
Blood ACE Phenotyping for Personalized Medicine: Revelation of Patients with Conformationally Altered ACE
 , ,
, , 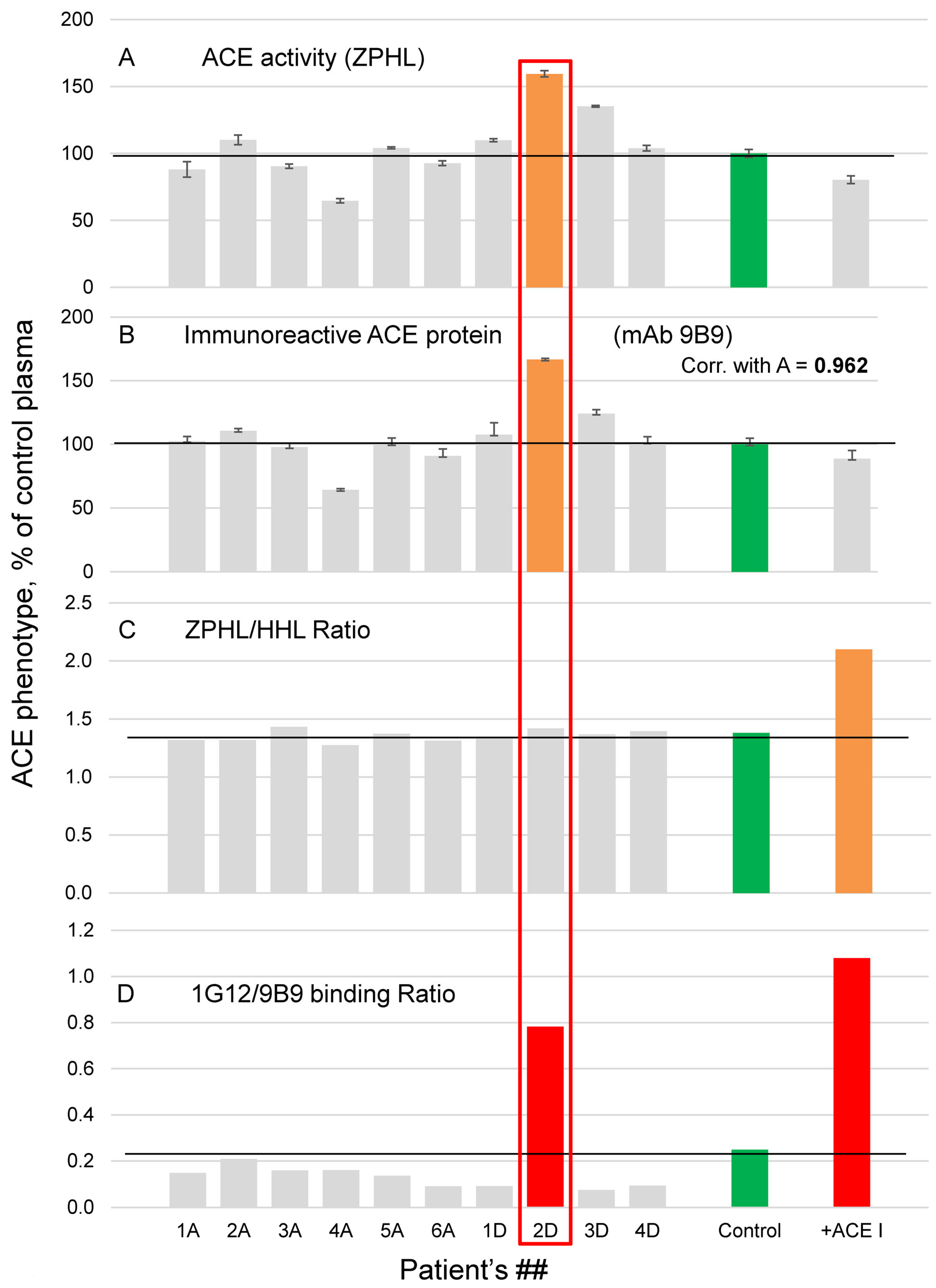{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Characterization of ACE in Donor 2D (with Conformationally Changed ACE)
3.2. ABCG2 Mutation in Donor 2D
3.3. Molecular Modeling of Bilirubin Binding to ACE
3.4. Endogenous ACE Inhibitors in Plasma of Donor 2D
3.5. Blood ACE Phenotyping in Patients with Gilbert and Crigler-Najjar Syndromes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sturrock, E.D.; Anthony, C.S.; Danilov, S.M. Peptidyl-dipeptidase A/Angiotensin I-converting enzyme. In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings, N., Salvesen, G., Eds.; Academic Press: Oxford, UK, 2012; Chapter 98, pp. 480–494. [Google Scholar]
- Bernstein, K.E.; Ong, F.S.; Blackwell, W.-L.B.; Shah, K.H.; Giani, J.F.; Gonzalez-Villalobos, R.A.; Shen, X.Z.; Fuchs, S. A Modern Understanding of the Traditional and Nontraditional Biological Functions of Angiotensin-Converting Enzyme. Pharmacol. Rev. 2012, 65, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Ching, S.F.; Hayes, L.W.; Slakey, L.L. Angiotensin-converting enzyme in cultured endothelial cells. Synthesis, degradation, and transfer to culture medium. Arteriosclerosis 1983, 3, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Metzger, R.; Franke, F.; Bohle, R.-M.; Alhenc-Gelas, F.; Danilov, S.M. Heterogeneous distribution of Angiotensin I-converting enzyme (CD143) in the human and rat vascular systems: Vessels, organs and species specificity. Microvasc. Res. 2011, 82, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Hooper, N.; Keen, J.; Pappin, D.; Turner, A. Pig kidney angiotensin converting enzyme. Purification and characterization of amphipatic and hydrophilic forms of the enzyme establishes C-terminal anchorage to the plasma membrane. Biochem. J. 1987, 247, 85–93. [Google Scholar] [CrossRef]
- Wei, L.; Gelas, F.A.; Soubrier, F.; Michaud, A.; Corvol, P.; Clauser, E. Expression and characterization of recombinant human angiotensin I-converting enzyme. Evidence for a C-terminal transmembrane anchor and for a proteolytic processing of the secreted recombinant and plasma enzymes. J. Biol. Chem. 1991, 266, 5540–5546. [Google Scholar] [CrossRef]
- Ehlers, M.; Gordon, K.; Schwager, S.; Sturrock, E. Shedding the load of hypertension: The proteolytic processing of angiotensin-converting enzyme. S. Afr. Med. J. 2017, 102, 461–464. [Google Scholar] [CrossRef]
- Alhenc-Gelas, F.; Richard, J.; Courbon, D.; Warnet, J.M.; Corvol, P. Distribution of plasma angiotensin I-converting enzyme levels in healthy men: Relationship to environmental and hormonal parameters. J. Lab. Clin. Med. 1991, 117, 33–39. [Google Scholar]
- Lieberman, J. Elevation of serum angiotensin-converting enzyme level in sarcoidosis. Am. J. Med. 1975, 59, 365–372. [Google Scholar] [CrossRef]
- Lieberman, J.; Beutler, E. Elevation of angiotensin-converting enzyme in Gaucher’s disease. N. Engl. J. Med. 1976, 294, 1442–1444. [Google Scholar] [CrossRef]
- Romer, F. Clinical and biochemical aspects of sarcoidosis. With special reference to angiotensin-converting enzyme (ACE). Acta Med. Scand. Suppl. 1984, 690, 3–96. [Google Scholar]
- Sawahata, M.; Sugiyama, Y.; Nakamura, Y.; Nakayama, M.; Mato, N.; Yamasawa, H.; Bando, M. Age-related and historical changes in the clinical characteristics of sarcoidosis in Japan. Resp. Med. 2015, 109, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Sznajder, J.; Ciechanover, A. Personalized Medicine. The Road Ahead. Am. J. Respir. Crit. Care Med. 2012, 186, 945–947. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.; Lew, D. Reconciling evidence-based medicine and precision medicine in the era of big data: Challenges and opportunities. Genome Med. 2016, 8, 134. [Google Scholar] [CrossRef]
- König, I.R.; Fuchs, O.; Hansen, G.; von Mutius, E.; Kopp, M.V. What is precision medicine? Eur. Respir. J. 2017, 50, 1700391. [Google Scholar] [CrossRef]
- Danser, A.J.; Batenburg, W.W.; Meiracker, A.H.V.D.; Danilov, S.M. ACE phenotyping as a first step toward personalized medicine for ACE inhibitors. Why does ACE genotyping not predict the therapeutic efficacy of ACE inhibition. Pharmacol. Ther. 2007, 113, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Petrov, M.N.; Shilo, V.Y.; Tarasov, A.V.; Schwartz, D.E.; Garcia, J.G.N.; Kost, O.A.; Danilov, S.M. Conformational changes of blood ACE in uremia. PLoS ONE 2012, 7, e49290. [Google Scholar] [CrossRef]
- Danilov, S.M.; Tovsky, S.I.; Schwartz, D.E.; Dull, R. ACE Phenotyping as a guide toward personalized therapy with ACE inhibitors. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 374–386. [Google Scholar] [CrossRef]
- Danilov, S.M.; Tikhomirova, V.E.; Kryukova, O.V.; Balatsky, A.V.; Bulaeva, N.I.; Golukhova, E.Z.; Bokeria, L.A.; Samokhodskaya, L.M.; Kost, O.A. Conformational fingerprint of blood and tissue ACEs: Personalized approach. PLoS ONE 2018, 13, e0209861. [Google Scholar] [CrossRef]
- Danilov, S.M.; Jain, M.S.; Petukhov, P.A.; Goldman, C.; DiSanto-Rose, M.; Vancavage, R.; Francuzevitch, L.Y.; Samokhodskaya, L.M.; Kamalov, A.A.; Arbieva, Z.H.; et al. Novel ACE mutations mimicking sarcoidosis by increasing blood ACE Levels. Transl. Res. 2021, 230, 5–20. [Google Scholar] [CrossRef]
- Samokhodskaya, L.M.; Jain, M.S.; Kurilova, O.V.; Bobkov, A.P.; Kamalov, A.A.; Dudek, S.M.; Danilov, S.M. Blood ACE phenotyping: A necessary approach for precision medicine. J. Appl. Lab. Med. 2021, 6, 1179–1191. [Google Scholar] [CrossRef]
- Umeukeje, E.M.; Young, B.A. Genetics and ESKD disparities in African-Americans. Am. J. Kidney Dis. 2019, 74, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Watchko, J.F.; Tiribelli, C. Bilirubin-induced neurologic damage-mechanisms and management approaches. N. Engl. J. Med. 2013, 369, 2021–2030. [Google Scholar] [CrossRef]
- Danilov, S.M.; Balyasnikova, I.V.; Danilova, A.S.; Naperova, I.A.; Arablinskaya, N.E.; Borisov, S.E.; Metzger, R.; Franke, F.E.; Schwartz, D.E.; Gachok, I.V.; et al. Conformational fingerprinting of the angiotensin-converting enzyme (ACE): Application in sarcoidosis. J. Proteome Res. 2010, 9, 5782–5793. [Google Scholar] [CrossRef]
- Kryukova, O.V.; Tikhomirova, V.E.; Golukhova, E.Z.; Evdokimov, V.V.; Kalantarov, G.F.; Trakht, I.N.; Schwartz, D.E.; Dull, R.O.; Gusakov, A.V.; Uporov, I.V.; et al. Tissue Specificity of human angiotensin I-converting enzyme. PLoS ONE 2015, 10, e0143455. [Google Scholar] [CrossRef] [PubMed]
- Danilov, S.; Savoie, F.; Lenoir, B.; Jeunemaitre, X.; Azizi, M.; Tarnow, L.; Alhenc-Gelas, F. Development of enzyme-linked immunoassays for human angiotensin I converting enzyme suitable for large-scale studies. J. Hypertens. 1996, 14, 719–727. [Google Scholar] [CrossRef]
- Danilov, S.M.; Balyasnikova, I.V.; Albrecht, R.F.; Kost, O.A. Simultaneous determination of ACE activity with two substrates provide an information on the nativity of somatic ACE and allow to detect ACE inhibitors in the human blood. J. Cardiovasc. Pharmacol. 2008, 52, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Kost, O.A.; Grinshtein, S.V.; Nikolskaya, I.; Shevchenko, A.; Binevski, P.V. Purification of soluble and membrane forms of somatic angiotensin-converting enzyme by cascade affinity chromatography. Biochemistry 1997, 62, 321–328. [Google Scholar]
- Danilov, S.M.; Lünsdorf, H.; Akinbi, H.T.; Nesterovitch, A.B.; Epshtein, Y.; Letsiou, E.; Kryukova, O.V.; Piegeler, T.; Golukhova, E.Z.; Schwartz, D.E.; et al. Lysozyme and bilirubin bind to ACE and regulate ACE conformation and shedding. Sci. Rep. 2016, 6, 34913. [Google Scholar] [CrossRef]
- Dumoulin, M.; Kumita, J.R.; Dobson, C.M. Normal and aberrant biological self-assembly: Insights from studies of human lysozyme and its amyloidogenic variants. Acc. Chem. Res. 2006, 39, 603–610. [Google Scholar] [CrossRef]
- Bosma, P.J.; Chowdhury, J.R.; Bakker, C.; Gantla, S.; de Boer, A.; Oostra, B.A.; Lindhout, D.; Tytgat, G.N.; Jansen, P.L.; Elferink, R.P.; et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. N. Engl. J. Med. 1995, 333, 1171–1175. [Google Scholar] [CrossRef]
- Okuneva, E.G.; Kozina, A.A.; Baryshnikova, N.V.; Krasnenko, A.Y.; Tsukanov, K.; Klimchuk, O.I.; Surkova, E.I.; Ilinsky, V.V. A novel elastin gene frameshift mutation in a Russian family with cutis laxa: A case report. BMC Dermatol. 2019, 19, 4. [Google Scholar] [CrossRef] [PubMed]
- Anthony, C.S.; Corradi, H.R.; Schwager, S.L.; Redelinghuys, P.; Georgiadis, D.; Dive, V.; Acharya, K.R.; Sturrock, E.D. The N domain of human angiotensin-I-converting enzyme: The role of N-glycosylation and the crystal structure in complex with an N domain-specific phosphinic inhibitor, RXP407. J. Biol. Chem. 2010, 285, 35685–35693. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), 2019.0101; Chemical Computing Group Inc.: Montreal, QC, Canada, 2019.
- Gerber, P.; Muller, K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput. Aided Mol. Des. 2015, 9, 251–268. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theor. Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Balyasnikova, I.V.; Skirgello, O.E.; Binevski, P.V.; Nesterovitch, A.B.; Albrecht, R.F.; Kost, O.A.; Danilov, S.M. Monoclonal antibodies 1G12 and 6A12 to the N-domain of human angiotensin-converting enzyme: Fine epitope mapping and antibody-based method for revelation and quantification of ACE inhibitors in the human blood. J. Proteome Res. 2007, 6, 1580–1594. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.; Balyasnikova, I.V.; Nesterovitch, A.B.; Schwartz, D.E.; Sturrock, E.D.; Danilov, S.M. Fine epitope mapping of monoclonal antibodies 9B9 and 3G8, to the N domain of human angiotensin I-converting enzyme (ACE) defines a region involved in regulating ACE dimerization and shedding. Tissue Antigens 2010, 75, 136–150. [Google Scholar] [CrossRef]
- Danilov, S.M.; Tikhomirova, V.E.; Metzger, R.; Naperova, I.A.; Bukina, T.M.; Goker-Alpan, O.; Tayebi, N.; Gayfullin, N.M.; Schwartz, D.E.; Samokhodskaya, L.M.; et al. ACE phenotyping in Gaucher disease. Mol. Genet Metab. 2018, 123, 501–510. [Google Scholar] [CrossRef]
- Danilov, S.; Jaspard, E.; Churakova, T.; Towbin, H.; Savoie, F.; Wei, L.; Alhenc-Gelas, F. Structure-function analysis of angiotensin I-converting enzyme using monoclonal antibodies. J. Biol. Chem. 1994, 269, 26806–26814. [Google Scholar] [CrossRef]
- Popova, I.A.; Lubbe, L.; Petukhov, P.A.; Kalantarov, G.F.; Trakht, I.N.; Chernykh, E.R.; Leplina, O.Y.; Lyubimov, A.V.; Garcia, J.G.; Dudek, S.M.; et al. Epitope mapping of novel monoclonal antibodies to human angiotensin I-converting enzyme. Protein Sci. 2021, 30, 1577–1593. [Google Scholar] [CrossRef]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.J. Hypertension: Renin-angiotensin-aldosteron system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef]
- Satou, R.; Penrose, H.; Navar, L. Inflammation as a regulator of the renin-angiotensin system and blood pressure. Curr. Hypertens. Rep. 2018, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, A.F. Ex uno plures; the concealed complexity of bilirubin species in the neonatal blood samples. Pediatrics 2006, 118, 1185–1187. [Google Scholar] [CrossRef] [PubMed]
- Skirgello, O.E.; Balyasnikova, I.V.; Binevski, P.V.; Sun, Z.-L.; Baskin, I.I.; Palyulin, V.A.; Nesterovitch, A.B.; Albrecht, R.F.; Kost, O.A.; Danilov, S.M. Inhibitory antibodies to human angiotensin-converting enzyme: Fine epitope mapping and mechanism of action. Biochemistry 2006, 45, 4831–4847. [Google Scholar] [CrossRef] [PubMed]
- Danilov, S.M.; Gordon, K.; Nesterovitch, A.B.; Lünsdorf, H.; Chen, Z.; Castellon, M.; Popova, I.A.; Kalinin, S.; Mendonca, E.; Petukhov, P.A.; et al. An angiotensin I-converting enzyme mutation (Y465D) causes a dramatic increase in blood ACE via accelerated ACE shedding. PLoS ONE 2011, 6, e25952. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-J.; Zhang, F.-B.; Liu, S.-Y.; Tian, Y.-H.; Le, F.; Wang, L.-Y.; Lou, H.-Y.; Xu, X.-R.; Huang, H.-F.; Jin, F. Human sperm devoid of germinal angiotensin-converting enzyme is responsible for total fertilization failure and lower fertilization rates by conventional in vitro fertilization. Biol. Reprod. 2014, 90, 125. [Google Scholar] [CrossRef]
- Bosma, P.J. Inherited disorders of bilirubin metabolism. J. Hepatol. 2003, 38, 107–117. [Google Scholar] [CrossRef]
- Sticova, E.; Jirsa, M. New insights in bilirubin metabolism and their clinical implications. World J. Gastroenterol. 2013, 19, 6398–6407. [Google Scholar] [CrossRef]
- Memon, N.; Weinberger, B.J.; Hegy, T.; Aleksunes, L.M. Inherited disorders of bilirubin clearance. Pediatric Res. 2016, 79, 378–386. [Google Scholar] [CrossRef]
- Minucci, G.C.A.; Zuppi, C.; Capoluongo, E. Gilbert and Crigler-Najjar syndromes: An update of the UDP-glucoronosyltransferase 1a1 (UGT1A1) gene mutation database. Blood Cells Mol. Dis. 2013, 50, 273–280. [Google Scholar]
- Bosma, P.; Seppen, J.; Goldhoorn, B.; Bakker, C.; Elferink, R.O.; Chowdhury, J.; Jansen, P. Bilirubin UDP-glucuronosyltransferase 1 is the only relevant bilirubin glucuronidating isoform in man. J. Biol. Chem. 1994, 269, 17960–17964. [Google Scholar] [CrossRef]
- Horsey, A.J.; Cox, M.H.; Sarwat, S.; Kerr, I.D. The multidrug transporter ABCG2: Still more questions than answers. Biochem. Soc. Trans. 2016, 44, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Vlaming, M.L.; Pala, Z.; van Esch, A.; Wagenaar, E.; de Waart, D.R.; van de Wetering, K.; van der Kruijssen, C.M.; Oude Elferink, R.P.; van Tellingen, O.; Schinkel, A.H. Functionally overlapping roles of Abcg2 (Bcrp1) and Abcc2 (Mrp2) in the elimination of methotrexate and its main toxic metabolite 7-hydroxymethotrexate in vivo. Clin. Cancer Res. 2009, 15, 3084–3093. [Google Scholar] [CrossRef] [PubMed]
- Zámbó, B.; Bartos, Z.; Mózner, O.; Szabó, E.; Várady, G.; Poór, G.; Pálinkás, M.; Andrikovics, H.; Hegedűs, T.; Homolya, L.; et al. Clinically relevant mutations in the ABCG2 transporter uncovered by genetic analysis linked to erythrocyte membrane protein expression. Sci. Rep. 2018, 8, 7487. [Google Scholar] [CrossRef]
- Nakagawa, H.; Wakabayashi-Nakao, K.; Tamura, A.; Toyoda, Y.; Koshiba, S.; Ishikawa, T. Disruption of N-linked glycosylation enhances ubiquitin-mediated proteasomal degradation of the human ATP-binding cassette transporter ABCG2. FEBS J. 2009, 276, 7237–7252. [Google Scholar] [CrossRef]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin binding to PPARα inhibits lipid accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef] [PubMed]
- Zunszain, P.; Ghuman, J.; McDonagh, A.; Curry, S. Crystallographic analysis of human serum albumin complexed with 4Z,15E-bilirubin-IXalpha. J. Mol. Biol. 2008, 381, 394–406. [Google Scholar] [CrossRef]
- Ahlfors, C.E.; Vreman, H.J.; Wong, R.J.; Bender, G.J.; Oh, W.; Morris, B.H.; Stevenson, D.K.; Subcommittee, T.P. Effects of sample dilution, peroxidase concentration, and chloride ion on the measurement of unbound bilirubin in premature newborns. Clin. Biochem. 2007, 40, 261–267. [Google Scholar] [CrossRef]
- Wennberg, R.P.; Ahlfors, C.E.; Bhutani, V.K.; Johnson, L.H.; Shapiro, S.M. Toward understanding kernicterus: A challenge to improve the management of jaundiced newborns. Pediatrics 2006, 117, 474–485. [Google Scholar] [CrossRef]
- McDonagh, A.F.; Vreman, H.J.; Wong, R.J.; Stevenson, D.K. Photoisomers: Obfuscating factors in clinical peroxidase measurements of unbound bilirubin? Pediatrics 2009, 123, 67–76. [Google Scholar] [CrossRef]
- Kumagai, A.; Ando, R.; Miyatake, H.; Greimel, P.; Kobayashi, T.; Hirabayashi, Y.; Shimogori, T.; Miyawaki, A. A bilirubin-inducible fluorescent protein from eel muscle. Cell 2009, 153, 1602–1611. [Google Scholar] [CrossRef]
- Huber, A.H.; Zhu, B.; Kwan, T.; Kampf, J.P.; Hegyi, T.; Kleinfeld, A.M. Fluorescence sensor for the quantification of unbound bilirubin concentration. Clin. Chem. 2012, 58, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, T.; Kleinfeld, A.; Huber, A.; Weinberger, B.; Memon, N.; Shih, W.J.; Carayannopoulos, M.; Oh, W. Effects of Soybean Lipid Infusion on Unbound Free Fatty Acids and Unbound Bilirubin in Preterm Infants. J. Pediatr. 2017, 184, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Iwatani, S.; Yamana, K.; Nakamura, H.; Nishida, K.; Morisawa, T.; Mizobuchi, M. A Novel Method for Measuring Serum Unbound Bilirubin Levels Using Glucose Oxidase-Peroxidase and Bilirubin-Inducible Fluorescent Protein (UnaG): No Influence of Direct Bilirubin. Int. J. Mol. Sci. 2020, 21, 6778. [Google Scholar] [CrossRef]
- Temme, E.H.; Zhang, J.; Schouten, E.; Kesteloot, H. Serum bilirubin and 10-year mortality risk in a Belgian population. Cancer Causes Control. 2001, 12, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Ip, S.; Chung, M.; Kulig, J.; O’Brien, R.; Sege, R.; Glicken, S.; Maisels, M.J.; Lau, J. An evidenced-based review of important issues concerning neonatal hyperbilirubinemia. Pediatrics 2004, 114, 130–153. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danilov, S.M.; Jain, M.S.; A. Petukhov, P.; Kurilova, O.V.; Ilinsky, V.V.; Trakhtman, P.E.; Dadali, E.L.; Samokhodskaya, L.M.; Kamalov, A.A.; Kost, O.A. Blood ACE Phenotyping for Personalized Medicine: Revelation of Patients with Conformationally Altered ACE. Biomedicines 2023, 11, 534. https://doi.org/10.3390/biomedicines11020534
Danilov SM, Jain MS, A. Petukhov P, Kurilova OV, Ilinsky VV, Trakhtman PE, Dadali EL, Samokhodskaya LM, Kamalov AA, Kost OA. Blood ACE Phenotyping for Personalized Medicine: Revelation of Patients with Conformationally Altered ACE. Biomedicines. 2023; 11(2):534. https://doi.org/10.3390/biomedicines11020534
Chicago/Turabian StyleDanilov, Sergei M., Mark S. Jain, Pavel A. Petukhov, Olga V. Kurilova, Valery V. Ilinsky, Pavel E. Trakhtman, Elena L. Dadali, Larisa M. Samokhodskaya, Armais A. Kamalov, and Olga A. Kost. 2023. "Blood ACE Phenotyping for Personalized Medicine: Revelation of Patients with Conformationally Altered ACE" Biomedicines 11, no. 2: 534. https://doi.org/10.3390/biomedicines11020534