
Exploring the Phospholipid Transport Mechanism of ATP8A1-CDC50

{kind=link}
{kind=link}
{kind=link}
{kind=link}
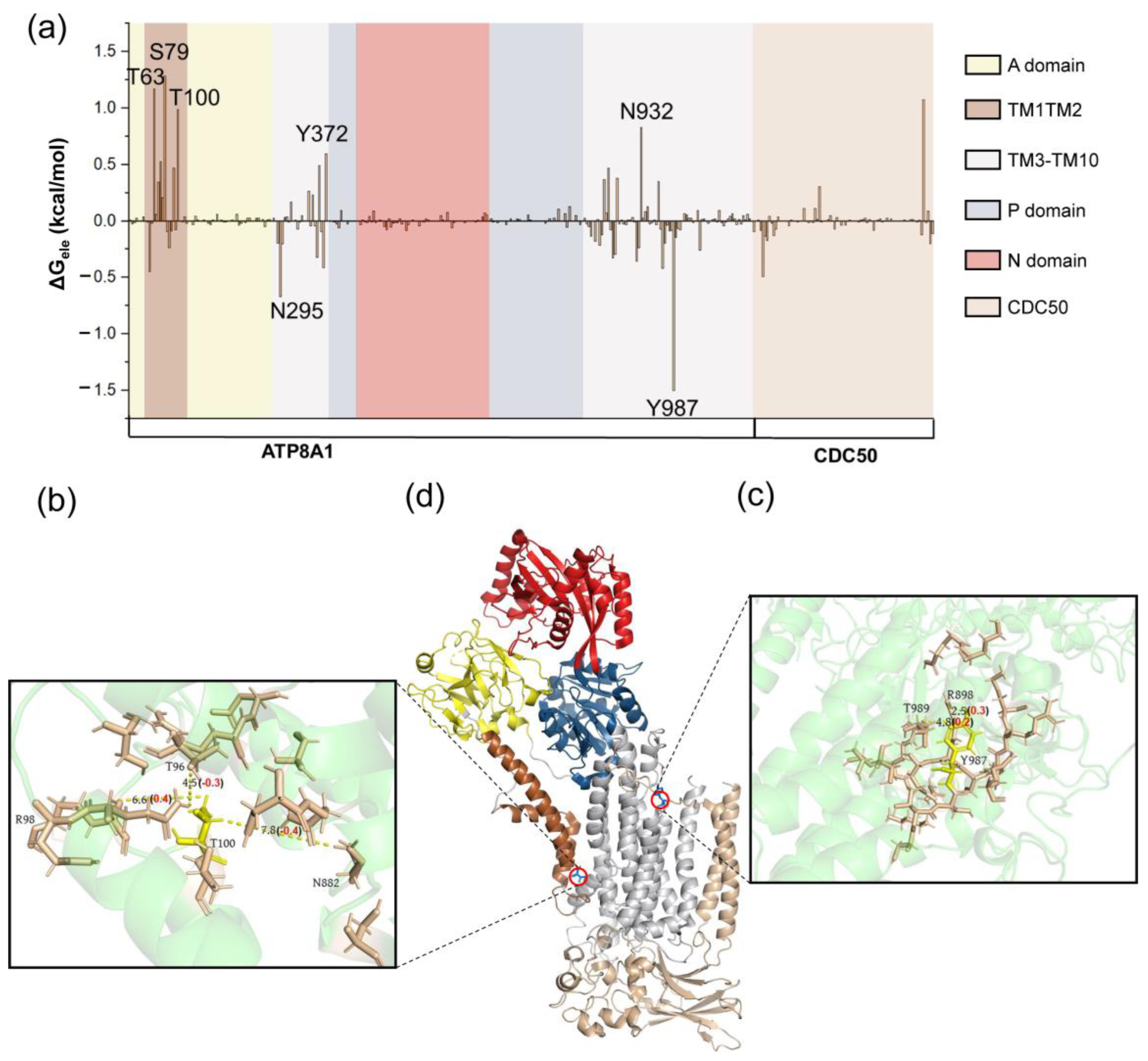{kind=link}
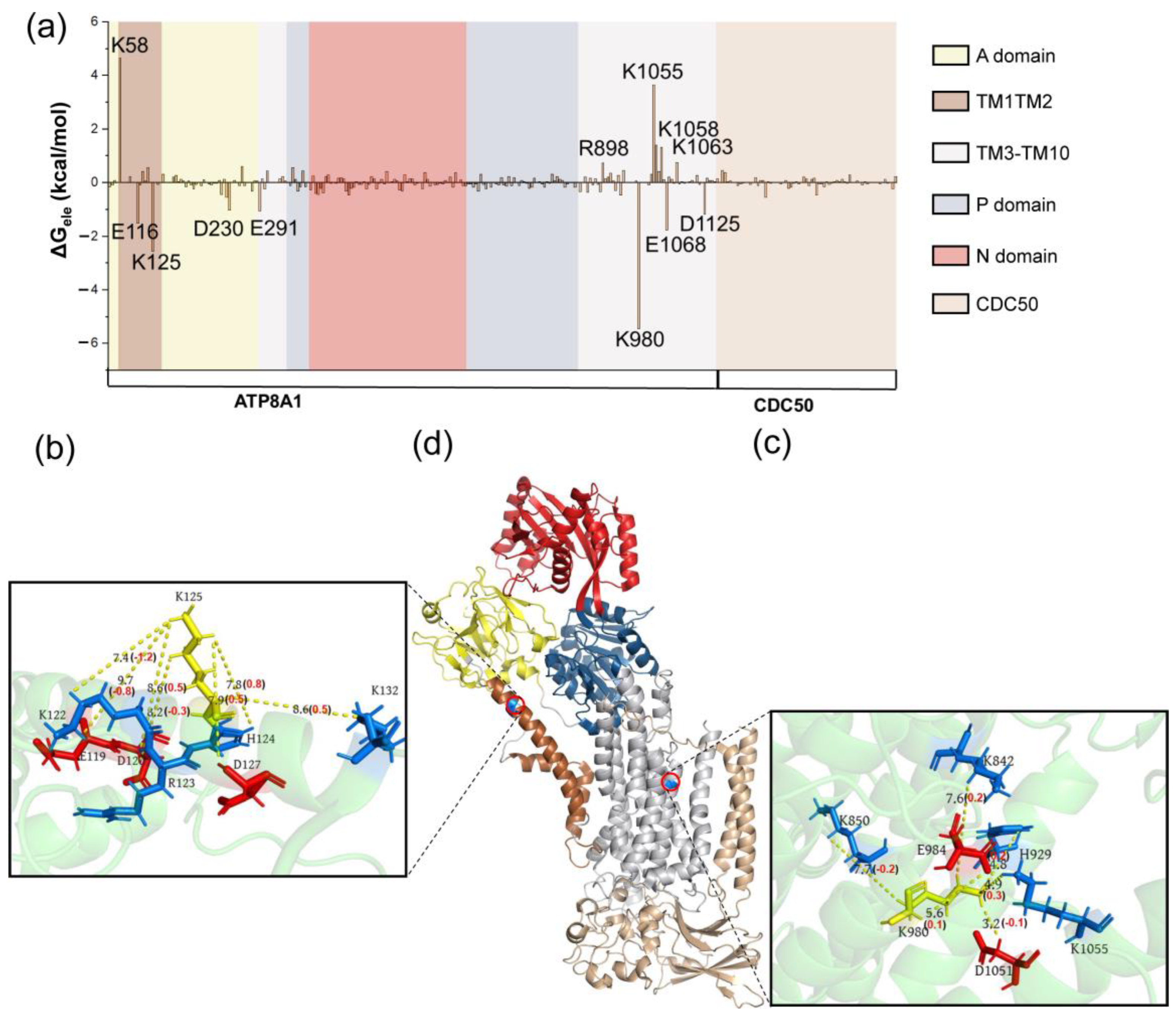{kind=link}
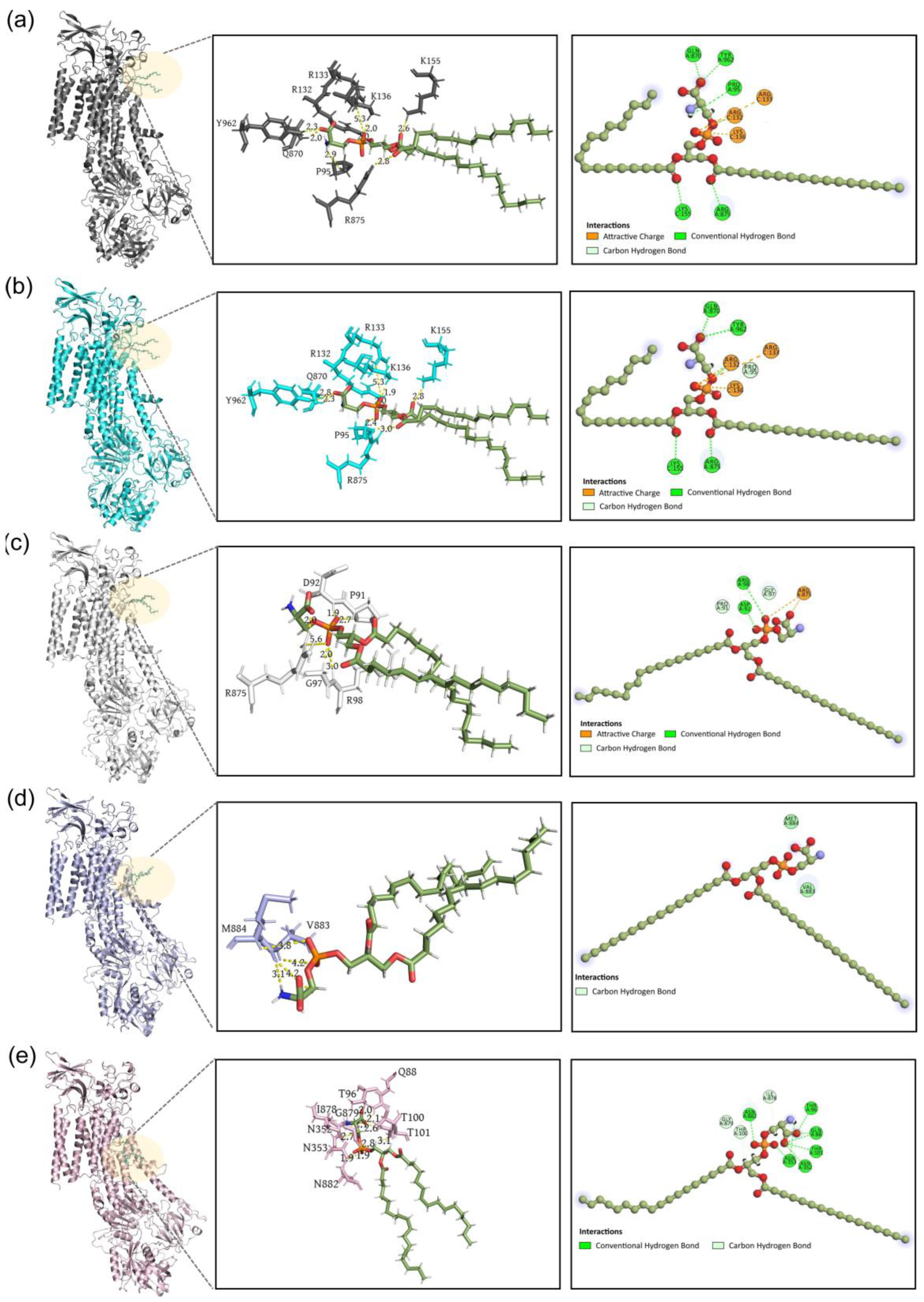{kind=link}
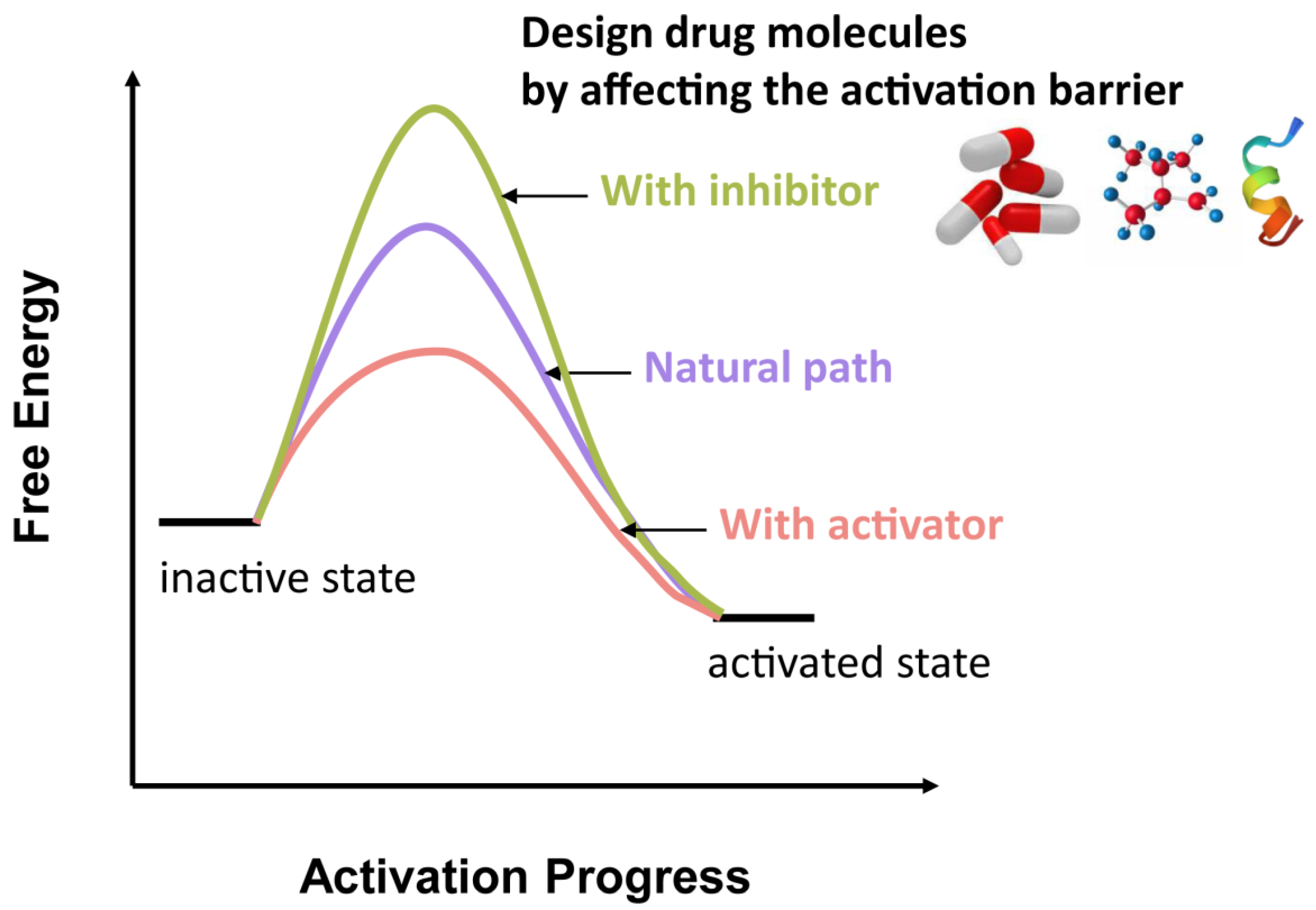{kind=link}
Abstract
:1. Introduction
2. Method
2.1. Coarse-Grained Model
2.2. PDLD/S-LRA/β
2.3. Targeted Molecular Simulations (TMD)
3. Results
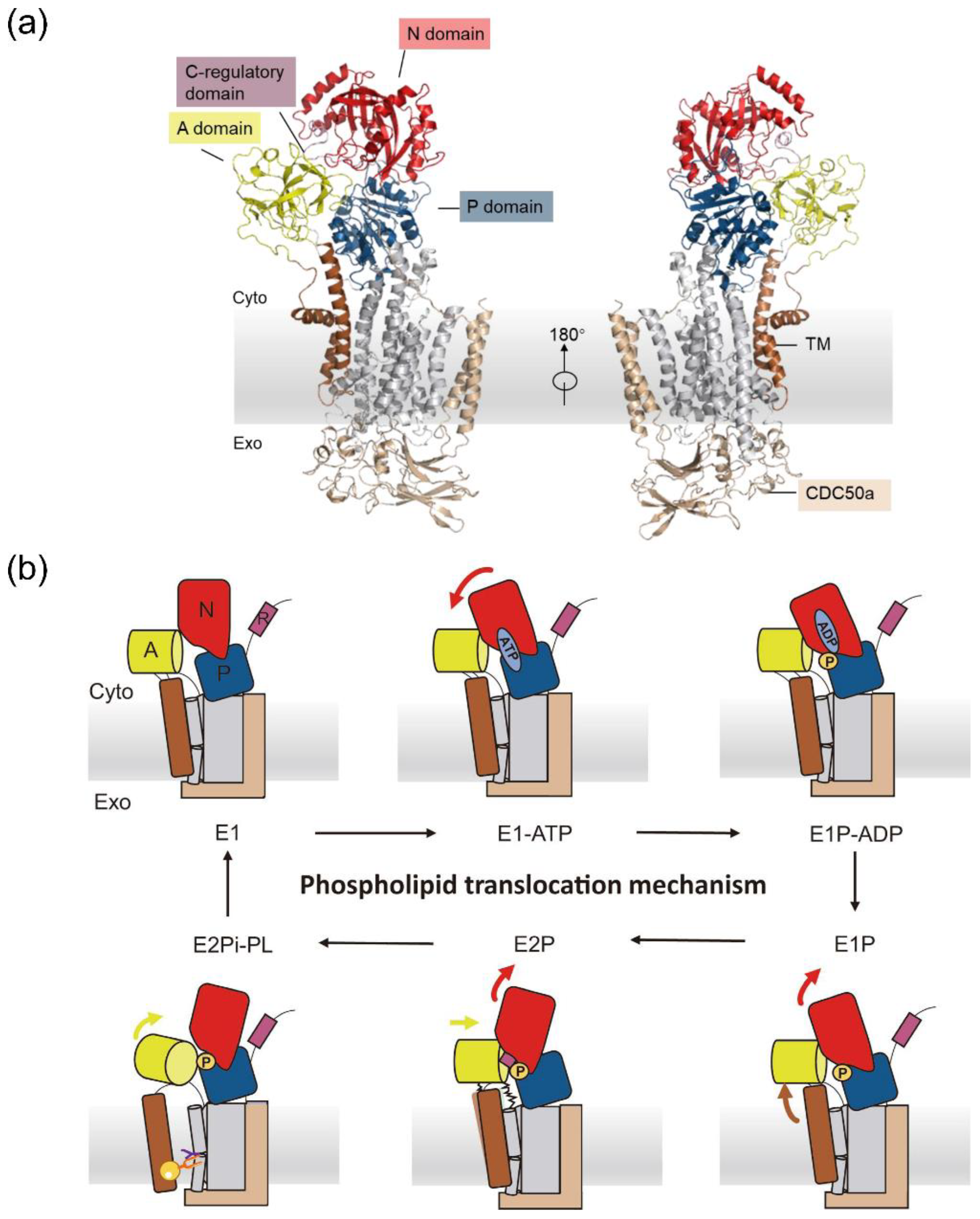
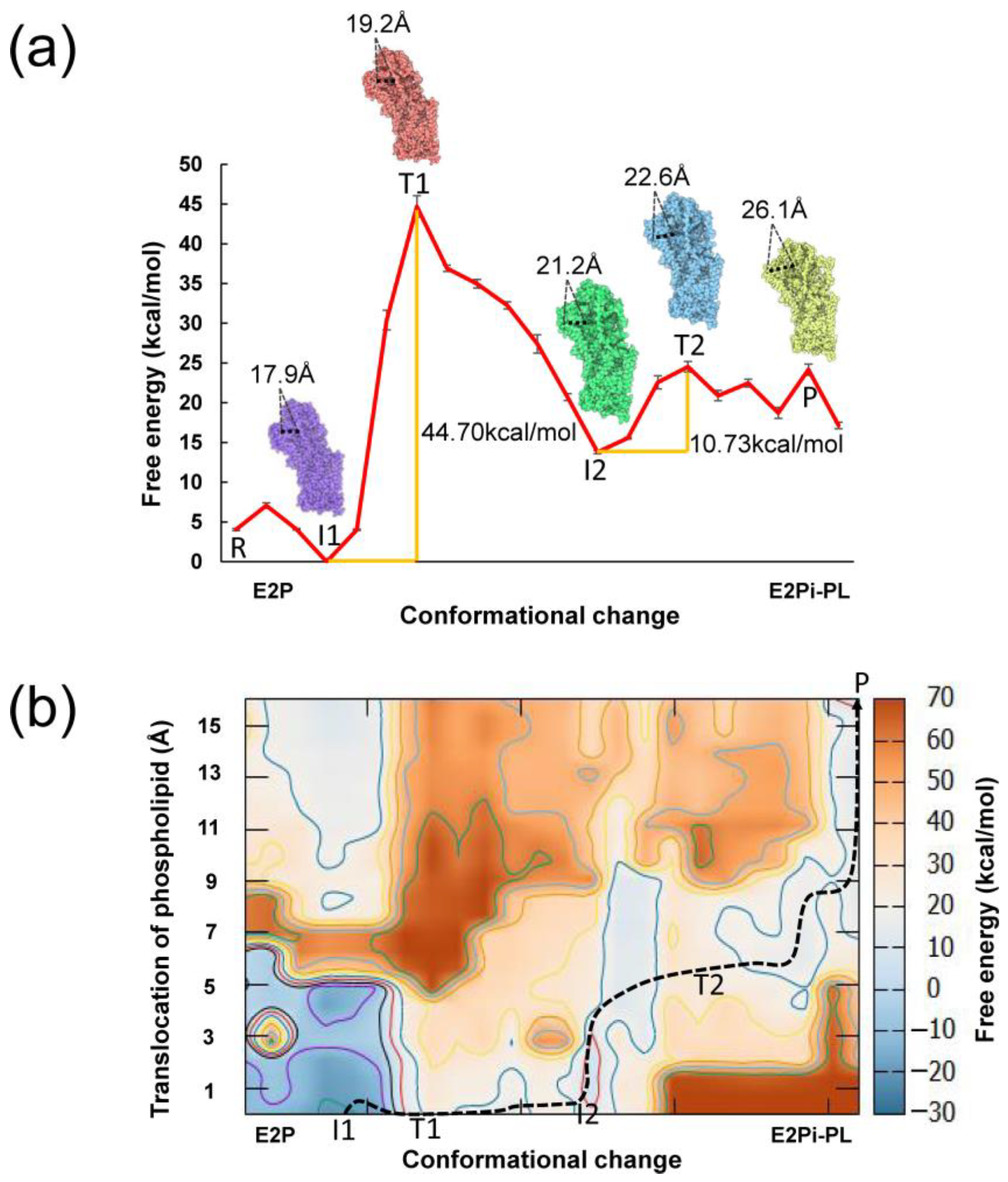
3.1. Coarse-Grained Free Energy Profile for Conformational Changes
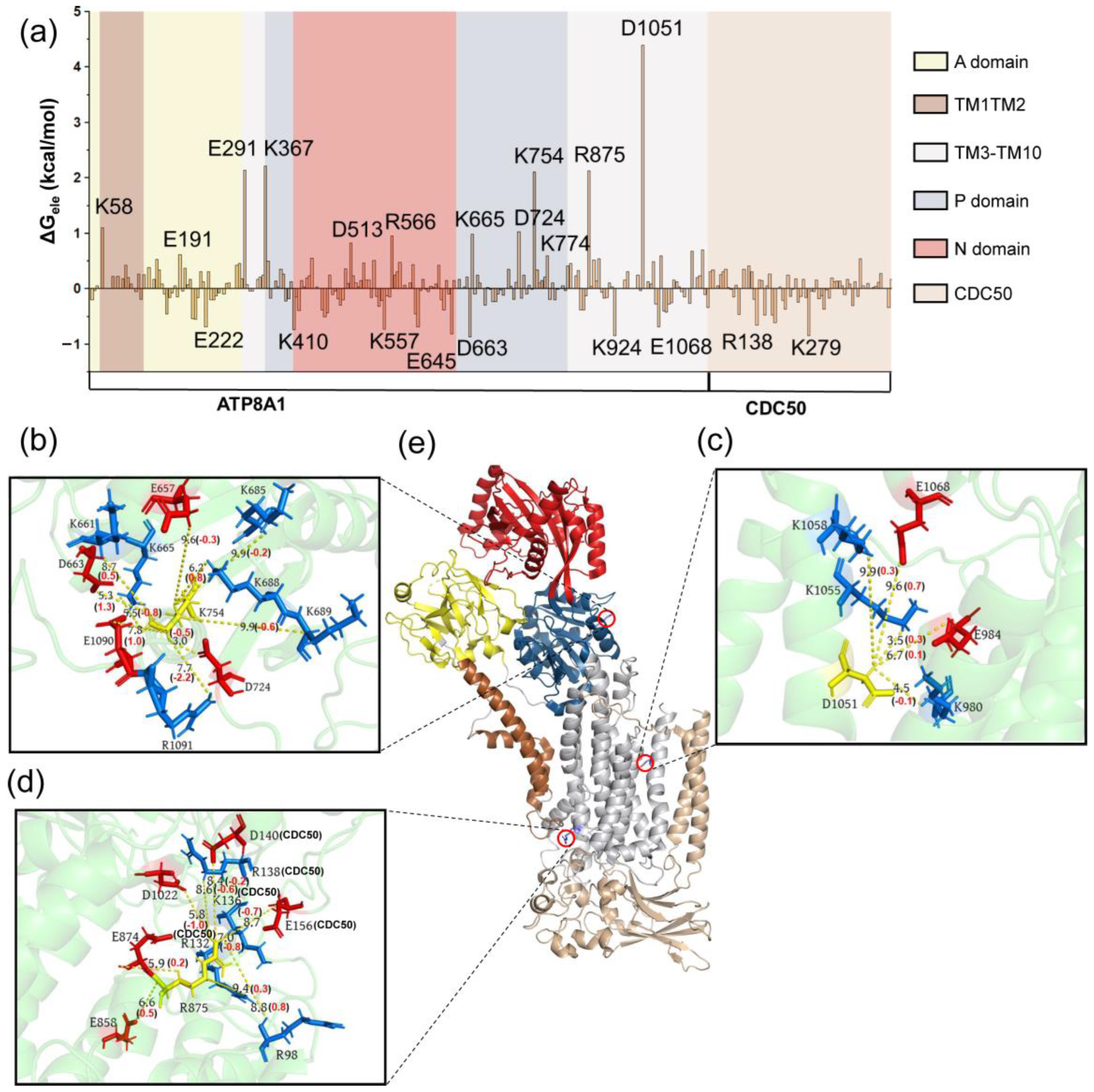
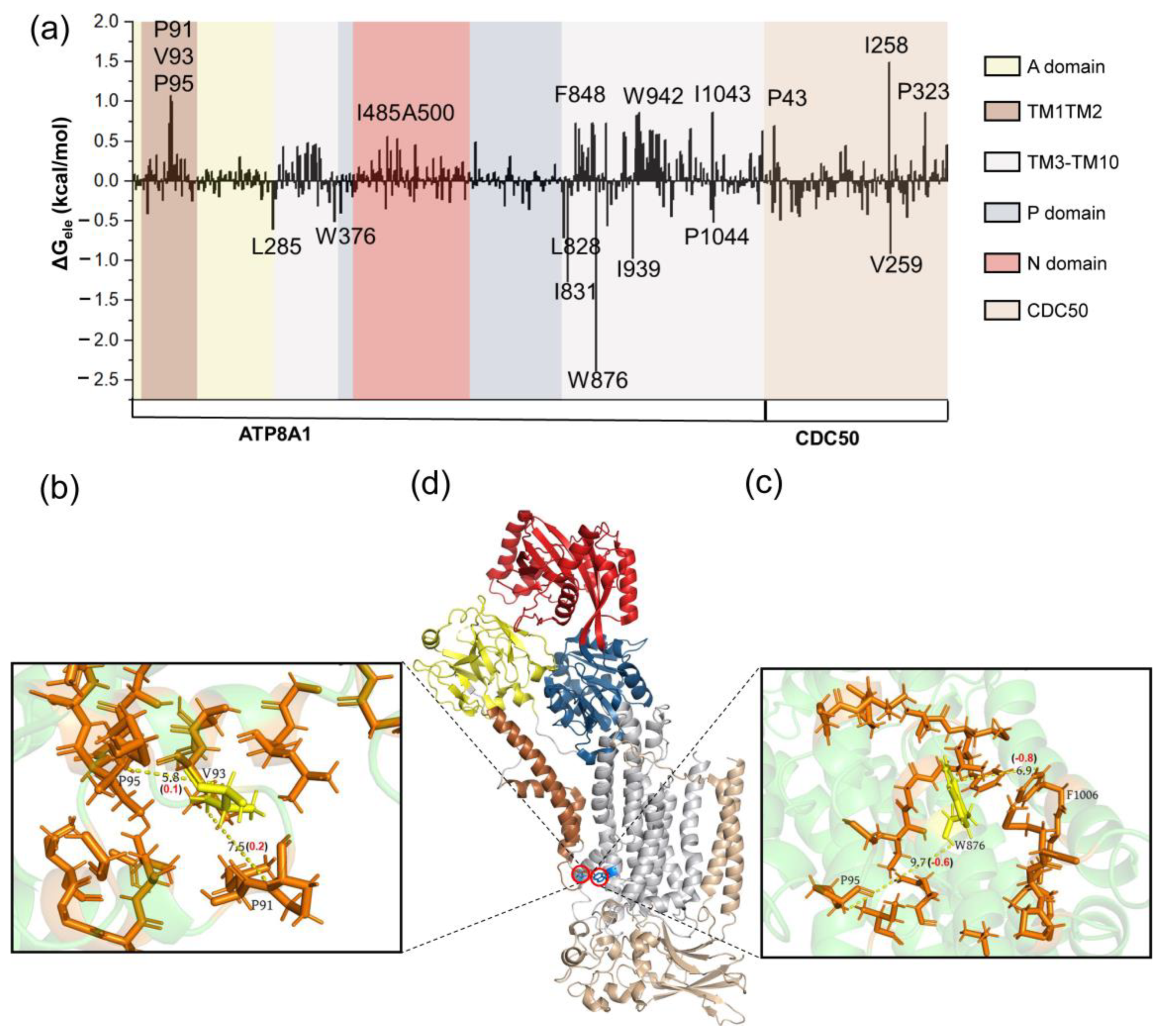
3.2. Conformational Changes Coupled with the Phospholipid Transport
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doktorova, M.; Symons, J.L.; Levental, I. Structural and functional consequences of reversible lipid asymmetry in living membranes. Nat. Chem. Biol. 2020, 16, 1321–1330. [Google Scholar] [CrossRef]
- Nintemann, S.J.; Palmgren, M.; López-Marqués, R.L. Catch You on the Flip Side: A Critical Review of Flippase Mutant Phenotypes. Trends Plant Sci. 2019, 24, 468–478. [Google Scholar] [CrossRef]
- Lopez-Marques, R.L.; Poulsen, L.R.; Bailly, A.; Geisler, M.; Pomorski, T.G.; Palmgren, M.G. Structure and mechanism of ATP-dependent phospholipid transporters. Biochim. Biophys. Acta 2015, 1850, 461–475. [Google Scholar] [CrossRef]
- Muthusamy, B.P.; Natarajan, P.; Zhou, X.; Graham, T.R. Linking phospholipid flippases to vesicle-mediated protein transport. Biochim. Biophys. Acta 2009, 1791, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Leventis, P.A.; Grinstein, S. The Distribution and Function of Phosphatidylserine in Cellular Membranes. Annu. Rev. Biophys. 2010, 39, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.J.; Hossain, K.R.; Cao, K. Physiological roles of transverse lipid asymmetry of animal membranes. Biochim. Et Biophys. Acta-Biomembr. 2020, 1862, 183382. [Google Scholar] [CrossRef]
- Kerr, D.J.; Guariglia, S.R.; Marsillo, A.; Kascsak, R.; Wen, G.Y.; Chauhan, A.; Banerjee, P. The role of Atp8a1 in neurotransmission, brain development, and autistic behavior. J. Neurochem. 2013, 125, 262. [Google Scholar]
- Mark, V.; Elferink, R.; Paulusma, C.C. P4 ATPases: Flippases in Health and Disease. Int. J. Mol. Sci. 2013, 14, 7897–7922. [Google Scholar] [CrossRef]
- Takada, N.; Naito, T.; Inoue, T.; Nakayama, K.; Takatsu, H.; Shin, H.W. Phospholipid-flipping activity of P4-ATPase drives membrane curvature. EMBO J. 2018, 37, e97705. [Google Scholar] [CrossRef]
- Sebastian, T.T.; Baldridge, R.D.; Xu, P.; Graham, T.R. Phospholipid flippases: Building asymmetric membranes and transport vesicles. Biochim. Biophys. Acta 2012, 1821, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, A.L.; Coleman, J.A.; Lemmin, T.; Mikkelsen, S.A.; Molday, L.L.; Vilsen, B.; Molday, R.S.; Dal Peraro, M.; Andersen, J.P. Critical roles of isoleucine-364 and adjacent residues in a hydrophobic gate control of phospholipid transport by the mammalian P4-ATPase ATP8A2. Proc. Natl. Acad. Sci. USA 2014, 111, E1334–E1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.T.; Chen, Y.; Chen, Z.P.; Liu, X.; Zhang, Z.; Chen, Y.; Hou, W.T.; Zhou, C.Z. Structural insights into the activation of autoinhibited human lipid flippase ATP8B1 upon substrate binding. Proc. Natl. Acad. Sci. USA 2022, 119, e2118656119. [Google Scholar] [CrossRef] [PubMed]
- Timcenko, M.; Dieudonné, T.; Montigny, C.; Boesen, T.; Lyons, J.; Lenoir, G.; Nissen, P. Structural Basis of Substrate-Independent Phosphorylation in a P4-ATPase Lipid Flippase. J. Mol. Biol. 2021, 433, 167062. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Nishizawa, T.; Segawa, K.; Nureki, O.; Fujiyoshi, Y.; Nagata, S.; Abe, K. Transport Cycle of Plasma Membrane Flippase ATP11C by Cryo-EM. Cell Rep. 2020, 32, 108208. [Google Scholar] [CrossRef]
- Hiraizumi, M.; Yamashita, K.; Nishizawa, T.; Nureki, O. Cryo-EM structures capture the transport cycle of the P4-ATPase flippase. Science 2019, 365, 1149–1155. [Google Scholar] [CrossRef]
- Albers, R.W. Biochemical Aspects of Active Transport. Annu. Rev. Biochem. 1967, 36, 727–756. [Google Scholar] [CrossRef]
- Olesen, C.; Picard, M.; Winther, A.-M.L.; Gyrup, C.; Morth, J.P.; Oxvig, C.; Møller, J.V.; Nissen, P. The structural basis of calcium transport by the calcium pump. Nature 2007, 450, 1036–1042. [Google Scholar] [CrossRef]
- Vicatos, S.; Rychkova, A.; Mukherjee, S.; Warshel, A. An effective Coarse-grained model for biological simulations: Recent refinements and validations. Proteins 2014, 82, 1168–1185. [Google Scholar] [CrossRef]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef]
- Baaden, M.; Marrink, S.J. Coarse-grain modelling of protein-protein interactions. Curr. Opin. Struct. Biol. 2013, 23, 878–886. [Google Scholar] [CrossRef]
- Messer, B.M.; Roca, M.; Chu, Z.T.; Vicatos, S.; Kilshtain, A.V.; Warshel, A. Multiscale simulations of protein landscapes: Using coarse-grained models as reference potentials to full explicit models. Proteins Struct. Funct. Bioinform. 2010, 78, 1212–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamerlin, S.C.L.; Vicatos, S.; Dryga, A.; Warshel, A. Coarse-Grained (Multiscale) Simulations in Studies of Biophysical and Chemical Systems. Annu. Rev. Phys. Chem. 2011, 62, 41–64. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Warshel, A. Revisiting the protomotive vectorial motion of F0-ATPase. Proc. Natl. Acad. Sci. USA 2019, 116, 19484–19489. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Wang, J.; Mondal, D.; Du, Y.; Ye, R.; Warshel, A. Exploring the Activation Process of the β2AR-G(s) Complex. J. Am. Chem. Soc. 2021, 143, 11044–11051. [Google Scholar] [CrossRef] [PubMed]
- Alhadeff, R.; Warshel, A. Reexamining the origin of the directionality of myosin V. Proc. Natl. Acad. Sci. USA 2017, 114, 10426–10431. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Schlitter, J.; Engels, M.; Krüger, P. Targeted molecular dynamics: A new approach for searching pathways of conformational transitions. J. Mol. Graphics 1994, 12, 84–89. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- El Malki, Z.; Bouzzine, S.M.; Bejjit, L.; Haddad, M.; Hamidi, M.; Bouachrine, M. Density functional theory [B3LYP/6-311G(d,p)] study of a new copolymer based on carbazole and (3,4-Ethylenedioxythiophene) in their aromatic and polaronic states. J. Appl. Polym. Sci. 2011, 122, 3351–3360. [Google Scholar] [CrossRef]
- Dryga, A.; Chakrabarty, S.; Vicatos, S.; Warshel, A. Coarse grained model for exploring voltage dependent ion channels. Biochim. Et Biophys. Acta 2012, 1818, 303–317. [Google Scholar] [CrossRef]
- Lee, F.S.; Chu, Z.T.; Warshel, A. Microscopic and semimicroscopic calculations of electrostatic energies in proteins by the POLARIS and ENZYMIX programs. J. Comput. Chem. 1993, 14, 161–185. [Google Scholar] [CrossRef]
- Vorobyov, I.; Kim, I.; Chu, Z.T.; Warshel, A. Refining the treatment of membrane proteins by coarse-grained models. Proteins 2016, 84, 92–117. [Google Scholar] [CrossRef] [PubMed]
- Beroza, P.; Fredkin, D.R.; Okamura, M.Y.; Feher, G. Protonation of interacting residues in a protein by a Monte Carlo method: Application to lysozyme and the photosynthetic reaction center of Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA 1991, 88, 5804–5808. [Google Scholar] [CrossRef]
- Fan, Z.; Hwang, J.-K.; Warshel, A.J.T.C.A. Using simplified protein representation as a reference potential for all-atom calculations of folding free energy. Theor. Chem. Acc. 1999, 103, 77–80. [Google Scholar] [CrossRef]
- Warshel, A.; Russell, S.T. Calculations of electrostatic interactions in biological systems and in solutions. Q. Rev. Biophys. 1984, 17, 283–422. [Google Scholar] [CrossRef]
- Kim, I.; Chakrabarty, S.; Brzezinski, P.; Warshel, A. Modeling gating charge and voltage changes in response to charge separation in membrane proteins. Proc. Natl. Acad. Sci. USA 2014, 111, 11353–11358. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Warshel, A. Absolute binding free energy calculations: On the accuracy of computational scoring of protein-ligand interactions. Proteins 2010, 78, 1705–1723. [Google Scholar] [CrossRef]
- Lee, F.S.; Chu, Z.-T.; Bolger, M.B.; Warshel, A.J.P.E. Calculations of antibody-antigen interactions: Microscopic and semi-microscopic evaluation of the free energies of binding of phosphorylcholine analogs to McPC603. Protein Eng. Des. Sel. 1992, 5, 215–228. [Google Scholar] [CrossRef]
- Xiao, X.; Zeng, X.; Yuan, Y.; Gao, N.; Guo, Y.; Pu, X.; Li, M. Understanding the conformation transition in the activation pathway of β2 adrenergic receptor via a targeted molecular dynamics simulation. Phys. Chem. Chem. Phys. 2014, 17, 2512–2522. [Google Scholar] [CrossRef]
- Ovchinnikov, V.; Karplus, M. Analysis and elimination of a bias in targeted molecular dynamics simulations of conformational transitions: Application to calmodulin. J. Phys. Chem. B 2012, 116, 8584–8603. [Google Scholar] [CrossRef]
- Fiorin, G.; Klein, M.L.; Hénin, J. Using collective variables to drive molecular dynamics simulations. Mol. Phys. 2013, 111, 3345–3362. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- Ristovski, M.; Farhat, D.; Bancud, S.E.M.; Lee, J.-Y. Lipid Transporters Beam Signals from Cell Membranes. Membranes 2021, 11, 562. [Google Scholar] [CrossRef]
- López-Marqués, R.L.; Davis, J.A.; Harper, J.F.; Palmgren, M. Dynamic membranes: The multiple roles of P4 and P5 ATPases. Plant Physiol. 2021, 185, 619–631. [Google Scholar] [CrossRef]
- Folmer, D.E.; Elferink, R.P.O.; Paulusma, C.C.J.B.e.B.A.-M.; Lipids, C.B.O. P4 ATPases-lipid flippases and their role in disease. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2009, 1791, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Kanai, R.; Cornelius, F.; Ogawa, H.; Motoyama, K.; Vilsen, B.; Toyoshima, C. Binding of cardiotonic steroids to Na(+),K(+)-ATPase in the E2P state. Proc. Natl. Acad. Sci. USA 2021, 118, e2020438118. [Google Scholar] [CrossRef] [PubMed]
- Gadsby, D.C.; Bezanilla, F.; Rakowski, R.F.; De Weer, P.; Holmgren, M. The dynamic relationships between the three events that release individual Na(+) ions from the Na(+)/K(+)-ATPase. Nat. Commun. 2012, 3, 669. [Google Scholar] [CrossRef]
- Saffioti, N.A.; de Sautu, M.; Riesco, A.S.; Ferreira-Gomes, M.S.; Rossi, J.; Mangialavori, I.C. Conformational changes during the reaction cycle of plasma membrane Ca2+-ATPase in the autoinhibited and activated states. Biochem. J. 2021, 478, 2019–2034. [Google Scholar] [CrossRef]
- Dyla, M.; Terry, D.S.; Kjaergaard, M.; Sørensen, T.L.M.; Lauwring Andersen, J.; Andersen, J.P.; Rohde Knudsen, C.; Altman, R.B.; Nissen, P.; Blanchard, S.C. Dynamics of P-type ATPase transport revealed by single-molecule FRET. Nature 2017, 551, 346–351. [Google Scholar] [CrossRef]
- Xu, J.; He, Y.; Wu, X.; Li, L. Conformational changes of a phosphatidylcholine flippase in lipid membranes. Cell Rep. 2022, 38, 110518. [Google Scholar] [CrossRef]
- Dieudonne, T.; Herrera, S.A.; Laursen, M.J.; Lejeune, M.; Stock, C.; Slimani, K.; Jaxel, C.; Lyons, J.A.; Montigny, C.; Pomorski, T.G.; et al. Autoinhibition and regulation by phosphoinositides of ATP8B1, a human lipid flippase associated with intrahepatic cholestatic disorders. Elife 2022, 11, e75272. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Asadi, M.; Warshel, A. The catalytic dwell in ATPases is not crucial for movement against applied torque. Nat. Chem. 2020, 12, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Ueno, H.; Shaikh, A.R.; Umemura, M.; Kamiya, M.; Ito, Y.; Ikeguchi, M.; Komoriya, Y.; Iino, R.; Noji, H. Molecular Mechanism of ATP Hydrolysis in F1-ATPase Revealed by Molecular Simulations and Single-Molecule Observations. J. Am. Chem. Soc. 2012, 134, 8447–8454. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ojeda-May, P.; Nagaraju, M.; Pu, J. Toward Determining ATPase Mechanism in ABC Transporters: Development of the Reaction Path-Force Matching QM/MM Method. Methods Enzymol. 2016, 577, 185–212. [Google Scholar] [PubMed]
- Van Der Velden, L.M.; Van De Graaf, S.F.; Klomp, L.W. Biochemical and cellular functions of P4 ATPases. Biochem. J. 2010, 431, 1–11. [Google Scholar] [CrossRef]
- Theorin, L.; Faxén, K.; Sørensen, D.M.; Migotti, R.; Dittmar, G.; Schiller, J.; Daleke, D.L.; Palmgren, M.; López-Marqués, R.L.; Günther Pomorski, T. The lipid head group is the key element for substrate recognition by the P4 ATPase ALA2: A phosphatidylserine flippase. Biochem. J. 2019, 476, 783–794. [Google Scholar] [CrossRef]
- Timcenko, M.; Lyons, J.A.; Januliene, D.; Ulstrup, J.J.; Dieudonné, T.; Montigny, C.; Ash, M.-R.; Karlsen, J.L.; Boesen, T.; Kühlbrandt, W.; et al. Structure and autoregulation of a P4-ATPase lipid flippase. Nature 2019, 571, 366–370. [Google Scholar] [CrossRef]
- Bai, L.; You, Q.; Jain, B.K.; Duan, H.D.; Kovach, A.; Graham, T.R.; Li, H. Transport mechanism of P4 ATPase phosphatidylcholine flippases. eLife 2020, 9, e62163. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, M.; Kádková, A.; Scheutzow, A.; Malsam, J.; Söllner, T.H.; Sørensen, J.B. An Electrostatic Energy Barrier for SNARE-Dependent Spontaneous and Evoked Synaptic Transmission. Cell Rep. 2019, 26, 2340–2352.e2345. [Google Scholar] [CrossRef]
- Nakanishi, H.; Irie, K.; Segawa, K.; Hasegawa, K.; Fujiyoshi, Y.; Nagata, S.; Abe, K. Crystal structure of a human plasma membrane phospholipid flippase. J. Biol. Chem. 2020, 295, 10180–10194. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.A.; Vestergaard, A.L.; Molday, R.S.; Vilsen, B.; Andersen, J.P. Critical role of a transmembrane lysine in aminophospholipid transport by mammalian photoreceptor P4-ATPase ATP8A2. Proc. Natl. Acad. Sci. USA 2012, 109, 1449–1454. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.S.; Costa, S.R.; Duelli, A.S.; Andersen, P.A.; Poulsen, L.R.; Stanchev, L.D.; Gourdon, P.; Palmgren, M.; Gunther Pomorski, T.; Lopez-Marques, R.L. Phospholipid flipping involves a central cavity in P4 ATPases. Sci. Rep. 2017, 7, 17621. [Google Scholar] [CrossRef]
- Roland, B.P.; Graham, T.R. Decoding P4-ATPase substrate interactions. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 513–527. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery studioVisualizer, v. 16.1.0.15350; Dassault Systèmes: San Diego, CA, USA, 2016. Available online: https://discover.3ds.com/discovery-studio-visualizer-download(accessed on 27 December 2022).
- Schutz, C.N.; Warshel, A. What are the dielectric "constants" of proteins and how to validate electrostatic models? Proteins 2001, 44, 400–417. [Google Scholar] [CrossRef]
- Perutz, M.F. Electrostatic Effects in Proteins. Science 1978, 201, 1187–1191. [Google Scholar] [CrossRef]
- Xie, W.J.; Xia, S.; Warshel, A.; Wu, H. Electrostatic influence on IL-1 transport through the GSDMD pore. Proc. Natl. Acad. Sci. USA 2022, 119, e2120287119. [Google Scholar] [CrossRef]
- Oanca, G.; Asadi, M.; Saha, A.; Ramachandran, B.; Warshel, A. Exploring the Catalytic Reaction of Cysteine Proteases. J. Phys. Chem. B 2020, 124, 11349–11356. [Google Scholar] [CrossRef]
- Zhou, J.; Saha, A.; Huang, Z.; Warshel, A. Fast and Effective Prediction of the Absolute Binding Free Energies of Covalent Inhibitors of SARS-CoV-2 Main Protease and 20S Proteasome. J. Am. Chem. Soc. 2022, 144, 7568–7572. [Google Scholar] [CrossRef]
- Andersen, J.P.; Vestergaard, A.L.; Mikkelsen, S.A.; Mogensen, L.S.; Chalat, M.; Molday, R.S. P4-ATPases as Phospholipid Flippases-Structure, Function, and Enigmas. Front. Physiol. 2016, 7, 275. [Google Scholar] [CrossRef]
- Lopez-Marques, R.L.; Theorin, L.; Palmgren, M.G.; Physiology, T.G.P.J.P.A.-E.J.o. P4-ATPases: Lipid flippases in cell membranes. Pflügers Arch. —Eur. J. Physiol. 2014, 466, 1227–1240. [Google Scholar] [CrossRef]
- Coleman, J.A.; Molday, R.S. Critical role of the beta-subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the P4-ATPase ATP8A2. J. Biol. Chem. 2011, 286, 17205–17216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenoir, G.; Williamson, P.; Puts, C.F.; Holthuis, J.C. Cdc50p Plays a Vital Role in the ATPase Reaction Cycle of the Putative Aminophospholipid Transporter Drs2p. J. Biol. Chem. 2009, 284, 17956–17967. [Google Scholar] [CrossRef]
- Stone, A.; Williamson, P. Outside of the box: Recent news about phospholipid translocation by P4 ATPases. J. Chem. Biol. 2012, 5, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Jain, B.K.; Roland, B.P.; Graham, T.R. Exofacial membrane composition and lipid metabolism regulates plasma membrane P4-ATPase substrate specificity. J. Biol. Chem. 2020, 295, 17997–18009. [Google Scholar] [CrossRef] [PubMed]
- Li, D.W.; Xu, T.; Wang, X.G.; Ma, X.B.; Liu, T.T.; Wang, Y.G.; Jiang, S.J. The role of ATP8A1 in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2017, 10, 7760–7766. [Google Scholar]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Zhang, Y.; Xu, P.; Bai, C. Exploring the Phospholipid Transport Mechanism of ATP8A1-CDC50. Biomedicines 2023, 11, 546. https://doi.org/10.3390/biomedicines11020546
Zhang H, Zhang Y, Xu P, Bai C. Exploring the Phospholipid Transport Mechanism of ATP8A1-CDC50. Biomedicines. 2023; 11(2):546. https://doi.org/10.3390/biomedicines11020546
Chicago/Turabian StyleZhang, Honghui, Yue Zhang, Peiyi Xu, and Chen Bai. 2023. "Exploring the Phospholipid Transport Mechanism of ATP8A1-CDC50" Biomedicines 11, no. 2: 546. https://doi.org/10.3390/biomedicines11020546