | RBD, insomnia, chronic diffuse CVD, |
| 32 | M | 71 | 63 | PD | No | MCI, RBD, lacunar CVD |
| 33 | F | 67 | 60 | PD | No | MCI, previous stroke, chronic diffuse CVD |
| Allele 1 | Allele 2 | Sex | Age at Onset (Years) | Ocular Abnormalities | Cognitive Deficit | Epilepsy | Myopathy | Movement Disorder | Peripheral Neuropathy | Hepatic Involvement | Other Signs | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Y831C | - | F | 6 | Optic atrophy, ophthalmoplegia, retinitis pigmentosa | Cognitive impairment | + | Exercise intolerance, persistent muscle weakness, diffuse myalgia, respiratory distress | - | - | - | Severe sensorineural hearing loss, nephrotic syndrome, severe mtDNA depletion, mtDNA multiple deletions and heteroplasmic point mutations | [26] |
| Y831C | - | F | 28 | PEO | - | - | - | Parkinsonism | + | - | Hypertension and gonadal dysgenesis, mtDNA multiple deletions | [25] |
| Y831C Q1236H | - | F | 70 | - | - | NA | - | PD | - | NA | Ischemic heart disease, atrial fibrillation, hypertension | [34] |
| Y831C | - | F | 0.1 | NA | NA | NA | NA | NA | NA | NA | - | [29] |
| Y831C | - | M | 0.1 | - | DD | + | Hypotonia | - | - | - | - | [29] |
| Y831C | - | M | 1.8 | - | - | - | Hypotonia | - | - | - | Elevated lactate level, autism | [29] |
| Y831C | - | F | 9 | - | DD | + | - | - | - | - | - | [29] |
| Y831C | - | F | 14 | NA | NA | NA | NA | NA | NA | NA | - | [29] |
| Y831C | - | M | 19 | PEO | DD | - | Muscle weakness, fatigue | Ataxia | + | - | Hearing loss, short stature | [29] |
| Y831C | H1134R | F | 0.3 | NA | NA | NA | NA | NA | NA | Mildly icteric with hepatomegaly | Anorexia, failure to thrive syndrome, drowsiness, peripheral edema, mtDNA depletion | [35] |
| Y831C | R722H | F | 56 | - | NA | NA | NA | PD | NA | NA | Hyperreflexia | [36] |
| Y831C | - | F | 34 | C-PEO | NA | + | + | Ataxia | + | + | Dysphagia/dysarthria, gastric and small bowel dysfunction, sensorineural hearing loss, palatal insufficiency, mtDNA multiple deletions | [30] |
| Y831C | - | F | 46 | Ptosis | NA | + | Myokymia, muscle weakness, myalgia | - | - | + | Migraine | [30] |
| Y831C | - | F | 59 | Visual deficit | Mild cognitive impairment | - | Fatigue | - | - | - | Ischemic stroke in the left occipital and parietal lobes, hypertension, hyperlipidaemia, intermittent limb paraesthesia, migraine-like headache, episodic confusion, dysarthria | [31] |
| Y831C | - | M | 72 | - | - | - | Axial myopathy | - | - | - | - | [32] |
| Y831C | - | F | 59 | Ptosis | - | - | - | - | - | - | Thyreopathy, hearing loss | [32] |
| Y831C | H1134R | NA | 0.1 | Visual deficit | DD | - | Hypotonia | - | - | + | Failure to thrive syndrome, altered growth, renal dysfunction, MCHS | [37] |
| Y831C | Q52dup | M | 3 | NA | DD | + | NA | NA | NA | NA | Epileptic encephalopathy with posterior electrical and neuroimaging abnormalities, SRPX2 mutation | [38] |
| Y831C | - | M | 50 | NA | NA | NA | NA | NA | NA | NA | Arrhythmogenic cardiomyopathy and left ventricular fibrosis | [33] |
| Study Population | Frequency | Reference | |
|---|---|---|---|
| Patients | Controls | ||
| Healthy Polish population | - | 2.25% (3/133) | [27] |
| British and Italian idiopathic patients with Parkinson’s disease | 2.14% (3/140 British) | 3.33% (3/90 British) * | [39] |
| 0.00% (0/279 Italian) | 0.00% (0/285 Italian) | ||
| Finnish patients with Parkinson’s disease | 0.71% (1/140) | 3.94% (5/127) * | [34] |
| Chinese sporadic patients with Parkinson’s disease | 8.14% (28/344) | 11.04% (17/154) * | [40] |
| Japanese patients with bipolar disorder | 0.00% (0/796) | 0.00% (0/767) | [41] |
| Present study | 6.06% (2/33) | 0.00% (0/100) | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borgione, E.; Lo Giudice, M.; Santa Paola, S.; Giuliano, M.; Lanza, G.; Cantone, M.; Ferri, R.; Scuderi, C. The Y831C Mutation of the POLG Gene in Dementia. Biomedicines 2023, 11, 1172. https://doi.org/10.3390/biomedicines11041172
Borgione E, Lo Giudice M, Santa Paola S, Giuliano M, Lanza G, Cantone M, Ferri R, Scuderi C. The Y831C Mutation of the POLG Gene in Dementia. Biomedicines. 2023; 11(4):1172. https://doi.org/10.3390/biomedicines11041172
Chicago/Turabian StyleBorgione, Eugenia, Mariangela Lo Giudice, Sandro Santa Paola, Marika Giuliano, Giuseppe Lanza, Mariagiovanna Cantone, Raffaele Ferri, and Carmela Scuderi. 2023. "The Y831C Mutation of the POLG Gene in Dementia" Biomedicines 11, no. 4: 1172. https://doi.org/10.3390/biomedicines11041172