
Neuroinflammation, Energy and Sphingolipid Metabolism Biomarkers Are Revealed by Metabolic Modeling of Autistic Brains

Abstract
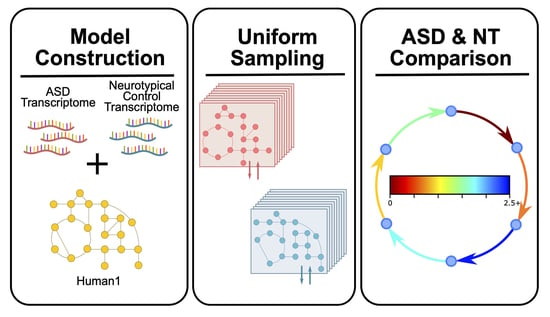
:
1. Introduction
2. Materials and Methods
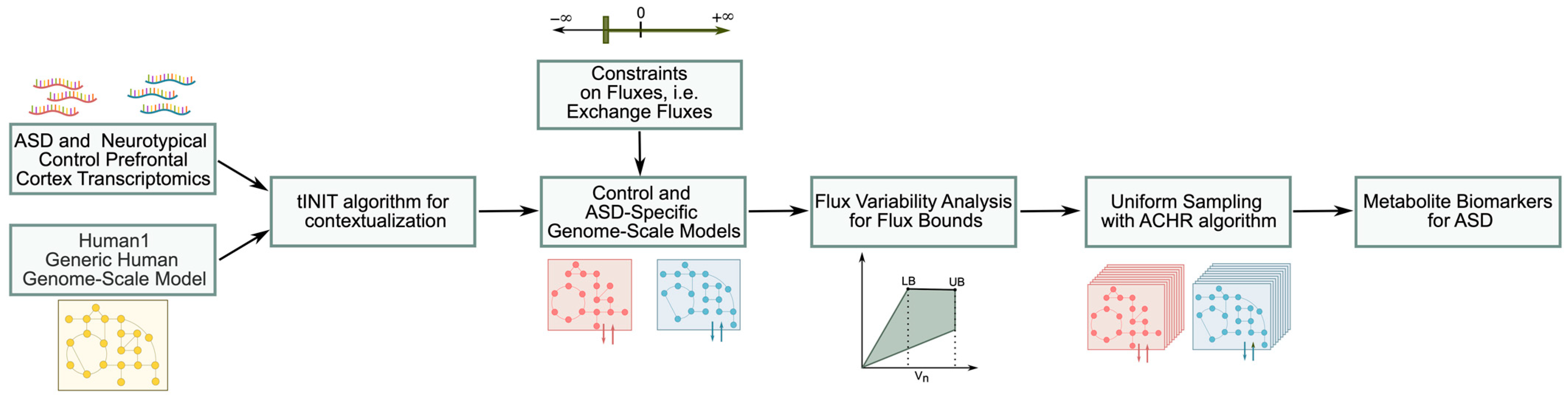
2.1. ASD-Specific Prefrontal Cortex GEM Construction
2.2. GEM Constraints
2.3. Model Simulations
2.4. Statistical Analysis
3. Results and Discussion
3.1. Model, Constraint, and Simulation Features
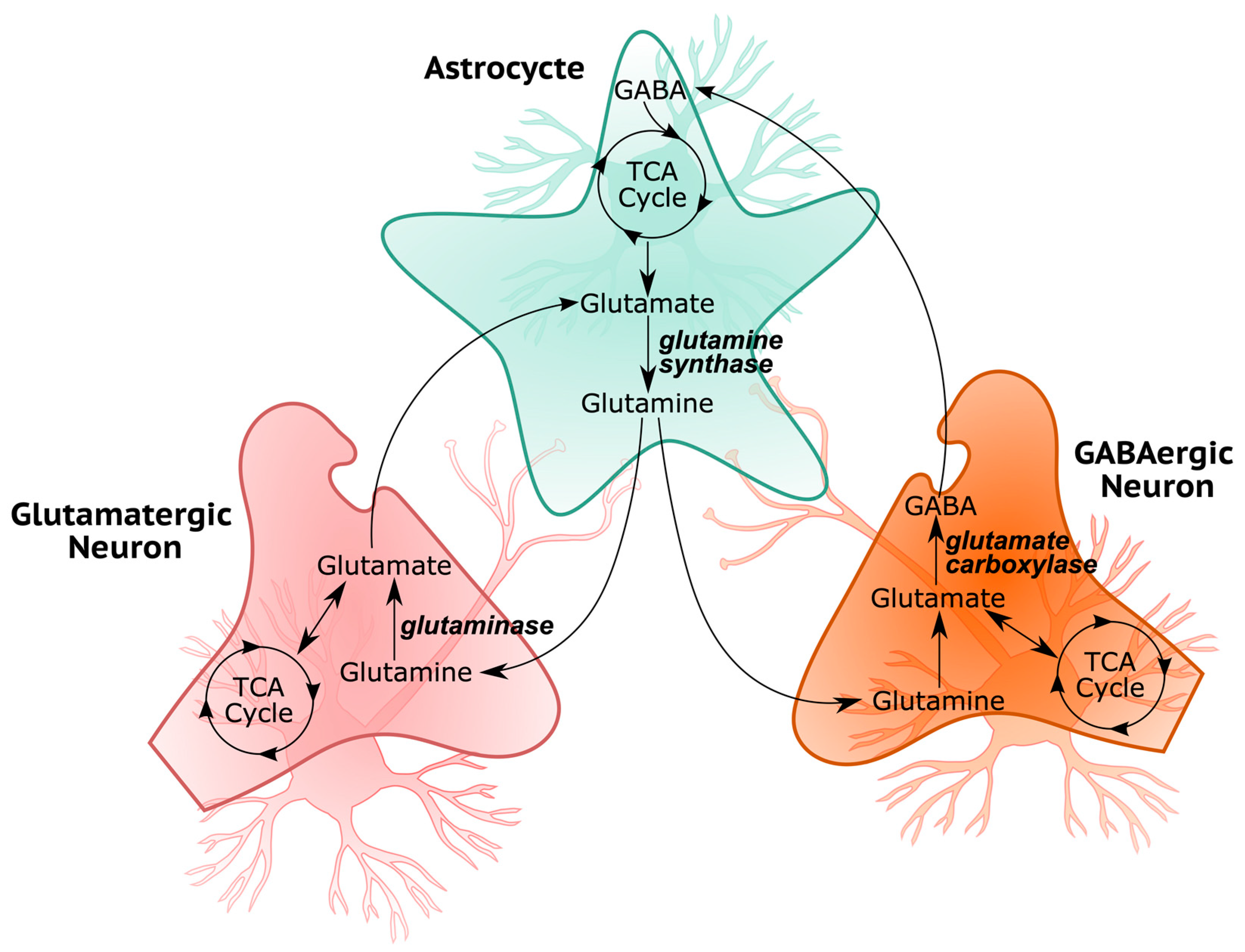
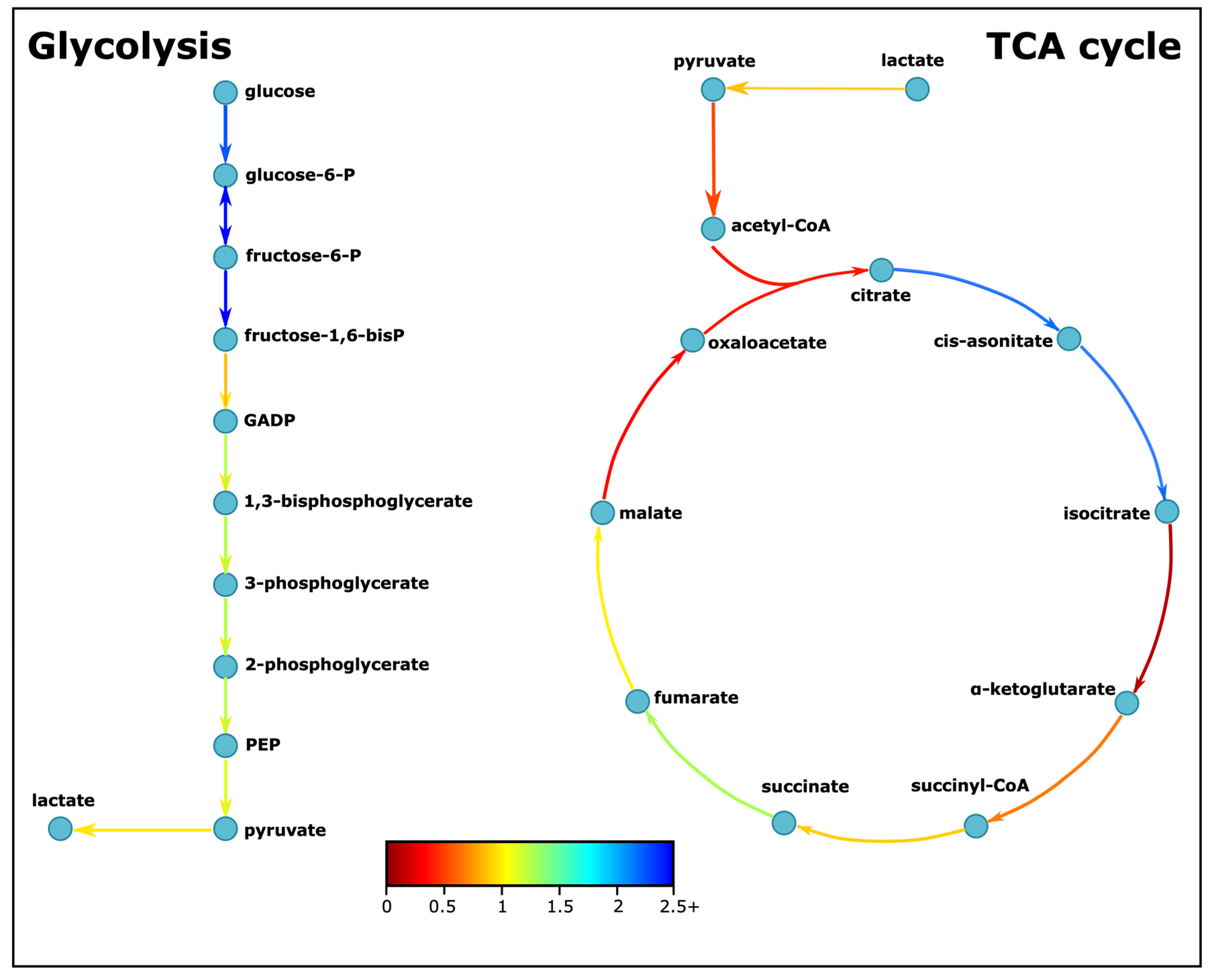
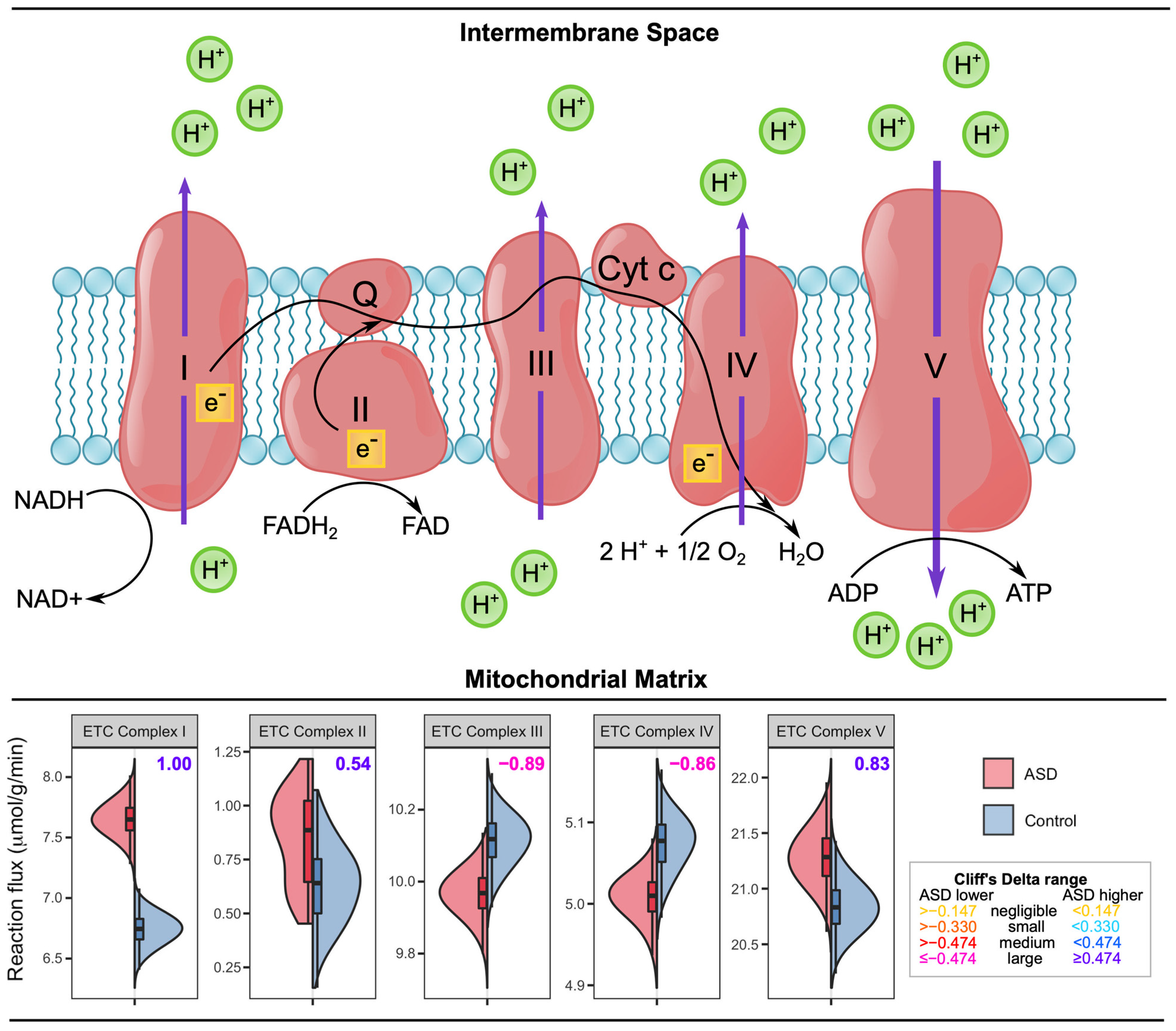
3.2. Cellular Respiration and Energy Metabolism Is Disturbed in ASD
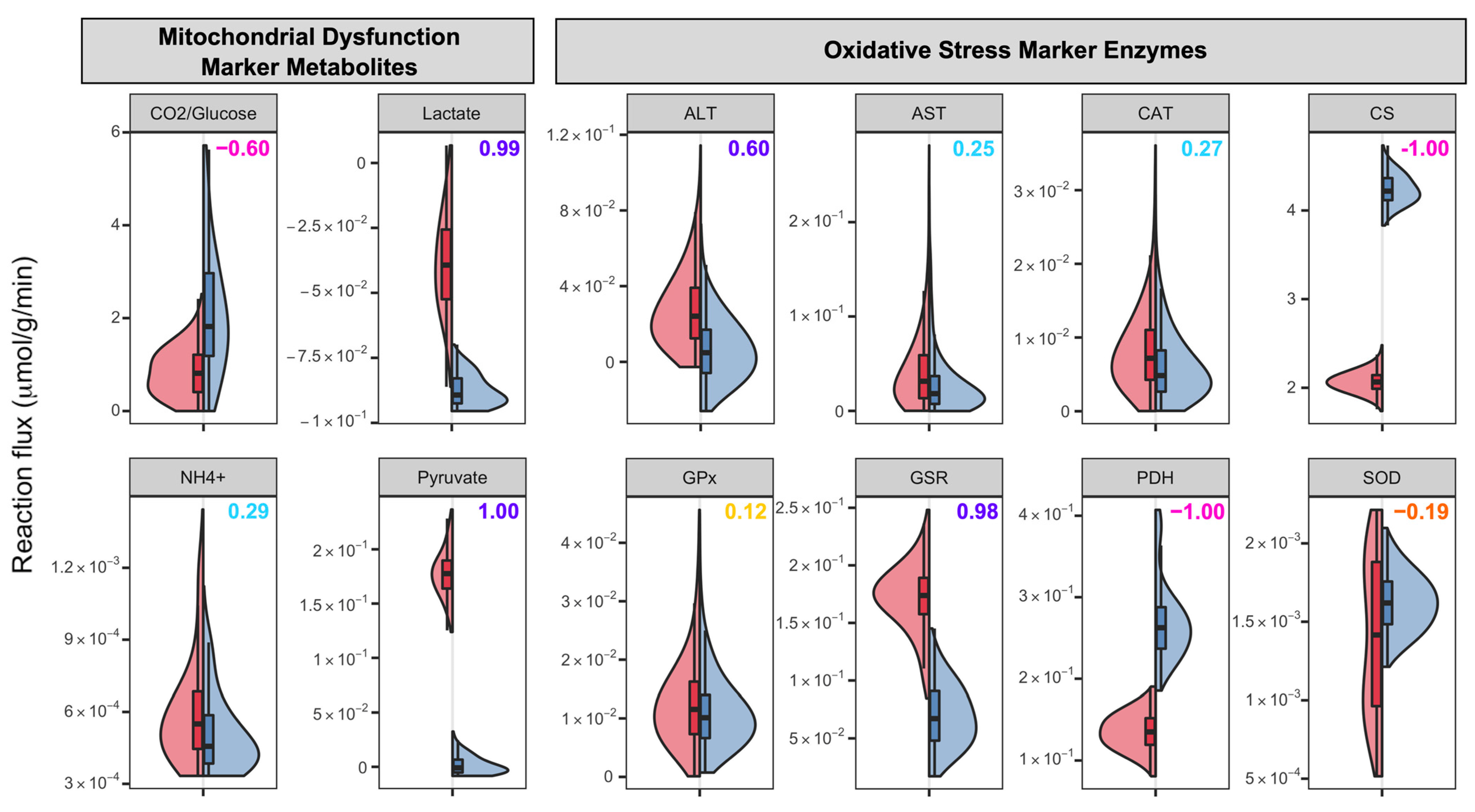
3.3. ASD-Related Changes in Mitochondrial Dysfunction and Oxidative Stress Related Markers
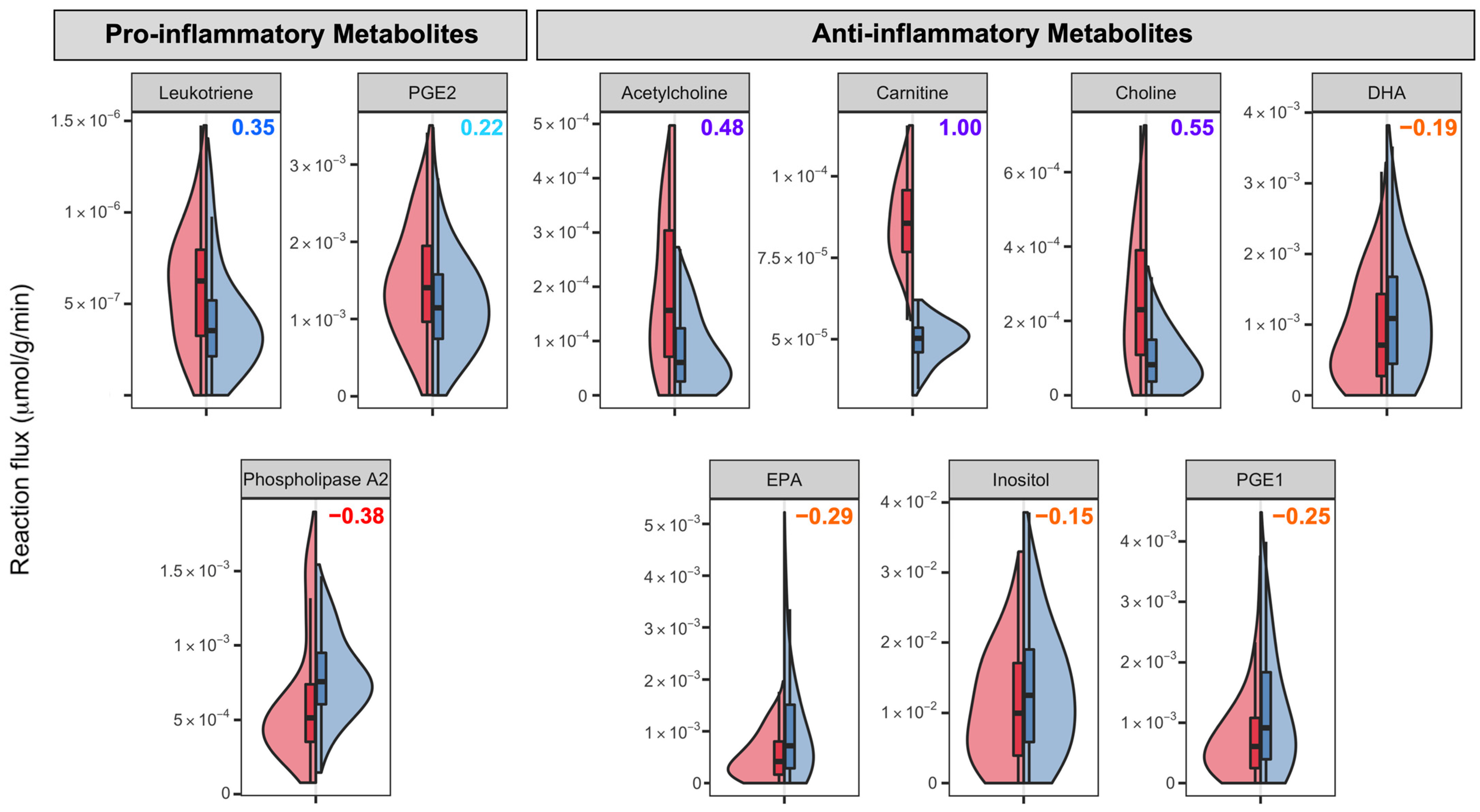
3.4. Neuroinflammatory Markers in ASD
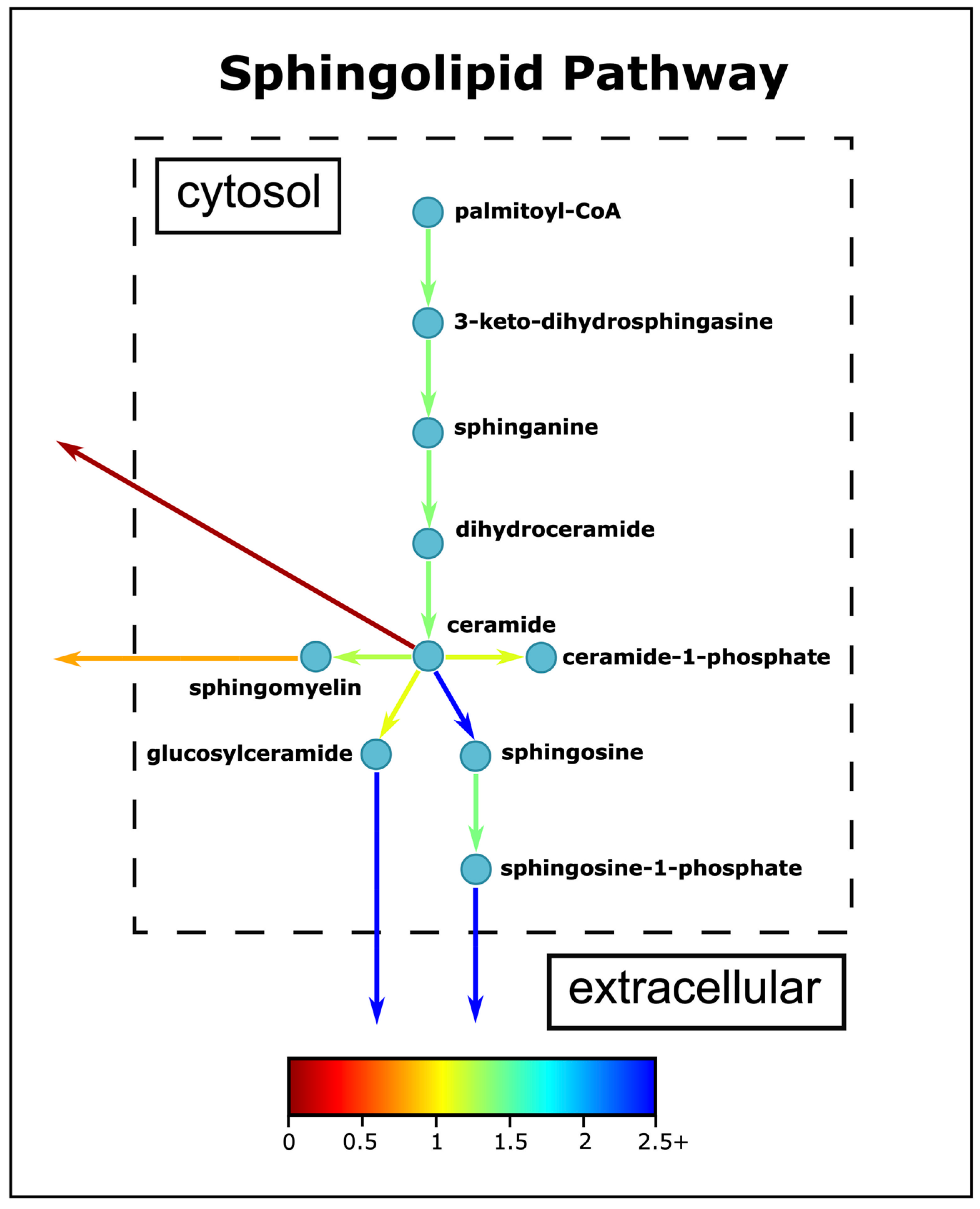
3.5. Sphingolipid Metabolism Changes in ASD
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism Spectrum Disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, J.; Fombonne, E.; Scorah, J.; Ibrahim, A.; Durkin, M.S.; Saxena, S.; Yusuf, A.; Shih, A.; Elsabbagh, M. Global Prevalence of Autism: A Systematic Review Update. Autism Res. 2022, 15, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Amaral, D.G.; Schumann, C.M.; Nordahl, C.W. Neuroanatomy of Autism. Trends Neurosci. 2008, 31, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, D.; Anagnostou, E.; Arango, C.; Auzias, G.; Behrmann, M.; Busatto, G.F.; Calderoni, S.; Daly, E.; Deruelle, C.; Di Martino, A.; et al. Cortical and Subcortical Brain Morphometry Differences between Patients with Autism Spectrum Disorder and Healthy Individuals across the Lifespan: Results from the ENIGMA ASD Working Group. Am. J. Psychiatry 2018, 175, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Carlisi, C.O.; Norman, L.J.; Lukito, S.S.; Radua, J.; Mataix-Cols, D.; Rubia, K. Comparative Multimodal Meta-Analysis of Structural and Functional Brain Abnormalities in Autism Spectrum Disorder and Obsessive-Compulsive Disorder. Biol. Psychiatry 2017, 82, 83–102. [Google Scholar] [CrossRef] [Green Version]
- Courchesne, E.; Pierce, K. Why the Frontal Cortex in Autism Might Be Talking Only to Itself: Local over-Connectivity but Long-Distance Disconnection. Curr. Opin. Neurobiol. 2005, 15, 225–230. [Google Scholar] [CrossRef]
- Courchesne, E.; Mouton, P.R.; Calhoun, M.E.; Semendeferi, K.; Ahrens-Barbeau, C.; Hallet, M.J.; Barnes, C.C.; Pierce, K. Neuron Number and Size in Prefrontal Cortex of Children With Autism. JAMA 2011, 306, 2001. [Google Scholar] [CrossRef]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial Activation and Increased Microglial Density Observed in the Dorsolateral Prefrontal Cortex in Autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef]
- Abraham, J.R.; Szoko, N.; Barnard, J.; Rubin, R.A.; Schlatzer, D.; Lundberg, K.; Li, X.; Natowicz, M.R. Proteomic Investigations of Autism Brain Identify Known and Novel Pathogenetic Processes. Sci. Rep. 2019, 9, 13118. [Google Scholar] [CrossRef] [Green Version]
- Park, D.I. Genomics, Transcriptomics, Proteomics and Big Data Analysis in the Discovery of New Diagnostic Markers and Targets for Therapy Development, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 173, ISBN 9780128212424. [Google Scholar]
- Rossignol, D.A.; Frye, R.E. A Review of Research Trends in Physiological Abnormalities in Autism Spectrum Disorders: Immune Dysregulation, Inflammation, Oxidative Stress, Mitochondrial Dysfunction and Environmental Toxicant Exposures. Mol. Psychiatry 2012, 17, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Siniscalco, D.; Schultz, S.; Brigida, A.; Antonucci, N. Inflammation and Neuro-Immune Dysregulations in Autism Spectrum Disorders. Pharmaceuticals 2018, 11, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, T.C.; Crewther, D.P. A Comprehensive Review of the 1H-MRS Metabolite Spectrum in Autism Spectrum Disorder. Front. Mol. Neurosci. 2016, 9, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Likhitweerawong, N.; Thonusin, C.; Boonchooduang, N.; Louthrenoo, O.; Nookaew, I.; Chattipakorn, N.; Chattipakorn, S.C. Profiles of Urine and Blood Metabolomics in Autism Spectrum Disorders. Metab. Brain Dis. 2021, 36, 1641–1671. [Google Scholar] [CrossRef]
- Bjørklund, G.; Meguid, N.A.; El-Bana, M.A.; Tinkov, A.A.; Saad, K.; Dadar, M.; Hemimi, M.; Skalny, A.V.; Hosnedlová, B.; Kizek, R.; et al. Oxidative Stress in Autism Spectrum Disorder. Mol. Neurobiol. 2020, 57, 2314–2332. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorders: A Systematic Review and Meta-Analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current Status and Applications of Genome-Scale Metabolic Models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.E.; Kemp, M.L. Integration of Machine Learning and Genome-Scale Metabolic Modeling Identifies Multi-Omics Biomarkers for Radiation Resistance. Nat. Commun. 2021, 12, 2700. [Google Scholar] [CrossRef]
- Moolamalla, S.T.R.; Vinod, P.K. Genome-Scale Metabolic Modelling Predicts Biomarkers and Therapeutic Targets for Neuropsychiatric Disorders. Comput. Biol. Med. 2020, 125, 103994. [Google Scholar] [CrossRef]
- Chow, M.L.; Li, H.-R.; Winn, M.E.; April, C.; Barnes, C.C.; Wynshaw-Boris, A.; Fan, J.-B.; Fu, X.-D.; Courchesne, E.; Schork, N.J. Genome-Wide Expression Assay Comparison across Frozen and Fixed Postmortem Brain Tissue Samples. BMC Genom. 2011, 12, 449. [Google Scholar] [CrossRef] [Green Version]
- Chow, M.L.; Winn, M.E.; Li, H.-R.; April, C.; Wynshaw-Boris, A.; Fan, J.-B.; Fu, X.-D.; Courchesne, E.; Schork, N.J. Preprocessing and Quality Control Strategies for Illumina DASL Assay-Based Brain Gene Expression Studies with Semi-Degraded Samples. Front. Genet. 2012, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Chow, M.L.; Pramparo, T.; Winn, M.E.; Barnes, C.C.; Li, H.-R.; Weiss, L.; Fan, J.-B.; Murray, S.; April, C.; Belinson, H.; et al. Age-Dependent Brain Gene Expression and Copy Number Anomalies in Autism Suggest Distinct Pathological Processes at Young Versus Mature Ages. PLoS Genet. 2012, 8, e1002592. [Google Scholar] [CrossRef] [Green Version]
- Agren, R.; Mardinoglu, A.; Asplund, A.; Kampf, C.; Uhlen, M.; Nielsen, J. Identification of Anticancer Drugs for Hepatocellular Carcinoma through Personalized Genome-scale Metabolic Modeling. Mol. Syst. Biol. 2014, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Kocabaş, P.; Wang, H.; Cholley, P.-E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An Atlas of Human Metabolism. Sci. Signal. 2020, 13, eaaz1482. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-Scale Metabolic Modelling of Hepatocytes Reveals Serine Deficiency in Patients with Non-Alcoholic Fatty Liver Disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [Green Version]
- Blais, E.M.; Rawls, K.D.; Dougherty, B.V.; Li, Z.I.; Kolling, G.L.; Ye, P.; Wallqvist, A.; Papin, J.A. Reconciled Rat and Human Metabolic Networks for Comparative Toxicogenomics and Biomarker Predictions. Nat. Commun. 2017, 8, 14250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D Enables a Three-Dimensional View of Gene Variation in Human Metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A Versatile Toolbox for Metabolic Network Reconstruction and a Case Study on Streptomyces Coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef] [Green Version]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and Analysis of Biochemical Constraint-Based Models Using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [Green Version]
- Gurobi Optimization, LLC. Gurobi Optimizer Reference Manual: Houston, Texas, USA. 2022. Available online: https://www.gurobi.com/wp-content/plugins/hd_documentations/documentation/9.0/refman.pdf (accessed on 8 January 2023).
- De Volder, A.; Bol, A.; Michel, C.; Congneau, M.; Goffinet, A.M. Brain Glucose Metabolism in Children with the Autistic Syndrome: Positron Tomography Analysis. Brain Dev. 1987, 9, 581–587. [Google Scholar] [CrossRef]
- Herold, S.; Frackowiak, R.S.J.; Le Couteur, A.; Rutter, M.; Howlin, P. Cerebral Blood Flow and Metabolism of Oxygen and Glucose in Young Autistic Adults. Psychol. Med. 1988, 18, 823–831. [Google Scholar] [CrossRef]
- Thorngren-Jerneck, K.; Ohlsson, T.; Sandell, A.; Erlandsson, K.; Strand, S.E.; Ryding, E.; Svenningsen, N.W. Cerebral Glucose Metabolism Measured by Positron Emission Tomography in Term Newborn Infants with Hypoxic Ischemic Encephalopathy. Pediatr. Res. 2001, 49, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwell, D.; Laughlin, S.B. An Energy Budget for Signaling in the Grey Matter of the Brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Hogstad, S.; Svenneby, G.; Torgner, I.A.; Kvamme, E.; Hertz, L.; Schousboe, A. Glutaminase in Neurons and Astrocytes Cultured from Mouse Brain: Kinetic Properties and Effects of Phosphate, Glutamate, and Ammonia. Neurochem. Res. 1988, 13, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Jeitner, T.M.; Cooper, A.J.L. Inhibition of Human Glutamine Synthetase by L-Methionine-S,R-Sulfoximine—Relevance to the Treatment of Neurological Diseases. Metab. Brain Dis. 2014, 29, 983–989. [Google Scholar] [CrossRef] [Green Version]
- Ureña-Guerrero, M.E.; López-Pérez, S.J.; Beas-Zárate, C. Neonatal Monosodium Glutamate Treatment Modifies Glutamic Acid Decarboxylase Activity during Rat Brain Postnatal Development. Neurochem. Int. 2003, 42, 269–276. [Google Scholar] [CrossRef]
- Perry, T.L.; Hansen, S.; Christie, R.G. Amino Compounds and Organic Acids in CSF, Plasma, and Urine of Autistic Children. Biol. Psychiatry 1978, 13, 575–586. [Google Scholar]
- Torchiano, Marco Effsize—A Package for Efficient Effect Size Computation. 2016. Available online: https://zenodo.org/record/196082#.Y-3ThnYzZPY (accessed on 8 January 2023).
- Hladky, S.B.; Barrand, M.A. Mechanisms of Fluid Movement into, through and out of the Brain: Evaluation of the Evidence. Fluids Barriers CNS 2014, 11, 26. [Google Scholar] [CrossRef] [Green Version]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The Glutamate/GABA-Glutamine Cycle: Aspects of Transport, Neurotransmitter Homeostasis and Ammonia Transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Rose, S.; Niyazov, D.M.; Rossignol, D.A.; Goldenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and Molecular Characteristics of Mitochondrial Dysfunction in Autism Spectrum Disorder. Mol. Diagnosis Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zhou, Y.; Ma, W.; Wang, J. An Integrated Transcriptomic Analysis of Autism Spectrum Disorder. Sci. Rep. 2019, 9, 11818. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, M.R.; Rubin, R.A.; Falcone, T.; Ting, A.H.; Natowicz, M.R. Brain Transcriptional and Epigenetic Associations with Autism. PLoS ONE 2012, 7, e44736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, S.; Bennuri, S.C.; Wynne, R.; Melnyk, S.; James, S.J.; Frye, R.E. Mitochondrial and Redox Abnormalities in Autism Lymphoblastoid Cells: A Sibling Control Study. FASEB J. 2017, 31, 904–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, R.M. Regulation of Mitochondrial Dehydrogenases by Calcium Ions. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Li, Y. Genetic Associations between Voltage-Gated Calcium Channels and Autism Spectrum Disorder: A Systematic Review. Mol. Brain 2020, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Ni, Y.; Feng, F.; Seyfi, M.; Sadler, T.; Frazier, T.W.; Eng, C. Distinct Alterations in Tricarboxylic Acid Cycle Metabolites Associate with Cancer and Autism Phenotypes in Cowden Syndrome and Bannayan-Riley-Ruvalcaba Syndrome. Am. J. Hum. Genet. 2019, 105, 813–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orozco, J.S.; Hertz-Picciotto, I.; Abbeduto, L.; Slupsky, C.M. Metabolomics Analysis of Children with Autism, Idiopathic-Developmental Delays, and Down Syndrome. Transl. Psychiatry 2019, 9, 243. [Google Scholar] [CrossRef] [Green Version]
- Rangel-Huerta, O.D.; Gomez-Fernández, A.; de la Torre-Aguilar, M.J.; Gil, A.; Perez-Navero, J.L.; Flores-Rojas, K.; Martín-Borreguero, P.; Gil-Campos, M. Metabolic Profiling in Children with Autism Spectrum Disorder with and without Mental Regression: Preliminary Results from a Cross-Sectional Case–Control Study. Metabolomics 2019, 15, 99. [Google Scholar] [CrossRef]
- Mahalaxmi, I.; Subramaniam, M.D.; Gopalakrishnan, A.V.; Vellingiri, B. Dysfunction in Mitochondrial Electron Transport Chain Complex I, Pyruvate Dehydrogenase Activity, and Mutations in ND1 and ND4 Gene in Autism Spectrum Disorder Subjects from Tamil Nadu Population, India. Mol. Neurobiol. 2021, 58, 5303–5311. [Google Scholar] [CrossRef]
- Filipek, P.A.; Juranek, J.; Nguyen, M.T.; Cummings, C.; Gargus, J.J. Relative Carnitine Deficiency in Autism. J. Autism Dev. Disord. 2004, 34, 615–623. [Google Scholar] [CrossRef]
- Saleem, T.H.; Shehata, G.A.; Toghan, R.; Sakhr, H.M.; Bakri, A.H.; Desoky, T.; Hamdan, F.R.A.; Mohamed, N.F.; Hassan, M.H. Assessments of Amino Acids, Ammonia and Oxidative Stress Among Cohort of Egyptian Autistic Children: Correlations with Electroencephalogram and Disease Severity [Corrigendum]. Neuropsychiatr. Dis. Treat. 2020, 16, 325–326. [Google Scholar] [CrossRef] [Green Version]
- Shahjadi, S.; Khan, A.S.; Ahmed, M.U. Mitochondrial Dysfunction in Early Diagnosed Autism Spectrum Disorder Children. J. Dhaka Med. Coll. 2017, 26, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.; Gu, F.; Essa, M.M.; Wegiel, J.; Kaur, K.; Brown, W.T.; Chauhan, V. Brain Region-Specific Deficit in Mitochondrial Electron Transport Chain Complexes in Children with Autism. J. Neurochem. 2011, 117, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Gu, F.; Chauhan, V.; Kaur, K.; Brown, W.T.; LaFauci, G.; Wegiel, J.; Chauhan, A. Alterations in Mitochondrial DNA Copy Number and the Activities of Electron Transport Chain Complexes and Pyruvate Dehydrogenase in the Frontal Cortex from Subjects with Autism. Transl. Psychiatry 2013, 3, e299. [Google Scholar] [CrossRef] [Green Version]
- Napoli, E.; Wong, S.; Hertz-Picciotto, I.; Giulivi, C. Deficits in Bioenergetics and Impaired Immune Response in Granulocytes From Children With Autism. Pediatrics 2014, 133, e1405–e1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, R.E.; Lionnard, L.; Singh, I.; Karim, M.A.; Chajra, H.; Frechet, M.; Kissa, K.; Racine, V.; Ammanamanchi, A.; McCarty, P.J.; et al. Mitochondrial Morphology Is Associated with Respiratory Chain Uncoupling in Autism Spectrum Disorder. Transl. Psychiatry 2021, 11, 527. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; Frye, R.E.; James, S.J. Evidence of Oxidative Damage and Inflammation Associated with Low Glutathione Redox Status in the Autism Brain. Transl. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef] [Green Version]
- Meguid, N.A.; Dardir, A.A.; Abdel-Raouf, E.R.; Hashish, A. Evaluation of Oxidative Stress in Autism: Defective Antioxidant Enzymes and Increased Lipid Peroxidation. Biol. Trace Elem. Res. 2011, 143, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Yorbik, O.; Sayal, A.; Akay, C.; Akbiyik, D.I.; Sohmen, T. Investigation of Antioxidant Enzymes in Children with Autistic Disorder. Prostaglandins Leukot. Essent. Fat. Acids 2002, 67, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Chauhan, V.; Chauhan, A. Impaired Synthesis and Antioxidant Defense of Glutathione in the Cerebellum of Autistic Subjects: Alterations in the Activities and Protein Expression of Glutathione-Related Enzymes. Free Radic. Biol. Med. 2013, 65, 488–496. [Google Scholar] [CrossRef]
- Söğüt, S.; Zoroğlu, S.S.; Özyurt, H.; Ramazan Yılmaz, H.; Özuğurlu, F.; Sivaslı, E.; Yetkin, Ö.; Yanık, M.; Tutkun, H.; Savaş, H.A.; et al. Changes in Nitric Oxide Levels and Antioxidant Enzyme Activities May Have a Role in the Pathophysiological Mechanisms Involved in Autism. Clin. Chim. Acta 2003, 331, 111–117. [Google Scholar] [CrossRef]
- Kondolot, M.; Ozmert, E.N.; Ascı, A.; Erkekoglu, P.; Oztop, D.B.; Gumus, H.; Kocer-Gumusel, B.; Yurdakok, K. Plasma Phthalate and Bisphenol a Levels and Oxidant-Antioxidant Status in Autistic Children. Environ. Toxicol. Pharmacol. 2016, 43, 149–158. [Google Scholar] [CrossRef]
- Altun, H.; Şahin, N.; Kurutaş, E.B.; Karaaslan, U.; Sevgen, F.H.; Fındıklı, E. Assessment of Malondialdehyde Levels, Superoxide Dismutase, and Catalase Activity in Children with Autism Spectrum Disorders. Psychiatry Clin. Psychopharmacol. 2018, 28, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Al-Gadani, Y.; El-Ansary, A.; Attas, O.; Al-Ayadhi, L. Metabolic Biomarkers Related to Oxidative Stress and Antioxidant Status in Saudi Autistic Children. Clin. Biochem. 2009, 42, 1032–1040. [Google Scholar] [CrossRef]
- Zoroglu, S.S.; Armutcu, F.; Ozen, S.; Gurel, A.; Sivasli, E.; Yetkin, O.; Meram, I. Increased Oxidative Stress and Altered Activities of Erythrocyte Free Radical Scavenging Enzymes in Autism. Eur. Arch. Psychiatry Clin. Neurosci. 2004, 254, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Ghezzo, A.; Visconti, P.; Abruzzo, P.M.; Bolotta, A.; Ferreri, C.; Gobbi, G.; Malisardi, G.; Manfredini, S.; Marini, M.; Nanetti, L.; et al. Oxidative Stress and Erythrocyte Membrane Alterations in Children with Autism: Correlation with Clinical Features. PLoS ONE 2013, 8, e66418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjbar, A.; Rashedi, V.; Rezaei, M. Comparison of Urinary Oxidative Biomarkers in Iranian Children with Autism. Res. Dev. Disabil. 2014, 35, 2751–2755. [Google Scholar] [CrossRef]
- Young, A.M.H.; Chakrabarti, B.; Roberts, D.; Lai, M.-C.; Suckling, J.; Baron-Cohen, S. From Molecules to Neural Morphology: Understanding Neuroinflammation in Autism Spectrum Condition. Mol. Autism 2016, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Matta, S.M.; Hill-Yardin, E.L.; Crack, P.J. The Influence of Neuroinflammation in Autism Spectrum Disorder. Brain. Behav. Immun. 2019, 79, 75–90. [Google Scholar] [CrossRef]
- Shirayama, Y.; Matsumoto, K.; Osone, F.; Hara, A.; Guan, S.; Hamatani, S.; Muneoka, K.; Sato, K.; Okada, A.; Yokokawa, T. The Lack of Alterations in Metabolites in the Medial Prefrontal Cortex and Amygdala, but Their Associations with Autistic Traits, Empathy, and Personality Traits in Adults with Autism Spectrum Disorder: A Preliminary Study. J. Autism Dev. Disord. 2022. [Google Scholar] [CrossRef]
- Hayek, J.; Cervellati, C.; Crivellari, I.; Pecorelli, A.; Valacchi, G. Lactonase Activity and Lipoprotein-Phospholipase A 2 as Possible Novel Serum Biomarkers for the Differential Diagnosis of Autism Spectrum Disorders and Rett Syndrome: Results from a Pilot Study. Oxid. Med. Cell. Longev. 2017, 2017, 5694058. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.G.; MacKinlay, E.E.; Dick, J.R.; MacDonald, D.J.; Boyle, R.M.; Glen, A.C.A. Essential Fatty Acids and Phospholipase A2 in Autistic Spectrum Disorders. Prostaglandins Leukot. Essent. Fat. Acids 2004, 71, 201–204. [Google Scholar] [CrossRef]
- Tostes, M.H.F.d.S.; Polonini, H.C.; Mendes, R.; Brandão, M.A.F.; Gattaz, W.F.; Raposo, N.R.B. Fatty Acid and Phospholipase A2 Plasma Levels in Children with Autism. Trends Psychiatry Psychother. 2013, 35, 76–80. [Google Scholar] [CrossRef] [Green Version]
- Fourrier, C.; Remus-Borel, J.; Greenhalgh, A.D.; Guichardant, M.; Bernoud-Hubac, N.; Lagarde, M.; Joffre, C.; Layé, S. Docosahexaenoic Acid-Containing Choline Phospholipid Modulates LPS-Induced Neuroinflammation in Vivo and in Microglia in Vitro. J. Neuroinflamm. 2017, 14, 170. [Google Scholar] [CrossRef] [Green Version]
- Orr, S.K.; Palumbo, S.; Bosetti, F.; Mount, H.T.; Kang, J.X.; Greenwood, C.E.; Ma, D.W.L.; Serhan, C.N.; Bazinet, R.P. Unesterified Docosahexaenoic Acid Is Protective in Neuroinflammation. J. Neurochem. 2013, 127, 378–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesei, A.; Crippa, A.; Ceccarelli, S.B.; Mauri, M.; Molteni, M.; Agostoni, C.; Nobile, M. The Potential Relevance of Docosahexaenoic Acid and Eicosapentaenoic Acid to the Etiopathogenesis of Childhood Neuropsychiatric Disorders. Eur. Child Adolesc. Psychiatry 2017, 26, 1011–1030. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and Inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Sreeramkumar, V.; Fresno, M.; Cuesta, N. Prostaglandin E2 and T Cells: Friends or Foes? Immunol. Cell Biol. 2012, 90, 579–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gezginci-Oktayoglu, S.; Orhan, N.; Bolkent, S. Prostaglandin-E 1 Has a Protective Effect on Renal Ischemia/Reperfusion-Induced Oxidative Stress and Inflammation Mediated Gastric Damage in Rats. Int. Immunopharmacol. 2016, 36, 142–150. [Google Scholar] [CrossRef]
- Fang, W.; Li, H.; Zhou, L.; Su, L.; Liang, Y.; Mu, Y. Effect of Prostaglandin E1 on TNF-Induced Vascular Inflammation in Human Umbilical Vein Endothelial Cells. Can. J. Physiol. Pharmacol. 2010, 88, 576–583. [Google Scholar] [CrossRef]
- Di Gennaro, A.; Haeggström, J.Z. Targeting Leukotriene B4 in Inflammation. Expert Opin. Ther. Targets 2014, 18, 79–93. [Google Scholar] [CrossRef]
- Qasem, H.; Al-Ayadhi, L.; Bjørklund, G.; Chirumbolo, S.; El-Ansary, A. Impaired Lipid Metabolism Markers to Assess the Risk of Neuroinflammation in Autism Spectrum Disorder. Metab. Brain Dis. 2018, 33, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- El-Ansary, A.; Al-Ayadhi, L. Lipid Mediators in Plasma of Autism Spectrum Disorders. Lipids Health Dis. 2012, 11, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigandi, S.; Shao, H.; Qian, S.; Shen, Y.; Wu, B.-L.; Kang, J. Autistic Children Exhibit Decreased Levels of Essential Fatty Acids in Red Blood Cells. Int. J. Mol. Sci. 2015, 16, 10061–10076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qasem, H.; Al-Ayadhi, L.; El-Ansary, A. Cysteinyl Leukotriene Correlated with 8-Isoprostane Levels as Predictive Biomarkers for Sensory Dysfunction in Autism. Lipids Health Dis. 2016, 15, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartke, N.; Hannun, Y.A. Bioactive Sphingolipids: Metabolism and Function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and Their Metabolism in Physiology and Disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Wang, H.; Liang, S.; Wang, M.; Gao, J.; Sun, C.; Wang, J.; Xia, W.; Wu, S.; Sumner, S.J.; Zhang, F.; et al. Potential Serum Biomarkers from a Metabolomics Study of Autism. J. Psychiatry Neurosci. 2016, 41, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Pardo, C.A.; Wheeler, D.; Vargas, D.L.; Haughey, N.J.; Zimmermann, A. Abnormalities in Cholesterol, Ceramides and Markers of Oxidative Stress Are Revealed by Lipidomic Analysis of Brain Tissues in Autism. In Proceedings of the International Meeting for Autism Research, London, UK, 15–17 May 2008. [Google Scholar]
- Yu, Q.; He, Z.; Zubkov, D.; Huang, S.; Kurochkin, I.; Yang, X.; Halene, T.; Willmitzer, L.; Giavalisco, P.; Akbarian, S.; et al. Lipidome Alterations in Human Prefrontal Cortex during Development, Aging, and Cognitive Disorders. Mol. Psychiatry 2020, 25, 2952–2969. [Google Scholar] [CrossRef] [Green Version]
- Arana, L.; Gangoiti, P.; Ouro, A.; Trueba, M.; Gómez-Muñoz, A. Ceramide and Ceramide 1-Phosphate in Health and Disease. Lipids Health Dis. 2010, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Snider, A.J.; Alexa Orr Gandy, K.; Obeid, L.M. Sphingosine Kinase: Role in Regulation of Bioactive Sphingolipid Mediators in Inflammation. Biochimie 2010, 92, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.E.; Srinivasan, S.; Lynch, K.R.; Proia, R.L.; Ferdek, P.; Hedrick, C.C. Sphingosine-1-Phosphate Induces an Antiinflammatory Phenotype in Macrophages. Circ. Res. 2008, 102, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Brodowicz, J.; Przegaliński, E.; Müller, C.P.; Filip, M. Ceramide and Its Related Neurochemical Networks as Targets for Some Brain Disorder Therapies. Neurotox. Res. 2018, 33, 474–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.S.; Sampson, E.L. Lipid Composition of the Normal Human Brain: Gray Matter, White Matter, and Myelin. J. Lipid Res. 1965, 6, 537–544. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ASD-Specific Markers Found in | Pathways/Metabolites | Main Findings (ASD/Control) | |
|---|---|---|---|
| Energy Metabolism | Glycolysis | ||
| TCA cycle | |||
| ETC I, II, V | |||
| ETC III, IV | |||
| Oxidative Stress | Ammonia, lactate, pyruvate | ||
| CO2/Glucose | |||
| Mitochondrial Dysfunction | ALT, AST, GSR | ||
| CS, PDH | |||
| Neuroinflammation | Pro-inflammatory | Leukotriene, PGE2 | |
| PLA2 | |||
| Anti-inflammatory | Carnitine, choline | ||
| DHA, EPA, inositol, PGE1 | |||
| Sphingolipid Metabolism | Glucosylceramide, S1P | ||
| Ceramide | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esvap, E.; Ulgen, K.O. Neuroinflammation, Energy and Sphingolipid Metabolism Biomarkers Are Revealed by Metabolic Modeling of Autistic Brains. Biomedicines 2023, 11, 583. https://doi.org/10.3390/biomedicines11020583
Esvap E, Ulgen KO. Neuroinflammation, Energy and Sphingolipid Metabolism Biomarkers Are Revealed by Metabolic Modeling of Autistic Brains. Biomedicines. 2023; 11(2):583. https://doi.org/10.3390/biomedicines11020583
Chicago/Turabian StyleEsvap, Elif, and Kutlu O. Ulgen. 2023. "Neuroinflammation, Energy and Sphingolipid Metabolism Biomarkers Are Revealed by Metabolic Modeling of Autistic Brains" Biomedicines 11, no. 2: 583. https://doi.org/10.3390/biomedicines11020583