
Non-alcoholic Fatty Liver Disease (NAFLD), Type 2 Diabetes, and Non-viral Hepatocarcinoma: Pathophysiological Mechanisms and New Therapeutic Strategies
 , , , , and
, , , , and 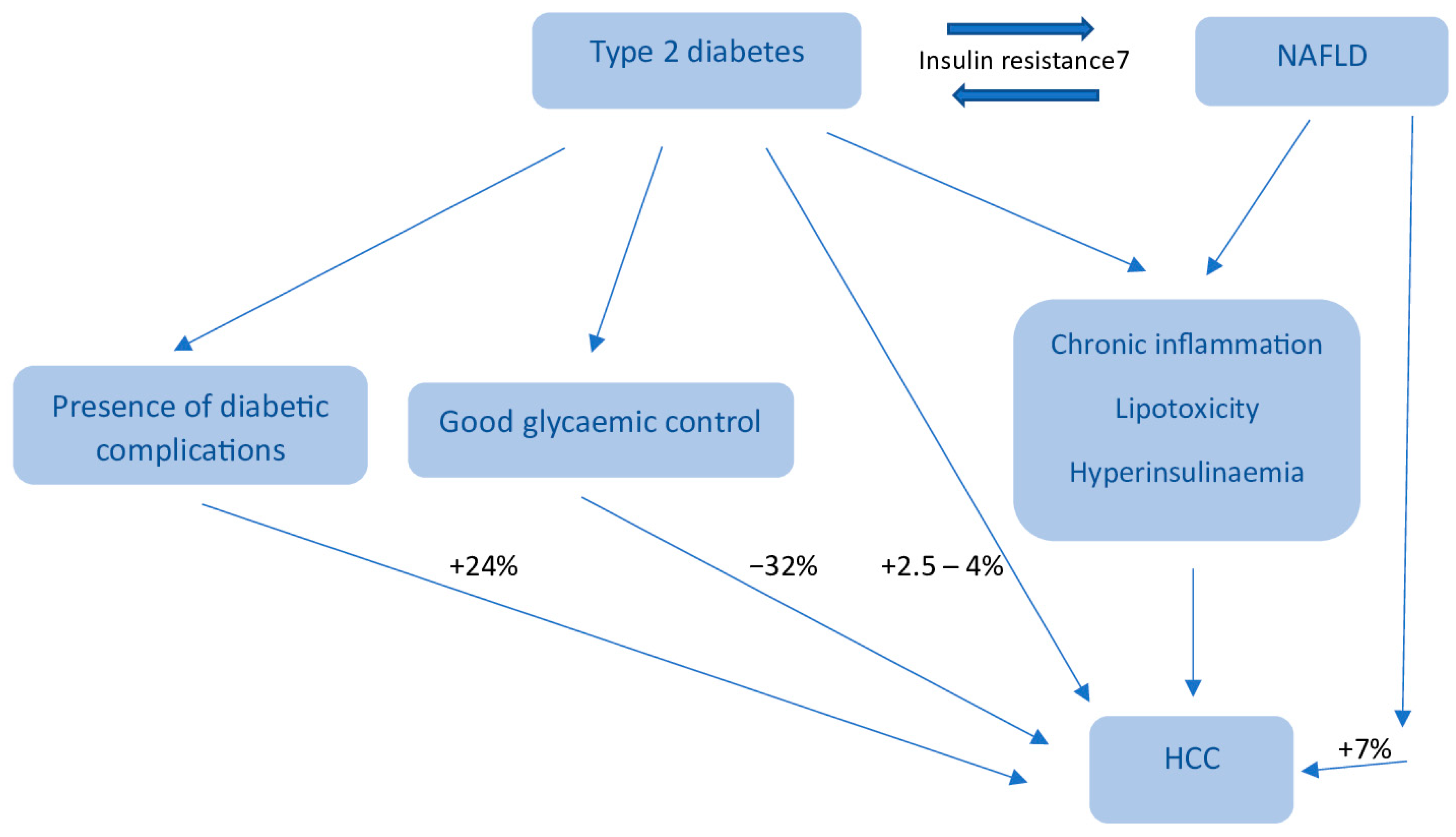{kind=link}
Abstract
:1. Introduction
2. Methods
3. NAFLD and NASH
4. NASH and HCC
5. NAFLD and Diabetes: A Single Combination Therapy?
6. Diabetes and HCC
7. HCC and Drug Treatment Update
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Metabolic Syndrome Pandemic. Arter. Thromb. Vasc. Biol. 2008, 28, 629–636. [Google Scholar] [CrossRef]
- Rinaldi, L.; Pafundi, P.C.; Galiero, R.; Caturano, A.; Morone, M.V.; Silvestri, C.F.; Giordano, M.; Salvatore, T.; Sasso, F.C. Mechanisms of Non-Alcoholic Fatty Liver Disease in the Metabolic Syndrome. A Narrative Review. Antioxidants 2021, 10, 270. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer Globocan. 2020. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/11-Liver-fact-sheet.pdf (accessed on 31 October 2022).
- Simon, T.G.; King, L.Y.; Chong, D.Q.; Nguyen, L.H.; Ma, Y.; VoPham, T.; Giovannucci, E.L.; Fuchs, C.S.; Meyerhardt, J.A.; Corey, K.E.; et al. Diabetes, metabolic comorbidities, and risk of hepatocellular carcinoma: Results from two prospective cohort studies. Hepatology 2017, 67, 1797–1806. [Google Scholar] [CrossRef]
- Kramer, J.R.; Natarajan, Y.; Dai, J.; Yu, X.; Li, L.; El-Serag, H.B.; Kanwal, F. Effect of diabetes medications and glycemic control on risk of hepatocellular cancer in patients with nonalcoholic fatty liver disease. Hepatology 2021, 75, 1420–1428. [Google Scholar] [CrossRef]
- Wu, X.; Chen, J.; Song, S.; Li, X.; Bian, D. Association of metabolic traits with occurrence of nonalcoholic fatty liver disease-related hepatocellular carcinoma: A systematic review and meta-analysis of longitudinal cohort studies. Saudi J. Gastroenterol. 2022, 28, 92–100. [Google Scholar] [CrossRef]
- Xia, M.-F.; Bian, H.; Gao, X. NAFLD and Diabetes: Two Sides of the Same Coin? Rationale for Gene-Based Personalized NAFLD Treatment. Front. Pharmacol. 2019, 10, 877. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated with Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005.e1. [Google Scholar] [CrossRef]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2014, 13, 643–654.e9. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Kwok, R.; Choi, K.C.; Wong, G.L.-H.; Zhang, Y.; Chan, H.L.-Y.; Luk, A.O.-Y.; Shu, S.S.-T.; Chan, A.W.-H.; Yeung, M.-W.; Chan, J.C.-N.; et al. Screening diabetic patients for non-alcoholic fatty liver disease with controlled attenuation parameter and liver stiffness measurements: A prospective cohort study. Gut 2015, 65, 1359–1368. [Google Scholar] [CrossRef]
- Koehler, E.M.; Plompen, E.P.; Schouten, J.N.; Hansen, B.E.; Murad, S.D.; Taimr, P.; Leebeek, F.W.; Hofman, A.; Stricker, B.H.; Castera, L.; et al. Presence of diabetes mellitus and steatosis is associated with liver stiffness in a general population: The Rotterdam study. Hepatology 2015, 63, 138–147. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; on behalf of the International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Xian, Y.-X.; Weng, J.-P.; Xu, F. MAFLD vs. NAFLD: Shared features and potential changes in epidemiology, pathophysiology, diagnosis, and pharmacotherapy. Chin. Med. J. 2020, 134, 8–19. [Google Scholar] [CrossRef]
- Huang, J.; Ou, W.; Wang, M.; Singh, M.; Liu, Y.; Liu, S.; Wu, Y.; Zhu, Y.; Kumar, R.; Lin, S. MAFLD Criteria Guide the Subtyping of Patients with Fatty Liver Disease. Risk Manag. Health Policy 2021, 14, 491–501. [Google Scholar] [CrossRef]
- Myers, S.; Neyroud-Caspar, I.; Spahr, L.; Gkouvatsos, K.; Fournier, E.; Giostra, E.; Magini, G.; Frossard, J.-L.; Bascaron, M.-E.; Vernaz, N.; et al. NAFLD and MAFLD as emerging causes of HCC: A populational study. JHEP Rep. 2021, 3, 100231. [Google Scholar] [CrossRef]
- Piscaglia, F.; Svegliati-Baroni, G.; Barchetti, A.; Pecorelli, A.; Marinelli, S.; Tiribelli, C.; Bellentani, S.; HCC-NAFLD Italian Study Group. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: A multicenter prospective study. Hepatology 2016, 63, 827–838. [Google Scholar] [CrossRef]
- Guo, F.; Estévez-Vázquez, O.; Benedé-Ubieto, R.; Maya-Miles, D.; Zheng, K.; Gallego-Durán, R.; Rojas, Á.; Ampuero, J.; Romero-Gómez, M.; Philip, K.; et al. A Shortcut from Metabolic-Associated Fatty Liver Disease (MAFLD) to Hepatocellular Carcinoma (HCC): C-MYC a Promising Target for Preventative Strategies and Individualized Therapy. Cancers 2021, 14, 192. [Google Scholar] [CrossRef]
- Du, D.; Liu, C.; Qin, M.; Zhang, X.; Xi, T.; Yuan, S.; Hao, H.; Xiong, J. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm. Sin. B 2021, 12, 558–580. [Google Scholar] [CrossRef]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef]
- Fabbrini, E.; Magkos, F. Hepatic Steatosis as a Marker of Metabolic Dysfunction. Nutrients 2015, 7, 4995–5019. [Google Scholar] [CrossRef]
- Paul, B.; Lewinska, M.; Andersen, J.B. Lipid alterations in chronic liver disease and liver cancer. JHEP Rep. 2022, 4, 100479. [Google Scholar] [CrossRef]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef]
- Diraison, F.; Moulin, P.; Beylot, M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003, 29, 478–485. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased De Novo Lipogenesis Is a Distinct Characteristic of Individuals with Nonalcoholic Fatty Liver Disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Chiu, S.; Mulligan, K.; Schwarz, J.-M. Dietary carbohydrates and fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 277–282. [Google Scholar] [CrossRef]
- Dentin, R.; Girard, J.; Postic, C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): Two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 2005, 87, 81–86. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Fraile, J.M.; Palliyil, S.; Barelle, C.J.; Porter, A.J.; Kovaleva, M. Non-Alcoholic Steatohepatitis (NASH)—A Review of a Crowded Clinical Landscape, Driven by a Complex Disease. Drug Des. Dev. Ther. 2021, 15, 3997–4009. [Google Scholar] [CrossRef]
- Mohammed, S.; Thadathil, N.; Selvarani, R.; Nicklas, E.H.; Wang, D.; Miller, B.F.; Richardson, A.; Deepa, S.S. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell 2021, 20, e13512. [Google Scholar] [CrossRef]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef]
- Mohamad, B.; Shah, V.; Onyshchenko, M.; ElShamy, M.; Aucejo, F.; Lopez, R.; Hanouneh, I.A.; Alhaddad, R.; Alkhouri, N. Characterization of hepatocellular carcinoma (HCC) in non-alcoholic fatty liver disease (NAFLD) patients without cirrhosis. Hepatol. Int. 2015, 10, 632–639. [Google Scholar] [CrossRef]
- Nevola, R.; Acierno, C.; Sasso, F.C.; Marrone, A.; Buffardi, F.; Adinolfi, L.E.; Rinaldi, L. Hepatocellular carcinoma and non alcoholic fatty liver disease; a dangerous liaison. WCRJ 2019, 6, e1299. [Google Scholar]
- Zoller, H.; Tilg, H. Nonalcoholic fatty liver disease and hepatocellular carcinoma. Metabolism 2016, 65, 1151–1160. [Google Scholar] [CrossRef]
- Global Burden of Disease Cancer Collaboration. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Otgonsuren, M.; Henry, L.; Venkatesan, C.; Mishra, A.; Erario, M.; Hunt, S. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015, 62, 1723–1730. [Google Scholar] [CrossRef]
- Stine, J.G.; Wentworth, B.J.; Zimmet, A.; Rinella, M.E.; Loomba, R.; Caldwell, S.H.; Argo, C.K. Systematic review with meta-analysis: Risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharmacol. Ther. 2018, 48, 696–703. [Google Scholar] [CrossRef]
- Gabbia, D.; Cannella, L.; De Martin, S. The Role of Oxidative Stress in NAFLD–NASH–HCC Transition—Focus on NADPH Oxidases. Biomedicines 2021, 9, 687. [Google Scholar] [CrossRef]
- Joachim, J.H.; Mehta, K.J. Hepcidin in hepatocellular carcinoma. Br. J. Cancer 2022, 127, 185–192. [Google Scholar] [CrossRef]
- Sorrentino, P.; D’Angelo, S.; Ferbo, U.; Micheli, P.; Bracigliano, A.; Vecchione, R. Liver iron excess in patients with hepatocellular carcinoma developed on non-alcoholic steato-hepatitis. J. Hepatol. 2009, 50, 351–357. [Google Scholar] [CrossRef]
- Mehta, K.J.; Farnaud, S.J.; Sharp, P.A. Iron and liver fibrosis: Mechanistic and clinical aspects. World J. Gastroenterol. 2019, 25, 521–538. [Google Scholar] [CrossRef]
- Qi, Y.; Xu, Z.; Zhu, Q.; Thomas, C.; Kumar, R.; Feng, H.; Dostal, D.E.; White, M.F.; Baker, K.M.; Guo, S. Myocardial loss of IRS1 and IRS2 causes heart failure and is controlled by p38α MAPK during insulin resistance. Diabetes 2013, 62, 3887–3900. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef]
- Cheng, Z.; Guo, S.; Copps, K.; Dong, X.; Kollipara, R.; Rodgers, J.T.; DePinho, R.A.; Puigserver, P.; White, M.F. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat. Med. 2009, 15, 1307–1311. [Google Scholar] [CrossRef]
- Altomonte, J.; Richter, A.; Harbaran, S.; Suriawinata, J.; Nakae, J.; Thung, S.N.; Meseck, M.; Accili, D.; Dong, H.H. Inhibition of Foxo1 function is associated with improved fasting glycemia in diabetic mice. Am. J. Physiol. Metab. 2003, 285, E718–E728. [Google Scholar] [CrossRef]
- Battiprolu, P.K.; Gillette, T.G.; Wang, Z.V.; Lavandero, S.; Hill, J.A. Diabetic Cardiomyopathy: Mechanisms and Therapeutic Targets. Drug Discov. Today Dis. Mech. 2010, 7, e135–e143. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.Q.; Liu, F. PDK2: The missing piece in the receptor tyrosine kinase signaling pathway puzzle. Am. J. Physiol. Metab. 2005, 289, E187–E196. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Gao, Z.; Yin, J.; Quon, M.J.; Ye, J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J. Biol. Chem. 2008, 283, 35375–35382. [Google Scholar] [CrossRef]
- Hendrikx, T.; Binder, C.J. Oxidation-Specific Epitopes in Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2020, 11, 607011. [Google Scholar] [CrossRef]
- Miura, K. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381–7391. [Google Scholar] [CrossRef]
- Ferreira, D.F.; Fiamoncini, J.; Prist, I.H.; Ariga, S.K.; de Souza, H.P.; de Lima, T.M. Novel role of TLR4 in NAFLD development: Modulation of metabolic enzymes expression. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2015, 1851, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Velayudham, A.; Hritz, I.; Petrasek, J.; Levin, I.; Lippai, D.; Catalano, D.; Mandrekar, P.; Dolganiuc, A.; Kurt-Jones, E.; et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am. J. Physiol. Liver Physiol. 2011, 300, G433–G441. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, S.; Kim, B.; Kwon, J. Toll-like receptor 7 affects the pathogenesis of non-alcoholic fatty liver disease. Sci. Rep. 2016, 6, 27849. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Zhou, Z.; Yu, Z.; He, M.; He, L.; Luo, Z.; Xiao, W.; Yang, Q.; Zhao, F.; Li, W.; et al. The Microbiota and It’s Correlation with Metabolites in the Gut of Mice with Nonalcoholic Fatty Liver Disease. Front. Cell. Infect. Microbiol. 2022, 12, 870785. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Török, N.J. NADPH Oxidases in Chronic Liver Diseases. Adv. Hepatol. 2014, 2014, 742931. [Google Scholar] [CrossRef]
- Gabbia, D.; Pozzo, L.; Zigiotto, G.; Roverso, M.; Sacchi, D.; Pozza, A.D.; Carrara, M.; Bogialli, S.; Floreani, A.; Guido, M.; et al. Dexamethasone counteracts hepatic inflammation and oxidative stress in cholestatic rats via CAR activation. PLoS ONE 2018, 13, e0204336. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Prieto, M.A.V.; Lanzi, C.R.; Miatello, R.M.; Haj, F.G.; Fraga, C.G.; Oteiza, P.I. (−)-Epicatechin mitigates high-fructose-associated insulin resistance by modulating redox signaling and endoplasmic reticulum stress. Free. Radic. Biol. Med. 2014, 72, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in NAFLD—Revisited after a Decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zou, J.; Li, S.; Guo, J.; Shi, W.; Wang, B.; Han, X.; Zhang, H.; Zhang, P.; Miao, Z.; et al. Farnesoid X receptor contributes to body weight-independent improvements in glycemic control after Roux-en-Y gastric bypass surgery in diet-induced obese mice. Mol. Metab. 2020, 37, 100980. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, F.; Guo, G.L. Tissue-specific function of farnesoid X receptor in liver and intestine. Pharmacol. Res. 2011, 63, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free. Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef]
- Jia, W.; Rajani, C. The Influence of Gut Microbial Metabolism on the Development and Progression of Non-alcoholic Fatty Liver Disease. Adv. Exp. Med. Biol. 2018, 1061, 95–110. [Google Scholar] [CrossRef]
- Yamada, T.; Murata, D.; Adachi, Y.; Itoh, K.; Kameoka, S.; Igarashi, A.; Kato, T.; Araki, Y.; Huganir, R.L.; Dawson, T.M.; et al. Mitochondrial Stasis Reveals p62-Mediated Ubiquitination in Parkin-Independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cell Metab. 2018, 28, 588–604.e5. [Google Scholar] [CrossRef]
- Yamada, S.; Takashina, Y.; Watanabe, M.; Nagamine, R.; Saito, Y.; Kamada, N.; Saito, H. Bile acid metabolism regulated by the gut microbiota promotes non-alcoholic steatohepatitis-associated hepatocellular carcinoma in mice. Oncotarget 2018, 9, 9925–9939. [Google Scholar] [CrossRef]
- Yu, L.-X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, N.; Ahlholm, N.; Luukkonen, P.K.; Porthan, K.; Sharpton, S.R.; Ajmera, V.; Kono, Y.; Dave, S.; Ahmed, A.; Sundaram, V.; et al. Risk of advanced fibrosis in first-degree relatives of patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2022, 132, e162513. [Google Scholar] [CrossRef] [PubMed]
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.-M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2020, 110, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Momtazi-Borojeni, A.A.; Banach, M.; Ruscica, M.; Sahebkar, A. The role of PCSK9 in NAFLD/NASH and therapeutic implications of PCSK9 inhibition. Expert Rev. Clin. Pharmacol. 2022, 15, 1199–1208. [Google Scholar] [CrossRef]
- Tziomalos, K.; Athyros, V.G.; Karagiannis, A. Non-alcoholic fatty liver disease in type 2 diabetes: Pathogenesis and treatment options. Curr. Vasc. Pharmacol. 2012, 10, 162–172. [Google Scholar] [CrossRef]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Sato, K.; Gosho, M.; Yamamoto, T.; Kobayashi, Y.; Ishii, N.; Ohashi, T.; Nakade, Y.; Ito, K.; Fukuzawa, Y.; Yoneda, M. Vitamin E has a beneficial effect on nonalcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Nutrition 2015, 31, 923–930. [Google Scholar] [CrossRef]
- Rinella, M.E.; Sanyal, A.J. Management of NAFLD: A stage-based approach. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 196–205. [Google Scholar] [CrossRef]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; Hartline, J.A.; et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009, 301, 39–51. [Google Scholar] [CrossRef]
- Schürks, M.; Glynn, R.J.; Rist, P.M.; Tzourio, C.; Kurth, T. Effects of vitamin E on stroke subtypes: Meta-analysis of randomised controlled trials. BMJ 2010, 341, c5702. [Google Scholar] [CrossRef] [Green Version]
- Lebovitz, H.E. Thiazolidinediones: The Forgotten Diabetes Medications. Curr. Diabetes Rep. 2019, 19, 151. [Google Scholar] [CrossRef]
- Vieira, R.; Souto, S.B.; Sánchez-López, E.; López Machado, A.; Severino, P.; Jose, S.; Santini, A.; Fortuna, A.; García, M.L.; Silva, A.M.; et al. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome—Review of Classical and New Compounds: Part-I. Pharmaceuticals 2019, 12, 152. [Google Scholar] [CrossRef]
- Bril, F.; Kalavalapalli, S.; Clark, V.C.; Lomonaco, R.; Soldevila-Pico, C.; Liu, I.-C.; Orsak, B.; Tio, F.; Cusi, K. Response to Pioglitazone in Patients with Nonalcoholic Steatohepatitis with vs without Type 2 Diabetes. Clin. Gastroenterol. Hepatol. 2018, 16, 558–566.e2. [Google Scholar] [CrossRef]
- Yuan, G.-J.; Zhang, M.-L.; Gong, Z. Effects of PPARg agonist pioglitazone on rat hepatic fibrosis. World J. Gastroenterol. 2004, 10, 1047–1051. [Google Scholar] [CrossRef]
- Shah, R.A.; Kowdley, K.V. Obeticholic acid for the treatment of nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 2020, 14, 311–321. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef]
- Hameed, B.; Terrault, N.A.; Gill, R.M.; Loomba, R.; Chalasani, N.; Hoofnagle, J.H.; Van Natta, M.L.; Crn, F.T.N. Clinical and metabolic effects associated with weight changes and obeticholic acid in non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2018, 47, 645–656. [Google Scholar] [CrossRef]
- Hindson, J. Obeticholic acid for the treatment of NASH. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 66. [Google Scholar] [CrossRef]
- Sanjay, K.; Vishwakarma, S.; Zope, B.R.; Mane, V.S.; Mohire, S.; Dhakshinamoorthy, S. ATP citrate lyase inhibitor Bempedoic Acid alleviate long term HFD induced NASH through improvement in glycemic control, reduction of hepatic triglycerides & total cholesterol, modulation of inflammatory & fibrotic genes and improvement in NAS score. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100051. [Google Scholar] [CrossRef]
- Gentilella, R.; Pechtner, V.; Corcos, A.; Consoli, A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: Are they all the same? Diabetes/Metab. Res. Rev. 2018, 35, e3070. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Petracca, G.; Beatrice, G.; Csermely, A.; Lonardo, A.; Targher, G. Glucagon-like Peptide-1 Receptor Agonists for Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: An Updated Meta-Analysis of Randomized Controlled Trials. Metabolites 2021, 11, 73. [Google Scholar] [CrossRef]
- Davies, M.J.; Aronne, L.J.; Caterson, I.D.; Thomsen, A.B.; Jacobsen, P.B.; Marso, S.P.; on behalf of the Satiety and Clinical Adiposity. Liraglutide and cardiovascular outcomes in adults with overweight or obesity: A post hoc analysis from SCALE randomized controlled trials. Diabetes, Obes. Metab. 2018, 20, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Kapodistria, K.; Tsilibary, E.; Kotsopoulou, E.; Moustardas, P.; Kitsiou, P. Liraglutide, a human glucagon-like peptide-1 analogue, stimulates AKT-dependent survival signalling and inhibits pancreatic β-cell apoptosis. J. Cell. Mol. Med. 2018, 22, 2970–2980. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Satoh, H.; Watanabe, T. Liraglutide enhances insulin sensitivity by activating AMP-activated protein kinase in male Wistar rats. Endocrinology 2014, 155, 3288–3301. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2017, 53, 362–376. [Google Scholar] [CrossRef]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef]
- Dickson, I. No anti-fibrotic effect of selonsertib in NASH. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 260. [Google Scholar] [CrossRef]
- Yamamoto, E.; Dong, Y.-F.; Kataoka, K.; Yamashita, T.; Tokutomi, Y.; Matsuba, S.; Ichijo, H.; Ogawa, H.; Kim-Mitsuyama, S. Olmesartan prevents cardiovascular injury and hepatic steatosis in obesity and diabetes, accompanied by apoptosis signal regulating kinase-1 inhibition. Hypertension 2008, 52, 573–580. [Google Scholar] [CrossRef]
- Cai, L.; Xiong, X.; Kong, X.; Xie, J. The Role of the Lysyl Oxidases in Tissue Repair and Remodeling: A Concise Review. Tissue Eng. Regen. Med. 2017, 14, 15–30. [Google Scholar] [CrossRef]
- Lipson, K.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenes. Tissue Repair 2012, 5 (Suppl. S1), S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, E.G.; McLaughlin, M.; Matthews, L.; Gharib, A.M.; Wood, B.J.; Levy, E.; Sinkus, R.; Virtaneva, K.; Sturdevant, D.; Martens, C.; et al. Simtuzumab treatment of advanced liver fibrosis in HIV and HCV-infected adults: Results of a 6-month open-label safety trial. Liver Int. 2016, 36, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Neuschwander-Tetri, B.A.; Wong, V.W.-S.; Abdelmalek, M.F.; Younossi, Z.M.; Yuan, J.; Pecoraro, M.L.; Seyedkazemi, S.; Fischer, L.; Bedossa, P.; et al. Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: AURORA Phase 3 study design. Contemp. Clin. Trials 2019, 89, 105922. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Cenicriviroc for the treatment of non-alcoholic steatohepatitis and liver fibrosis. Expert Opin. Investig. Drugs 2018, 27, 301–311. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2017, 67, 1754–1767. [Google Scholar] [CrossRef]
- Houghton, D.; Stewart, C.J.; Day, C.P.; Trenell, M. Gut Microbiota and Lifestyle Interventions in NAFLD. Int. J. Mol. Sci. 2016, 17, 447. [Google Scholar] [CrossRef]
- Plaza-Díaz, J.; Solís-Urra, P.; Rodríguez-Rodríguez, F.; Olivares-Arancibia, J.; Navarro-Oliveros, M.; Abadía-Molina, F.; Álvarez-Mercado, A. The Gut Barrier, Intestinal Microbiota, and Liver Disease: Molecular Mechanisms and Strategies to Manage. Int. J. Mol. Sci. 2020, 21, 8351. [Google Scholar] [CrossRef]
- Sikalidis, A.K.; Maykish, A. The Gut Microbiome and Type 2 Diabetes Mellitus: Discussing A Complex Relationship. Biomedicines 2020, 8, 8. [Google Scholar] [CrossRef]
- Guo, C.; Zhao, Z.; Deng, X.; Chen, Z.; Tu, Z.; Yuan, G. Regulation of angiopoietin-like protein 8 expression under different nutritional and metabolic status. Endocr. J. 2019, 66, 1039–1046. [Google Scholar] [CrossRef]
- Acierno, C.; Caturano, A.; Pafundi, P.C.; Nevola, R.; Adinolfi, L.E.; Sasso, F.C. Nonalcoholic fatty liver disease and type 2 diabetes: Pathophysiological mechanisms shared between the two faces of the same coin. Explor. Med. 2020, 1, 287–306. [Google Scholar] [CrossRef]
- Papamichou, D.; Panagiotakos, D.; Itsiopoulos, C. Dietary patterns and management of type 2 diabetes: A systematic review of randomised clinical trials. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, J.P.; Sacks, J.; Nieuwoudt, S. The essential role of exercise in the management of type 2 diabetes. Clevel. Clin. J. Med. 2017, 84 (Suppl. S1), S15–S21. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, L.; Nascimbeni, F.; Giordano, M.; Masetti, C.; Guerrera, B.; Amelia, A.; Fascione, M.C.; Ballestri, S.; Romagnoli, D.; Zampino, R.; et al. Clinical features and natural history of cryptogenic cirrhosis compared to hepatitis C virus-related cirrhosis. World J. Gastroenterol. 2017, 23, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Valente, G.; Rinaldi, L.; Sgambato, M.; Piai, G. Conversion from twice-daily to once-daily tacrolimus in stable liver transplant patients: Effectiveness in a real-world setting. Transplant. Proc. 2013, 45, 1273–1275. [Google Scholar] [CrossRef]
- Rinaldi, L.; Nevola, R.; Franci, G.; Perrella, A.; Corvino, G.; Marrone, A.; Berretta, M.; Morone, M.V.; Galdiero, M.; Giordano, M.; et al. Risk of Hepatocellular Carcinoma after HCV Clearance by Direct-Acting Antivirals Treatment Predictive Factors and Role of Epigenetics. Cancers 2020, 12, 1351. [Google Scholar] [CrossRef]
- Ascione, A.; Fontanella, L.; Imparato, M.; Rinaldi, L.; De Luca, M. Mortality from cirrhosis and hepatocellular carcinoma in Western Europe over the last 40 years. Liver Int. 2017, 37, 1193–1201. [Google Scholar] [CrossRef]
- Rinaldi, L.; Di Francia, R.; Coppola, N.; Guerrera, B.; Imparato, M.; Monari, C.; Nevola, R.; Rosato, V.; Fontanella, L.; Franci, G.; et al. Hepatocellular carcinoma in HCV cirrhosis after viral clearance with direct acting antiviral therapy: Preliminary evidence and possible meanings. WCRJ 2016, 3, e748. [Google Scholar]
- El-Serag, H.B.; Richardson, P.A.; Everhart, J.E. The role of diabetes in hepatocellular carcinoma: A case-control study among United States veterans. Am. J. Gastroenterol. 2001, 96, 2462–2467. [Google Scholar] [CrossRef]
- Davila, J.A.; Morgan, R.O.; Shaib, Y.; McGlynn, K.A.; El-Serag, H.B. Diabetes increases the risk of hepatocellular carcinoma in the United States: A population based case control study. Gut 2005, 54, 533–539. [Google Scholar] [CrossRef]
- Lai, S.; Quan, Z.; Hao, Y.; Liu, J.; Wang, Z.; Dai, L.; Dai, H.; He, S.; Tang, B. Long Non-Coding RNA LINC01572 Promotes Hepatocellular Carcinoma Progression via Sponging miR-195-5p to Enhance PFKFB4-Mediated Glycolysis and PI3K/AKT Activation. Front. Cell Dev. Biol. 2021, 9, 783088. [Google Scholar] [CrossRef] [PubMed]
- El–Serag, H.B.; Hampel, H.; Javadi, F. The association between diabetes and hepatocellular carcinoma: A systematic review of epidemiologic evidence. Clin. Gastroenterol. Hepatol. 2006, 4, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.-L.; Han, Y.; Pei, L.; Yue, Z.-H.; Liu, B.-Y.; Cui, J.-W.; Jia, M.; Wang, H. A Serum Metabolite Classifier for the Early Detection of Type 2 Diabetes Mellitus-Positive Hepatocellular Cancer. Metabolites 2022, 12, 610. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.C.P.; Jaffe, A.; Perry, R.; Marabotto, E.; Strazzabosco, M.; Giannini, E.G. Diabetes medications and risk of HCC. Hepatology 2022, 76, 1880–1897. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X.; Gong, G.; Ben, Q.; Qiu, W.; Chen, Y.; Li, G.; Wang, L. Increased risk of hepatocellular carcinoma in patients with diabetes mellitus: A systematic review and meta-analysis of cohort studies. Int. J. Cancer 2012, 130, 1639–1648. [Google Scholar] [CrossRef]
- Shi, Z.; Xiao, Z.; Hu, L.; Gao, Y.; Zhao, J.; Liu, Y.; Shen, G.; Xu, Q.; Huang, D. The genetic association between type 2 diabetic and hepatocellular carcinomas. Ann. Transl. Med. 2020, 8, 380. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-S.; Va, P.; Bray, F.; Gao, S.; Gao, J.; Li, H.-L.; Xiang, Y.-B. The role of pre-existing diabetes mellitus on hepatocellular carcinoma occurrence and prognosis: A meta-analysis of prospective cohort studies. PLoS ONE 2011, 6, e27326. [Google Scholar] [CrossRef]
- Doycheva, I.; Zhang, T.; Amjad, W.; Thuluvath, P.J. Diabetes and Hepatocellular Carcinoma: Incidence Trends and Impact of Liver Disease Etiology. J. Clin. Exp. Hepatol. 2019, 10, 296–303. [Google Scholar] [CrossRef]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef]
- Zaki, M.Y.W.; Mahdi, A.K.; Patman, G.L.; Whitehead, A.; Maurício, J.P.; McCain, M.V.; Televantou, D.; Abou-Beih, S.; Ramon-Gil, E.; Watson, R.; et al. Key features of the environment promoting liver cancer in the absence of cirrhosis. Sci. Rep. 2021, 11, 16727. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, S.; Yang, M. Hepatocellular Carcinoma and Obesity, Type 2 Diabetes Mellitus, Cardiovascular Disease: Causing Factors, Molecular Links, and Treatment Options. Front. Endocrinol. 2021, 12, 808526. [Google Scholar] [CrossRef]
- Ngo, M.-H.; Jeng, H.-Y.; Kuo, Y.-C.; Nanda, J.D.; Brahmadhi, A.; Ling, T.-Y.; Chang, T.-S.; Huang, Y.-H. The Role of IGF/IGF-1R Signaling in Hepatocellular Carcinomas: Stemness-Related Properties and Drug Resistance. Int. J. Mol. Sci. 2021, 22, 1931. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Albanese, G.; Di Martino, A.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Sasso, F.C. The Diabetic Cardiomyopathy: The Contributing Pathophysiological Mechanisms. Front. Med. 2021, 8, 695792. [Google Scholar] [CrossRef]
- Sasso, F.; Pafundi, P.C.; Gelso, A.; Bono, V.; Costagliola, C.; Marfella, R.; Sardu, C.; Rinaldi, L.; Galiero, R.; Acierno, C.; et al. Relationship between albuminuric CKD and diabetic retinopathy in a real-world setting of type 2 diabetes: Findings from No blind study. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 923–930. [Google Scholar] [CrossRef]
- Giordano, M.; Ciarambino, T.; Castellino, P.; Malatino, L.; Cataliotti, A.; Rinaldi, L.; Paolisso, G.; Adinolfi, L.E. Seasonal variations of hyponatremia in the emergency department: Age-related changes. Am. J. Emerg. Med. 2017, 35, 749–752. [Google Scholar] [CrossRef]
- Singh, M.K.; Das, B.K.; Choudhary, S.; Gupta, D.; Patil, U.K. Diabetes and hepatocellular carcinoma: A pathophysiological link and pharmacological management. Biomed. Pharmacother. 2018, 106, 991–1002. [Google Scholar] [CrossRef]
- Grecian, S.M.; McLachlan, S.; Fallowfield, J.A.; Kearns, P.K.A.; Hayes, P.C.; Guha, N.I.; Morling, J.R.; Glancy, S.; Williamson, R.M.; Reynolds, R.M.; et al. Non-invasive risk scores do not reliably identify future cirrhosis or hepatocellular carcinoma in Type 2 diabetes: The Edinburgh type 2 diabetes study. Liver Int. 2020, 40, 2252–2262. [Google Scholar] [CrossRef]
- Luo, X.; Sui, J.; Yang, W.; Sun, Q.; Ma, Y.; Simon, T.G.; Liang, G.; Meyerhardt, J.A.; Chan, A.T.; Giovannucci, E.L.; et al. Type 2 Diabetes Prevention Diet and Hepatocellular Carcinoma Risk in US Men and Women. Am. J. Gastroenterol. 2019, 114, 1870–1877. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, C.; Fang, L.; Zhao, H.-C.; Yao, S.-K. Metformin and reduced risk of hepatocellular carcinoma in diabetic patients: A meta-analysis. Scand. J. Gastroenterol. 2012, 48, 78–87. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.; Galiero, R.; Gjeloshi, K.; Masini, F.; Acierno, C.; Di Martino, A.; Albanese, G.; Alfano, M.; Rinaldi, L.; et al. Metformin: A Potential Therapeutic Tool for Rheumatologists. Pharmaceuticals 2020, 13, 234. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Kim, G.; Lee, Y.-H.; Lee, B.-W.; Cha, B.-S.; Nam, C.; Kang, E. Comparison of hepatocellular carcinoma risk between patients treated with glimepiride and gliclazide. Diabetes Metab. 2017, 45, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, C.; Franchi, M.; Nicotra, F.; Asciutto, R.; Merlino, L.; La Vecchia, C.; Corrao, G. Insulin and other antidiabetic drugs and hepatocellular carcinoma risk: A nested case-control study based on Italian healthcare utilization databases. Pharmacoepidemiol. Drug Saf. 2015, 24, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, P.P.; Singh, A.G.; Murad, M.H.; Sanchez, W. Anti-diabetic medications and the risk of hepatocellular cancer: A systematic review and meta-analysis. Am. J. Gastroenterol. 2013, 108, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, M.; Mitsui, H.; Akihisa, M.; Sekine, T.; Niitsu, Y.; Kobayashi, A.; Miyake, A.; Hashimoto, N.; Kawamura, M.; Ogawa, Y. Efficacy and safety of sitagliptin for the treatment of diabetes mellitus complicated by chronic liver injury. Springerplus 2015, 4, 346. [Google Scholar] [CrossRef]
- Nishina, S.; Yamauchi, A.; Kawaguchi, T.; Kaku, K.; Goto, M.; Sasaki, K.; Hara, Y.; Tomiyama, Y.; Kuribayashi, F.; Torimura, T.; et al. Dipeptidyl Peptidase 4 Inhibitors Reduce Hepatocellular Carcinoma by Activating Lymphocyte Chemotaxis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2018, 7, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Palmiero, G.; Cesaro, A.; Vetrano, E.; Pafundi, P.; Galiero, R.; Caturano, A.; Moscarella, E.; Gragnano, F.; Salvatore, T.; Rinaldi, L.; et al. Impact of SGLT2 Inhibitors on Heart Failure: From Pathophysiology to Clinical Effects. Int. J. Mol. Sci. 2021, 22, 5863. [Google Scholar] [CrossRef]
- Salvatore, T.; Caturano, A.; Galiero, R.; Di Martino, A.; Albanese, G.; Vetrano, E.; Sardu, C.; Marfella, R.; Rinaldi, L.; Sasso, F.C. Cardiovascular Benefits from Gliflozins: Effects on Endothelial Function. Biomedicines 2021, 9, 1356. [Google Scholar] [CrossRef]
- Gardini, A.C.; Marisi, G.; Scarpi, E.; Scartozzi, M.; Faloppi, L.; Silvestris, N.; Masi, G.; Vivaldi, C.; Brunetti, O.; Tamberi, S.; et al. Effects of metformin on clinical outcome in diabetic patients with advanced HCC receiving sorafenib. Expert Opin. Pharmacother. 2015, 16, 2719–2725. [Google Scholar] [CrossRef]
- Sharma, I.; Vakayil, V.; Einstein, M.; Elias, R.; Menendez, A.; Morgan, G.; Rochon, C.; Sotil, E.U.; Hong, T.; Sheiner, P.A.; et al. Disparities in treatment of liver lesions: NSQIP analysis of how race affects oncologic management. WCRJ 2021, 8, e2100. [Google Scholar]
- Jayakrishnan, T.T.; Bakalov, V.; Finley, G.; Monga, D.; Wegner, R.E. Influence of social determinants of health on timeliness to treatment for metastatic HCC and the impact of affordable care act. WCRJ 2021, 8, e2073. [Google Scholar] [CrossRef]
- Rinaldi, L.; Vetrano, E.; Rinaldi, B.; Galiero, R.; Caturano, A.; Salvatore, T.; Sasso, F.C. HCC and Molecular Targeting Therapies: Back to the Future. Biomedicines 2021, 9, 1345. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Dawkins, J.; Webster, R.M. The hepatocellular carcinoma market. Nat. Rev. Drug Discov. 2019, 18, 13–14. [Google Scholar] [CrossRef]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.; Meyer, T.; Nault, J.-C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 871–873. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Gordan, J.D.; Kennedy, E.B.; Abou-Alfa, G.K.; Beg, M.S.; Brower, S.T.; Gade, T.P.; Goff, L.; Gupta, S.; Guy, J.; Harris, W.P.; et al. Systemic Therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 4317–4345. [Google Scholar] [CrossRef]
- Bruix, J.; Chan, S.L.; Galle, P.R.; Rimassa, L.; Sangro, B. Systemic treatment of hepatocellular carcinoma: An EASL position paper. J. Hepatol. 2021, 75, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.V.; Merle, P.; et al. IMbrave150: Updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC). J. Clin. Oncol. 2021, 39, 267. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; Toni, E.N.D.; et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. J. Clin. Oncol. 2022, 40, 379. [Google Scholar] [CrossRef]
- Maestri, M.; Pallozzi, M.; Santopaolo, F.; Cerrito, L.; Pompili, M.; Gasbarrini, A.; Ponziani, F.R. Durvalumab: An investigational agent for unresectable hepatocellular carcinoma. Expert Opin. Investig. Drugs 2022, 31, 347–360. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Kelley, R.K.; Meyer, T.; Rimassa, L.; Merle, P.; Park, J.-W.; Yau, T.; Chan, S.L.; Blanc, J.-F.; Tam, V.C.; Tran, A.; et al. Serum Alpha-fetoprotein Levels and Clinical Outcomes in the Phase III CELESTIAL Study of Cabozantinib versus Placebo in Patients with Advanced Hepatocellular Carcinoma. Clin. Cancer Res. 2020, 26, 4795–4804. [Google Scholar] [CrossRef]
- Zhu, A.X.; Park, J.O.; Ryoo, B.-Y.; Yen, C.-J.; Poon, R.; Pastorelli, D.; Blanc, J.-F.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E.F.; et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as Second-Line Therapy in Patients with Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Qin, S.; Chen, Z.; Fang, W.; Ren, Z.; Xu, R.; Ryoo, B.-Y.; Meng, Z.; Bai, Y.; Chen, X.; Liu, X.; et al. Pembrolizumab versus Placebo as Second-Line Therapy in Patients from Asia with Advanced Hepatocellular Carcinoma: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2022, JCO2200620. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval to Nivolumab and Ipilimumab Combination for Hepatocellular Carcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-nivolumab-and-ipilimumab-combination-hepatoc (accessed on 31 October 2022).
- Qin, S.; Bi, F.; Gu, S.; Bai, Y.; Chen, Z.; Wang, Z.; Ying, J.; Lu, Y.; Meng, Z.; Pan, H.; et al. Donafenib Versus Sorafenib in First-Line Treatment of Unresectable or Metastatic Hepatocellular Carcinoma: A Randomized, Open-Label, Parallel-Controlled Phase II-III Trial. J. Clin. Oncol. 2021, 39, 3002–3011. [Google Scholar] [CrossRef]
- Feng, M.Y.; Chan, L.L.; Chan, S.L. Drug Treatment for Advanced Hepatocellular Carcinoma: First-Line and Beyond. Curr. Oncol. 2022, 29, 5489–5507. [Google Scholar] [CrossRef]
- Yang, T.K.; Yu, Y.F.; Tsai, C.L.; Li, H.J.; Yang, P.S.; Huang, K.W.; Cheng, J.C.H. Efficacy and safety of combined targeted therapy and immunotherapy versus targeted monotherapy in unresectable hepatocellular carcinoma: A systematic review and meta-analysis. BMC Cancer 2022, 22, 1085. [Google Scholar] [CrossRef]
- Sasso, F.C.; Pafundi, P.C.; Caturano, A.; Galiero, R.; Vetrano, E.; Nevola, R.; Petta, S.; Fracanzani, A.L.; Coppola, C.; Di Marco, V.; et al. Impact of direct acting antivirals (DAAs) on cardiovascular events in HCV cohort with pre-diabetes. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 2345–2353. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vetrano, E.; Rinaldi, L.; Mormone, A.; Giorgione, C.; Galiero, R.; Caturano, A.; Nevola, R.; Marfella, R.; Sasso, F.C. Non-alcoholic Fatty Liver Disease (NAFLD), Type 2 Diabetes, and Non-viral Hepatocarcinoma: Pathophysiological Mechanisms and New Therapeutic Strategies. Biomedicines 2023, 11, 468. https://doi.org/10.3390/biomedicines11020468
Vetrano E, Rinaldi L, Mormone A, Giorgione C, Galiero R, Caturano A, Nevola R, Marfella R, Sasso FC. Non-alcoholic Fatty Liver Disease (NAFLD), Type 2 Diabetes, and Non-viral Hepatocarcinoma: Pathophysiological Mechanisms and New Therapeutic Strategies. Biomedicines. 2023; 11(2):468. https://doi.org/10.3390/biomedicines11020468
Chicago/Turabian StyleVetrano, Erica, Luca Rinaldi, Andrea Mormone, Chiara Giorgione, Raffaele Galiero, Alfredo Caturano, Riccardo Nevola, Raffaele Marfella, and Ferdinando Carlo Sasso. 2023. "Non-alcoholic Fatty Liver Disease (NAFLD), Type 2 Diabetes, and Non-viral Hepatocarcinoma: Pathophysiological Mechanisms and New Therapeutic Strategies" Biomedicines 11, no. 2: 468. https://doi.org/10.3390/biomedicines11020468