
Lysosomal-Cleavable Peptide Linkers in Antibody–Drug Conjugates


Abstract
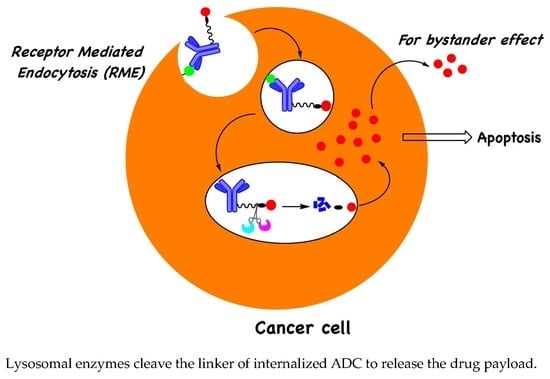
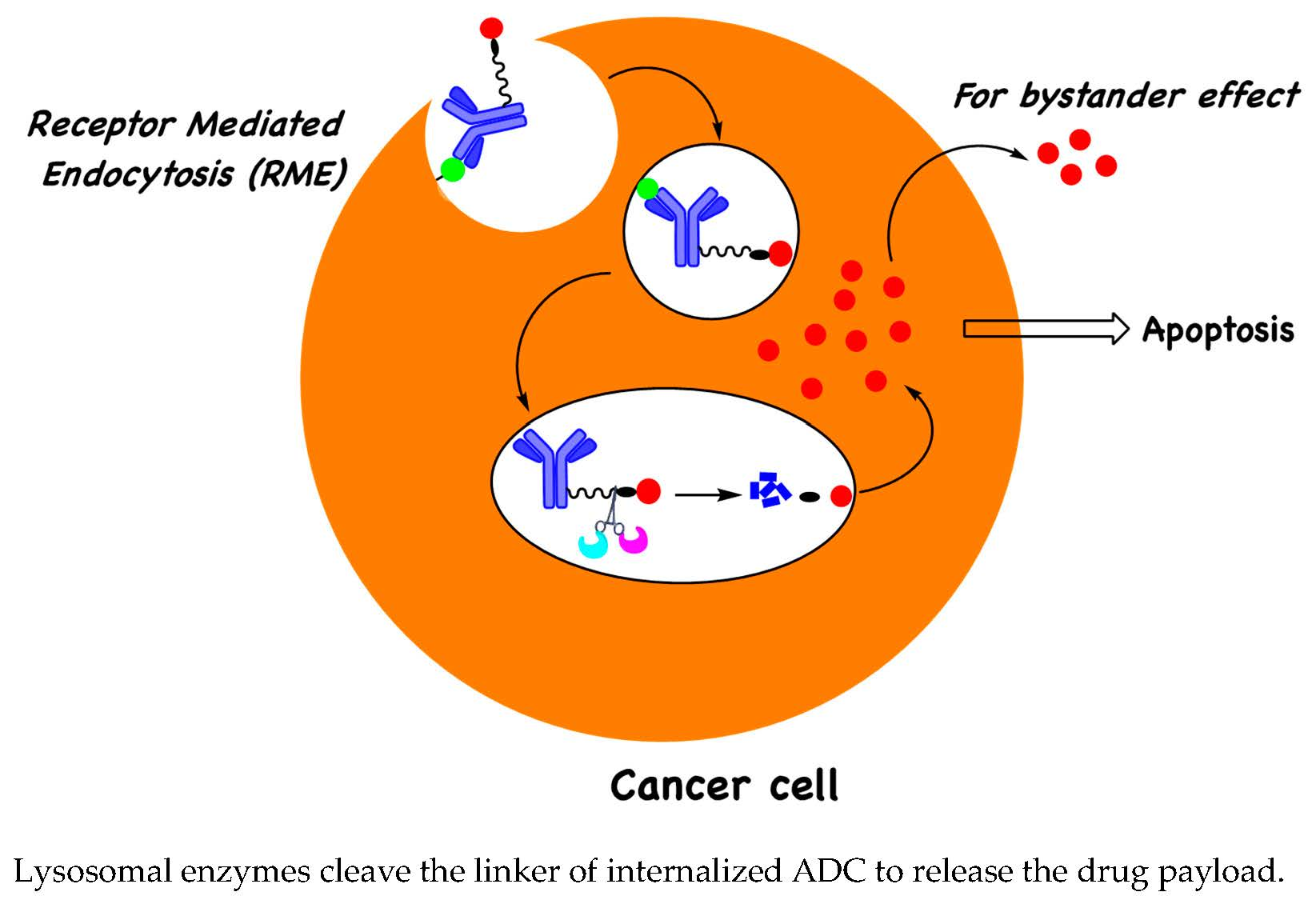
:
1. Introduction
2. Adding a P3 Polar Acidic Residue to the ValCitPABC Linker Increases Plasma Stability
3. Polar Basic Residue Substitution at the P1 Position of the ValCit-PABC Linker Improves Lysosomal Cleavage Activity
4. Peptidomimetic Substitution on the Cathepsin-Specific Dipeptide-PABC Linker
5. The Effect of Substitution on the PABC Benzene Ring
6. Tandem Cleavable Linkers
7. The GGFG Tetrapeptidyl-Aminomethoxy Linker
8. Legumain-Cleavable Linkers
9. Site of Conjugation on ADC Stability and Activity
10. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.T.; Harris, P.W.; Brimble, M.A.; Kavianinia, I. An insight into FDA approved antibody-drug conjugates for cancer therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R. Drawing lessons from the clinical development of antibody-drug conjugates. Drug Discov. Today Technol. 2018, 30, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Hughes, B. Antibody-drug conjugates for cancer: Poised to deliver? Highlighted by Genentech’s recent US regulatory submission for trastuzumab-DM1, antibody-drug conjugation technology could be heading for the mainstream in anticancer drug development. Nat. Rev. Drug Discov. 2010, 9, 665–668. [Google Scholar] [CrossRef]
- Carter, P.J.; Senter, P.D. Antibody-drug conjugates for cancer therapy. Cancer J. 2008, 14, 154–169. [Google Scholar] [CrossRef]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016, 8, 659–671. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotech. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Doronina, S.O.; Bovee, T.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.E.; Morris-Tilden, C.A.; Senter, P.D. Novel peptide linkers for highly potent antibody—Auristatin conjugate. Bioconjug. Chem. 2008, 19, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
- Caculitan, N.G.; Chuh, J.D.C.; Ma, Y.; Zhang, D.; Kozak, K.R.; Liu, Y.; Pillow, T.H.; Sadowsky, J.; Cheung, T.K.; Phung, Q.; et al. Cathepsin B is dispensable for cellular processing of cathepsin B-cleavable antibody-drug conjugates. Cancer Res. 2017, 77, 7027–7037. [Google Scholar] [CrossRef] [PubMed]
- Salomon, P.L.; Reid, E.E.; Archer, K.E.; Harris, L.; Maloney, E.K.; Wilhelm, A.J.; Miller, M.L.; Chari, R.V.; Keating, T.A.; Singh, R. Optimizing Lysosomal Activation of Antibody–Drug Conjugates (ADCs) by Incorporation of Novel Cleavable Dipeptide Linkers. Mol. Pharm. 2019, 16, 4817–4825. [Google Scholar] [CrossRef] [PubMed]
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.-T. Molecular basis of valine-citrulline-pabc linker instability in site-specific ADCs and its mitigation by linker designmolecular basis of VC-PABC linker instability. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Mahalingaiah, P.K.; Ciurlionis, R.; Durbin, K.R.; Yeager, R.L.; Philip, B.K.; Bawa, B.; Mantena, S.R.; Enright, B.P.; Liguori, M.J.; Van Vleet, T.R. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol. Ther. 2019, 200, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; De Vos, S. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. 2012, 30, 2183. [Google Scholar] [CrossRef]
- Zhao, H.; Gulesserian, S.; Malinao, M.C.; Ganesan, S.K.; Song, J.; Chang, M.S.; Williams, M.M.; Zeng, Z.; Mattie, M.; Mendelsohn, B.A. A potential mechanism for ADC-induced neutropenia: Role of neutrophils in their own demisemechanism for ADC-induced neutropenia. Mol. Cancer Ther. 2017, 16, 1866–1876. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Poreba, M. Protease-activated prodrugs: Strategies, challenges, and future directions. Febs J. 2020, 287, 1936–1969. [Google Scholar] [CrossRef]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody–drug conjugates: Recent advances in linker chemistry. Acta. Pharm. Sin. B 2021, 11, 3889–3907. [Google Scholar] [CrossRef]
- Mckertish, C.M.; Kayser, V. Advances and limitations of antibody drug conjugates for cancer. Biomedicines 2021, 9, 872. [Google Scholar] [CrossRef] [PubMed]
- Sheyi, R.; de la Torre, B.G.; Albericio, F. Linkers: An assurance for controlled delivery of antibody-drug conjugate. Pharmaceutics 2022, 14, 396. [Google Scholar] [CrossRef] [PubMed]
- Mort, J.S.; Buttle, D.J. Cathepsin b. Int. J. Biochem. Cell Biol. 1997, 29, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhao, M.; Yan, C.; Kong, W.; Lan, F.; Narengaowa; Zhao, S.; Yang, Q.; Bai, Z.; Qing, H. Cathepsin B in programmed cell death machinery: Mechanisms of execution and regulatory pathways. Cell Death Dis. 2023, 14, 255. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Anami, Y.; Yamazaki, C.M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid–valine–citrulline linkers ensure stability and efficacy of antibody–drug conjugates in mice. Nat. Commun. 2018, 9, 2512. [Google Scholar] [CrossRef]
- Poudel, Y.B.; Chowdari, N.S.; Cheng, H.; Iwuagwu, C.I.; King, H.D.; Kotapati, S.; Passmore, D.; Rampulla, R.; Mathur, A.; Vite, G. Chemical modification of linkers provides stable linker–payloads for the generation of antibody–drug conjugates. ACS Med. Chem. Lett. 2020, 11, 2190–2194. [Google Scholar] [CrossRef]
- Wei, B.; Gunzner-Toste, J.; Yao, H.; Wang, T.; Wang, J.; Xu, Z.; Chen, J.; Wai, J.; Nonomiya, J.; Tsai, S.P. Discovery of peptidomimetic antibody–drug conjugate linkers with enhanced protease specificity. J. Med. Chem. 2018, 61, 989–1000. [Google Scholar] [CrossRef]
- Poudel, Y.B.; Rao, C.; Kotapati, S.; Deshpande, M.; Thevanayagam, L.; Pan, C.; Cardarelli, J.; Chowdari, N.; Kaspady, M.; Samikannu, R. Design, synthesis and biological evaluation of phenol-linked uncialamycin antibody-drug conjugates. Bioorg. Med. Chem. Lett. 2020, 30, 126782. [Google Scholar] [CrossRef]
- Chuprakov, S.; Ogunkoya, A.O.; Barfield, R.M.; Bauzon, M.; Hickle, C.; Kim, Y.C.; Yeo, D.; Zhang, F.; Rabuka, D.; Drake, P.M. Tandem-cleavage linkers improve the in vivo stability and tolerability of antibody–drug conjugates. Bioconjug. Chem. 2021, 32, 746–754. [Google Scholar] [CrossRef]
- Agarwal, P.; Kudirka, R.; Albers, A.E.; Barfield, R.M.; de Hart, G.W.; Drake, P.M.; Jones, L.C.; Rabuka, D. Hydrazino-pictet-spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconjug. Chem. 2013, 24, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Rabuka, D.; Rush, J.S.; Dehart, G.W.; Wu, P.; Bertozzi, C.R. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 2012, 7, 1052–1067. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Walsh, S.J.; Ashman, N.; Isidro-Llobet, A.; Carroll, J.S.; Spring, D.R. A dual-enzyme cleavable linker for antibody–drug conjugates. Chem. Commun. 2021, 57, 3457–3460. [Google Scholar] [CrossRef] [PubMed]
- Nogusa, H.; Yano, T.; Okuno, S.; Hamada, H.; Inoue, K. Synthesis of Carboxymethylpullulan-Peptide-Doxorubicin Conjugates and Their Properties. Chem. Pharm. Bull. 1995, 43, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Shiose, Y.; Ochi, Y.; Kuga, H.; Yamashita, F.; Hashida, M. Relationship between Drug Release of DE-310, Macromolecular Prodrug of DX-8951f, and Cathepsins Activity in Several Tumors. Biol. Pharm. Bull. 2007, 30, 2365–2370. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, I.; Kumazawa, E.; Hirota, Y.; Aonuma, M.; Sugimori, M.; Ohsuki, S.; Uoto, K.; Ejima, A.; Terasawa, H.; Sato, K. A New Water-Soluble Camptothecin Derivative, DX-8951f, Exhibits Potent Antitumor Activity against Human Tumors in Vitro and in Vivo. Jpn. J. Cancer Res. 1995, 86, 776–782. [Google Scholar] [CrossRef]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Modi, S.; Park, H.; Murthy, R.K.; Iwata, H.; Tamura, K.; Tsurutani, J.; Moreno-Aspitia, A.; Doi, T.; Sagara, Y.; Redfern, C.; et al. Antitumor Activity and Safety of Trastuzumab Deruxtecan in Patients with HER2-Low–Expressing Advanced Breast Cancer: Results From a Phase Ib Study. J. Clin. Oncol. 2020, 38, 1887–1896. [Google Scholar] [CrossRef]
- Ishii, S. Legumain: Asparaginyl endopeptidase. Methods Enzymol. 1994, 244, 604–615. [Google Scholar]
- Chen, J.M.; Dando, P.M.; Stevens, R.A.E.; Fortunato, M.; Barrett, A.J. Cloning and expression of mouse legumain, a lysosomal endopeptidase. Biochem. J. 1998, 335, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-M.; Dando, P.M.; Rawlings, N.D.; Brown, M.A.; Young, N.E.; Stevens, R.A.; Hewitt, E.; Watts, C.; Barrett, A.J. Cloning, isolation, and characterization of mammalian legumain, an asparaginyl endopeptidase. J. Biol. Chem. 1997, 272, 8090–8098. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sun, C.; Huang, H.; Janda, K.; Edgington, T. Overexpression of legumain in tumors is significant for invasion/metastasis and a candidate enzymatic target for prodrug therapy. Cancer Res. 2003, 63, 2957–2964. [Google Scholar] [PubMed]
- Wu, W.; Luo, Y.; Sun, C.; Liu, Y.; Kuo, P.; Varga, J.; Xiang, R.; Reisfeld, R.; Janda, K.D.; Edgington, T.S. Targeting cell-impermeable prodrug activation to tumor microenvironment eradicates multiple drug-resistant neoplasms. Cancer Res. 2006, 66, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Bajjuri, K.M.; Liu, Y.; Liu, C.; Sinha, S.C. The legumain protease-activated auristatin prodrugs suppress tumor growth and metastasis without toxicity. ChemMedChem 2011, 6, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Li, J.; Tanaka, K.; Majumder, U.; Milinichik, A.Z.; Verdi, A.C.; Maddage, C.J.; Rybinski, K.A.; Fernando, S.; Fernando, D. MORAb-202, an antibody–drug conjugate utilizing humanized anti-human frα farletuzumab and the microtubule-targeting agent eribulin, has potent antitumor activityMORAb-202, an anti-FRA ADC utilizing eribulin as payload. Mol. Cancer Ther. 2018, 17, 2665–2675. [Google Scholar] [CrossRef]
- Lerchen, H.-G.; Stelte-Ludwig, B.; Sommer, A.; Berndt, S.; Rebstock, A.-S.; Johannes, S.; Mahlert, C.; Greven, S.; Dietz, L.; Jörißen, H. Tailored linker chemistries for the efficient and selective activation of ADCs with KSPi payloads. Bioconjug. Chem. 2020, 31, 1893–1898. [Google Scholar] [CrossRef]
- Lerchen, H.G.; Wittrock, S.; Stelte-Ludwig, B.; Sommer, A.; Berndt, S.; Griebenow, N.; Rebstock, A.S.; Johannes, S.; Cancho-Grande, Y.; Mahlert, C. Antibody–drug conjugates with pyrrole-based KSP inhibitors as the payload class. Angew. Chem. Int. Ed. 2018, 57, 15243–15247. [Google Scholar] [CrossRef]
- Karpov, A.S.; Nieto-Oberhuber, C.M.; Abrams, T.; Beng-Louka, E.; Blanco, E.; Chamoin, S.; Chene, P.; Dacquignies, I.; Daniel, D.; Dillon, M.P.; et al. Discovery of potent and selective antibody–drug conjugates with Eg5 inhibitors through linker and payload optimization. ACS Med. Chem. Lett. 2019, 10, 1674–1679. [Google Scholar] [CrossRef]
- Miller, J.T.; Vitro, C.N.; Fang, S.; Benjamin, S.R.; Tumey, L.N. Enzyme-agnostic lysosomal screen identifies new legumain-cleavable ADC linkers. Bioconjug. Chem. 2021, 32, 842–858. [Google Scholar] [CrossRef]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-Selective Modification Strategies in Antibody-Drug Conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.-Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.-F.; Mai, E.; et al. Conjugation Site Modulates the In Vivo Stability and Therapeutic Activity of Antibody-Drug Conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Dorywalska, M.; Strop, P.; Melton-Witt, J.A.; Hasa-Moreno, A.; Farias, S.E.; Galindo Casas, M.; Delaria, K.; Lui, V.; Poulsen, K.; Loo, C.; et al. Effect of Attachment Site on Stability of Cleavable Antibody Drug Conjugates. Bioconjugate Chem. 2015, 26, 650–659. [Google Scholar] [CrossRef]
- Kaempffe, A.; Dickgiesser, S.; Rasche, N.; Paoletti, A.; Bertotti, E.; De Salve, I.; Sirtori, F.R.; Kellner, R.; Könning, D.; Hecht, S.; et al. Effect of Conjugation Site and Technique on the Stability and Pharmacokinetics of Antibody-Drug Conjugates. J. Pharm. Sci. 2021, 110, 3776–3785. [Google Scholar] [CrossRef] [PubMed]
- Ashman, N.; Bargh, J.D.; Spring, D.R. Non-internalising antibody-drug conjugates. Chem. Soc. Rev. 2022, 51, 9182–9202. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | ADC | Linker System | Cleavage Mechanism | Payload | Company |
|---|---|---|---|---|---|
| 1 | Gemtuzumab ozogamicin (Mylotarg) | 4-(4-acetylphenoxy) butanoic acid | pH sensitive | Calicheamicin | Pfizer/Wyeth |
| 2 | Brentuximab vedotin (Adcetris) | mc-ValCitPABC | Lysosomal | MMAE | Seattle/Takeda |
| 3 | Trastuzumab emtansine (Kadcyla) | MCC | Non-cleavable | Maytansine DM1 | Genentech Roche |
| 4 | Inotuzumab ozogamicin (Besponsa) | Hydrazone | pH sensitive | Calicheamicin | Pfizer/Wyeth |
| 5 | Polatuzumab vedotin (Polivy) | mc-ValCitPABC | Lysosomal degradation | MMAE | Genentech Roche |
| 6 | Enfortumab vedotin (Padcev) | mc-ValCitPABC | Lysosomal degradation | MMAE | Astellas/Seattle Genetics |
| 7 | Trastuzumab deruxtecan (Enhertu) | mc-GGFG-aminomethoxy | Lysosomal degradation | Deruxtecan, Dxd | Daiichi-Sankyo/AstraZeneca |
| 8 | Sacituzumab govitecan (Trodelvy) | mc-PEG-carbonate | pH | SN-98 | Immunomedics |
| 9 | Belantamab mafodotin (Blenerp) * | mc-MMAF | Non-cleavable | MMAF | GSK |
| 10 | Loncastuximab tesirine (Zynlonta) | mc-ValCitPABC | Lysosomal degradation | SG3199, PDB dimer | ADC Therapeutics |
| 11 | Tisotumab vedotin (Tivdak) | mc-ValCitPABC | Lysosomal degradation | MMAE | Genmab and Seattle Genetics |
| 12 | Disitamab Vedotin (Aidixi) | mc-ValCitPABC | Lysosomal degradation | MMAE | RemeGen |
| 13 | Moxetumomab pasudotox (Lumoxiti) | mc-ValCitPABC | Lysosomal degradation | PE38 | AstraZeneca |
| 14 | Cetuximab sarotalocan (Akalux) | NA | NA | IRDye700DX | Rakuten Medical |
| 15 | Mirvetuximab Soravtansine (ELAHERE) | Disulfide-containing cleavable linker sulfo-SPDB | Glutathione cleavable | Maytansinoid DM4 | ImmunoGen |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balamkundu, S.; Liu, C.-F. Lysosomal-Cleavable Peptide Linkers in Antibody–Drug Conjugates. Biomedicines 2023, 11, 3080. https://doi.org/10.3390/biomedicines11113080
Balamkundu S, Liu C-F. Lysosomal-Cleavable Peptide Linkers in Antibody–Drug Conjugates. Biomedicines. 2023; 11(11):3080. https://doi.org/10.3390/biomedicines11113080
Chicago/Turabian StyleBalamkundu, Seetharamsing, and Chuan-Fa Liu. 2023. "Lysosomal-Cleavable Peptide Linkers in Antibody–Drug Conjugates" Biomedicines 11, no. 11: 3080. https://doi.org/10.3390/biomedicines11113080