
Homozygous Duplication in the CHRNE in a Family with Congenital Myasthenic Syndrome 4C: 18-Year Follow Up

Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects and Ethical Approval
2.2. Extraction of Genomic DNA and Quantification
2.3. Exome Sequencing
2.4. Sanger Sequencing and In Silico Analysis
3. Results
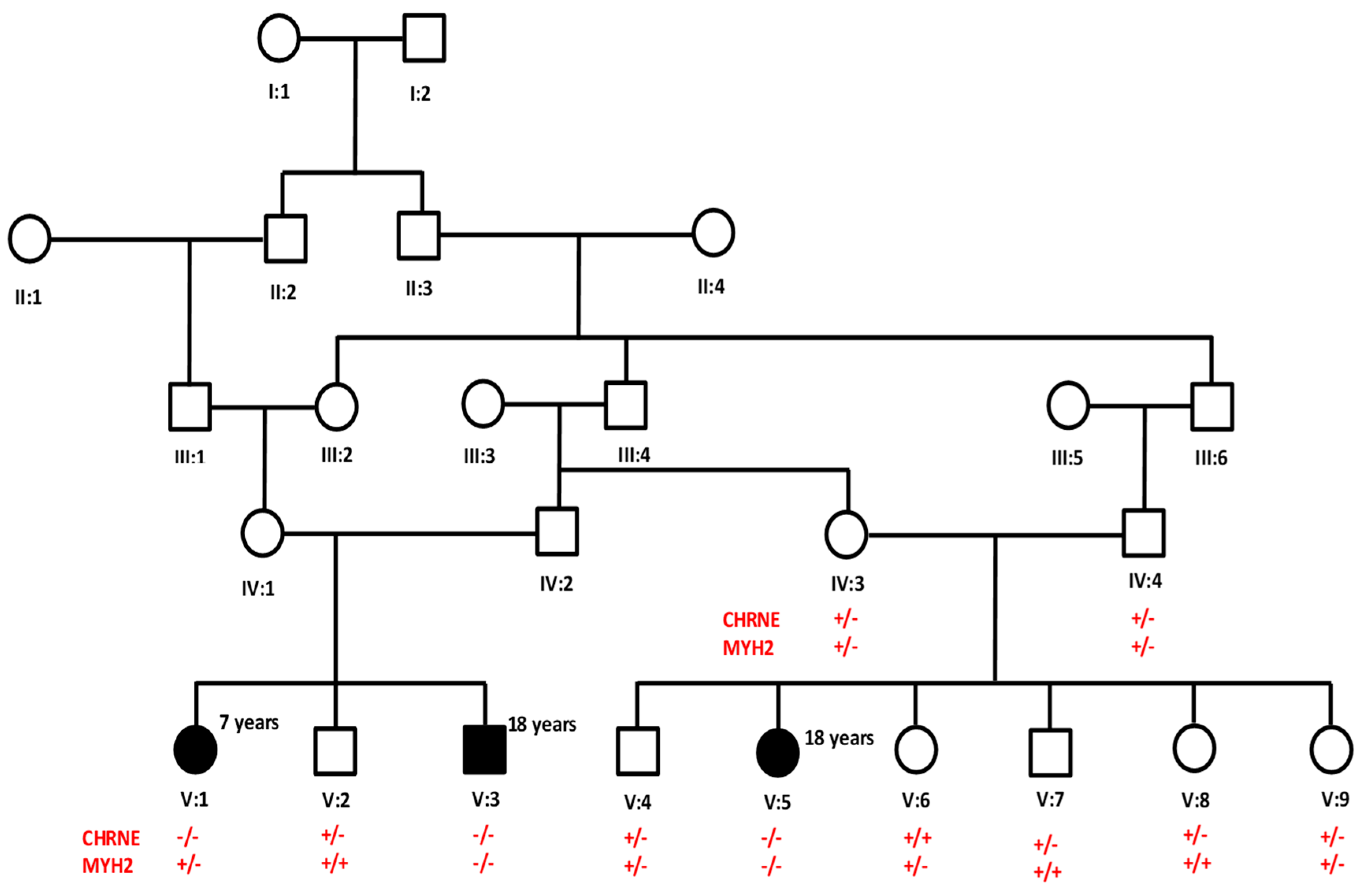
3.1. Clinical Assessment
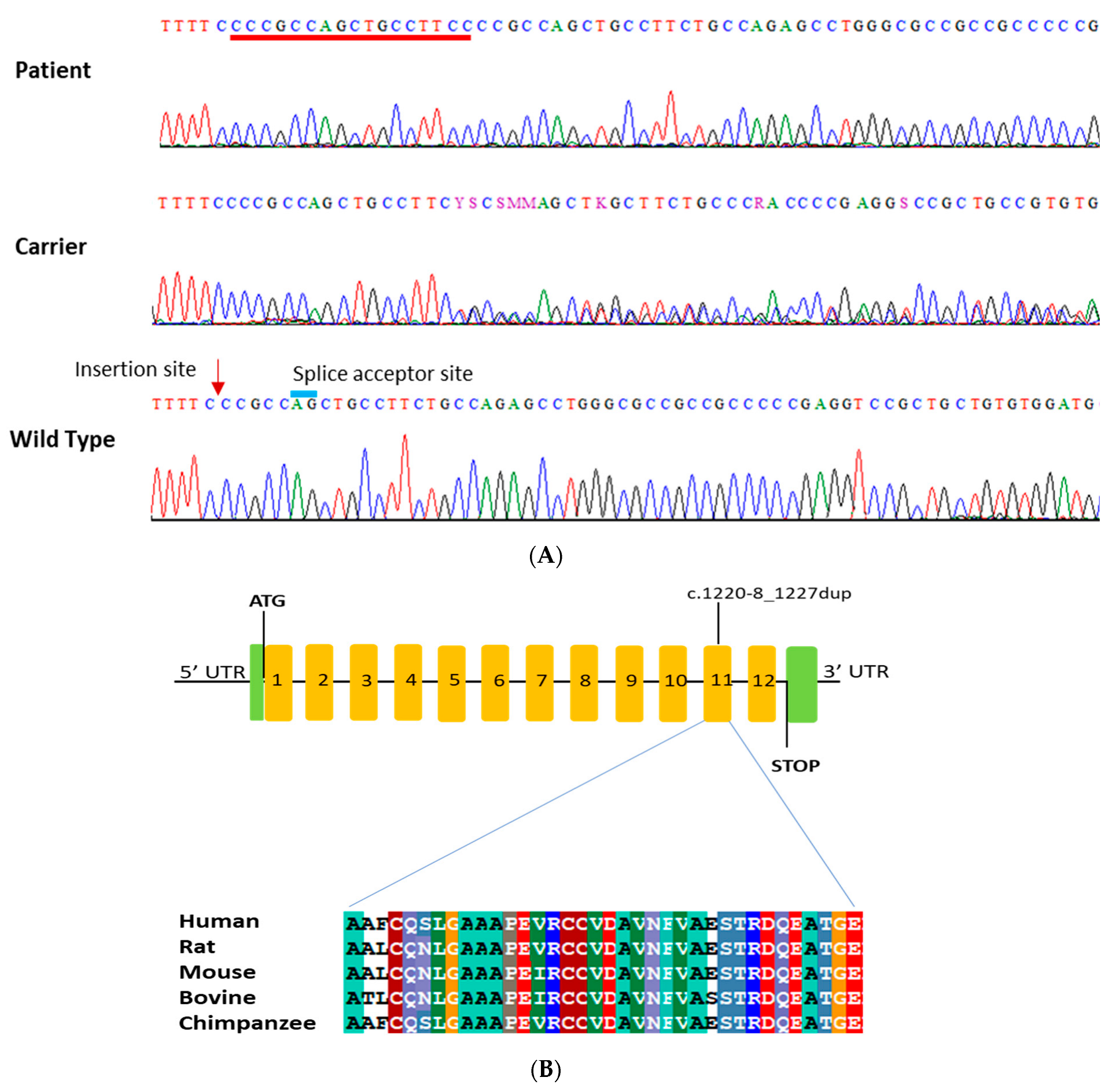
3.2. Genetic Analysis
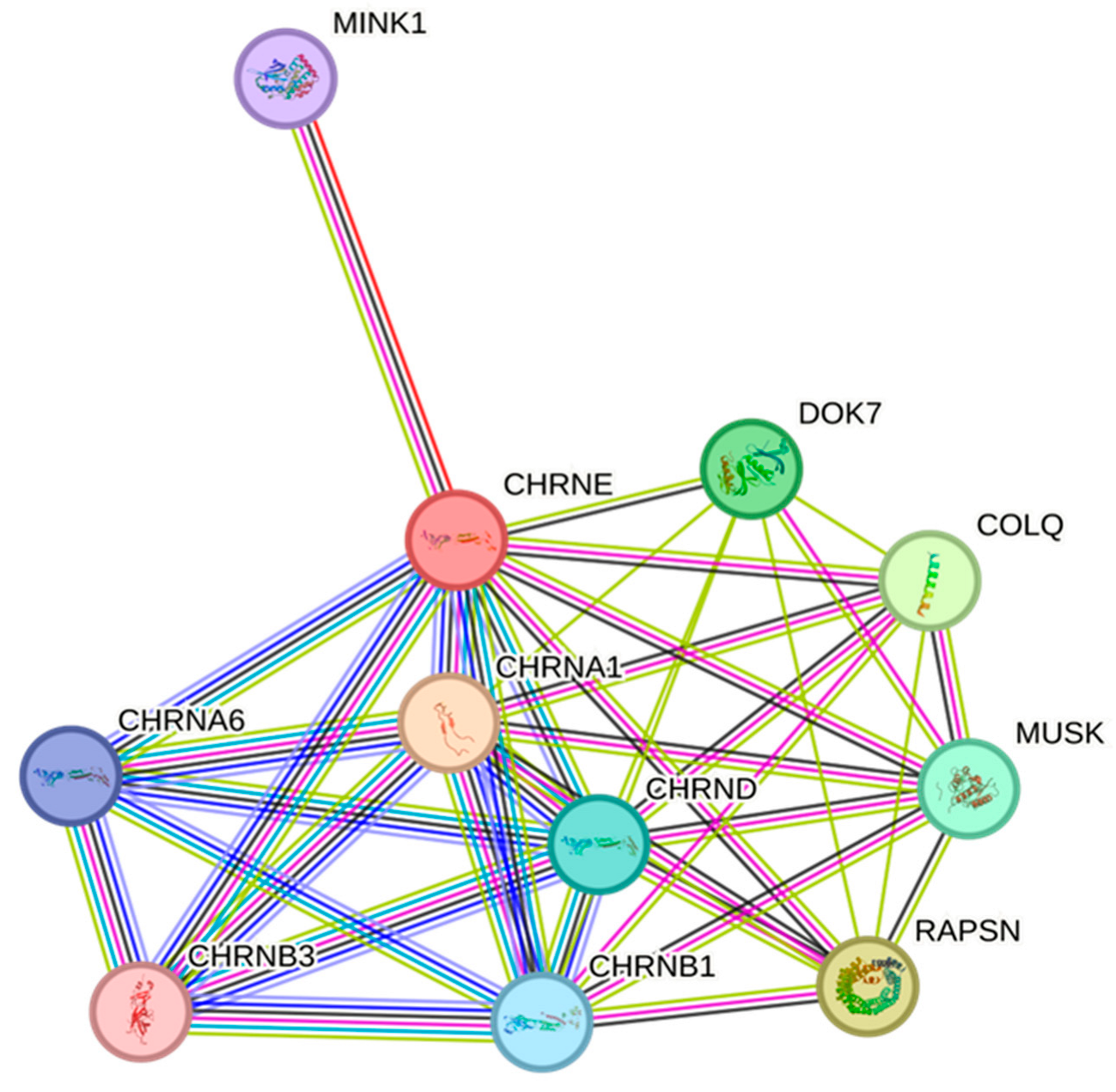
3.3. CHRNE Protein Conservation and Interaction Network Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodríguez Cruz, P.M.; Palace, J.; Beeson, D. Congenital myasthenic syndromes and the neuromuscular junction. Curr. Opin. Neurol. 2014, 27, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434, Erratum in Lancet Neurol. 2015, 14, 461. [Google Scholar] [CrossRef] [PubMed]
- McMacken, G.; Abicht, A.; Evangelista, T.; Spendiff, S.; Lochmüller, H. The Increasing Genetic and Phenotypical Diversity of Congenital Myasthenic Syndromes. Neuropediatrics 2017, 48, 294–308. [Google Scholar] [CrossRef]
- Rodríguez Cruz, P.M.; Palace, J.; Beeson, D. The Neuromuscular Junction and Wide Heterogeneity of Congenital Myasthenic Syndromes. Int. J. Mol. Sci. 2018, 19, 1677. [Google Scholar] [CrossRef]
- Finsterer, J. Congenital myasthenic syndromes. Orphanet J. Rare Dis. 2019, 14, 57. [Google Scholar] [CrossRef]
- Engel, A.G.; Lambert, E.H.; Gomez, M.R. A new myasthenic syndrome with end-plate acetylcholinesterase deficiency, small nerve terminals, and reduced acetylcholine release. Ann. Neurol. 1977, 1, 315–330. [Google Scholar] [CrossRef]
- Yang, K.; Cheng, H.; Yuan, F.; Meng, L.; Yin, R.; Zhang, Y.; Wang, S.; Wang, C.; Lu, Y.; Xi, J.; et al. CHRNE compound heterozygous mutations in congenital myasthenic syndrome: A case report. Medicine 2018, 97, e0347. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Milone, M.; Shen, X.M.; Engel, A.G. A frameshifting mutation in CHRNE unmasks skipping of the preceding exon. Hum. Mol. Genet. 2003, 12, 3055–3066. [Google Scholar] [CrossRef] [PubMed]
- Rinz, C.J.; Lennon, V.A.; James, F.; Thoreson, J.B.; Tsai, K.L.; Starr-Moss, A.N.; Humphries, H.D.; Guo, L.T.; Palmer, A.C.; Clark, L.A.; et al. A CHRNE frameshift mutation causes congenital myasthenic syndrome in young Jack Russell Terriers. Neuromuscul. Disord. 2015, 25, 921–927. [Google Scholar] [CrossRef]
- Salih, M.A.; Oystreck, D.T.; Al-Faky, Y.H.; Kabiraj, M.; Omer, M.I.; Subahi, E.M.; Beeson, D.; Abu-Amero, K.K.; Bosley, T.M. Congenital myasthenic syndrome due to homozygous CHRNE mutations: Report of patients in Arabia. J. Neuroophthalmol. 2011, 31, 42–47. [Google Scholar] [CrossRef]
- Sieb, J.P.; Kraner, S.; Rauch, M.; Steinlein, O.K. Immature end-plates and utrophin deficiency in congenital myasthenic syndrome caused by epsilon-AChR subunit truncating mutations. Hum. Genet. 2000, 107, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Gaudon, K.; Haddad, H.; Ammar, A.B.; Genin, E.; Bauché, S.; Paturneau-Jouas, M.; Müller, J.S.; Lochmüller, H.; Grid, D.; et al. The CHRNE 1293insG founder mutation is a frequent cause of congenital myasthenia in North Africa. Neurology 2008, 71, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Abicht, A.; Stucka, R.; Karcagi, V.; Herczegfalvi, A.; Horváth, R.; Mortier, W.; Schara, U.; Ramaekers, V.; Jost, W.; Brunner, J.; et al. A common mutation (epsilon1267delG) in congenital myasthenic patients of Gypsy ethnic origin. Neurology 1999, 53, 1564–1569. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Lispi, L.; Salvoro, C.; Mostacciuolo, M.L.; Vazza, G. Clinical and genetic characterization of an Italian family with slow-channel syndrome. Neurol. Sci. 2019, 40, 503–507. [Google Scholar] [CrossRef]
- Ardissone, A.; Moroni, I.; Bernasconi, P.; Brugnoni, R. Congenital myasthenic syndrome: Phenotypic variability in patients harbouring p.T159P mutation in CHRNE gene. Acta Myol. 2017, 36, 28–32. [Google Scholar]
- Almatrafi, A.M.; Hibshi, A.M.; Basit, S. Association of Homozygous PROP1 Mutation in a Saudi Family with Combined Pituitary Hormone Deficiency. Medicina 2023, 59, 474. [Google Scholar] [CrossRef]
- Hashmi, J.A.; Albarry, M.A.; Almatrafi, A.M.; Albalawi, A.M.; Mahmood, A.; Basit, S. Whole exome sequencing identified a novel single base pair insertion mutation in the EYS gene in a six generation family with retinitis pigmentosa. Congenit. Anom. 2018, 58, 10–15. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Durmus, H.; Shen, X.M.; Serdaroglu-Oflazer, P.; Kara, B.; Parman-Gulsen, Y.; Ozdemir, C.; Brengman, J.; Deymeer, F.; Engel, A.G. Congenital myasthenic syndromes in Turkey: Clinical clues and prognosis with long term follow-up. Neuromuscul. Disord. 2018, 28, 315–322, Erratum in Neuromuscul. Disord. 2018, 28, 896. [Google Scholar] [CrossRef]
- Vanhaesebrouck, A.E.; Beeson, D. The congenital myasthenic syndromes: Expanding genetic and phenotypic spectrums and refining treatment strategies. Curr. Opin. Neurol. 2019, 32, 696–703. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. CCDS Report for Consensus CDS. Available online: https://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi?REQUEST=CCDS&GO=MainBrowse&DATA=CCDS11058.1 (accessed on 22 July 2023).
- Webster, R.; Liu, W.W.; Chaouch, A.; Lochmüller, H.; Beeson, D. Fast-channel congenital myasthenic syndrome with a novel acetylcholine receptor mutation at the α-ε subunit interface. Neuromuscul. Disord. 2014, 24, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.M.; Okuno, T.; Milone, M.; Otsuka, K.; Takahashi, K.; Komaki, H.; Giles, E.; Ohno, K.; Engel, A.G. Mutations Causing Slow-Channel Myasthenia Reveal That a Valine Ring in the Channel Pore of Muscle AChR is Optimized for Stabilizing Channel Gating. Hum. Mutat. 2016, 37, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Maquat, L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467. [Google Scholar] [CrossRef]
- Hsu, M.K.; Lin, H.Y.; Chen, F.C. NMD Classifier: A reliable and systematic classification tool for nonsense-mediated decay events. PLoS ONE 2017, 12, e0174798. [Google Scholar] [CrossRef] [PubMed]
- Nadezhdin, K.D.; Neuberger, A.; Nikolaev, Y.A.; Murphy, L.A.; Gracheva, E.O.; Bagriantsev, S.N.; Sobolevsky, A.I. Extracellular cap domain is an essential component of the TRPV1 gating mechanism. Nat. Commun. 2021, 12, 2154. [Google Scholar] [CrossRef]
- Okada, K.; Inoue, A.; Okada, M.; Murata, Y.; Kakuta, S.; Jigami, T.; Kubo, S.; Shiraishi, H.; Eguchi, K.; Motomura, M.; et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science 2006, 312, 1802–1805. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, J.A.; Lara, M.; Díaz, J.; Campero, M.; Vázquez, J.; Maselli, R.A. Congenital Myasthenic Syndrome due to DOK7 mutations in a family from Chile. Eur. J. Transl. Myol. 2017, 27, 6832. [Google Scholar] [CrossRef]
- Hu, Y.; Leo, C.; Yu, S.; Huang, B.C.; Wang, H.; Shen, M.; Luo, Y.; Daniel-Issakani, S.; Payan, D.G.; Xu, X. Identification and functional characterization of a novel human misshapen/Nck interacting kinase-related kinase, hMINK beta. J. Biol. Chem. 2004, 279, 54387–54397, Erratum in J. Biol. Chem. 2005, 280, 5128. [Google Scholar] [CrossRef]
- DeChiara, T.M.; Bowen, D.C.; Valenzuela, D.M.; Simmons, M.V.; Poueymirou, W.T.; Thomas, S.; Kinetz, E.; Compton, D.L.; Rojas, E.; Park, J.S.; et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 1996, 85, 501–512. [Google Scholar] [CrossRef]
- Sadeh, M.; Shen, X.M.; Engel, A.G. Beneficial effect of albuterol in congenital myasthenic syndrome with epsilon-subunit mutations. Muscle Nerve 2011, 44, 289–291. [Google Scholar] [CrossRef]
- Finlayson, S.; Palace, J.; Belaya, K.; Walls, T.J.; Norwood, F.; Burke, G.; Holton, J.L.; Pascual-Pascual, S.I.; Cossins, J.; Beeson, D. Clinical features of congenital myasthenic syndrome due to mutations in DPAGT1. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1119–1125. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Clinical Features | Description |
|---|---|
| Early onset | Since birth |
| Fluctuating symptoms | Increasing viral infection and activity |
| Muscle fatigue and muscular weakness | Yes, improved with treatment |
| Breastfeeding difficulties chokes developed post-partum | Yes, improved gradually at 9 months |
| Ocular muscle impairment | Ptosis and sometime squint |
| Bulbar symptoms | Yes, improved with treatment |
| Scoliosis | Mild |
| Improvement with pyridostigmine | Up to 80% improvement |
| Mental development | Normal |
| Facial weakness | None |
| Little functional restriction | None |
| Delayed motor milestones | None |
| High-arched palate | None |
| Gene | Nucleotide Variant | Exon | Protein Variant | Zygosity | gnomAD Exome Freq | ACMG Franklin | VarSome | ClinVar |
|---|---|---|---|---|---|---|---|---|
| CHRNE | c.1220-8_1227dup | 11 | N/A | Homo | Not reported | Likely Pathogenic | Pathogenic | Pathogenic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almatrafi, A.M.; Alluqmani, M.M.; Basit, S. Homozygous Duplication in the CHRNE in a Family with Congenital Myasthenic Syndrome 4C: 18-Year Follow Up. Biomedicines 2023, 11, 2983. https://doi.org/10.3390/biomedicines11112983
Almatrafi AM, Alluqmani MM, Basit S. Homozygous Duplication in the CHRNE in a Family with Congenital Myasthenic Syndrome 4C: 18-Year Follow Up. Biomedicines. 2023; 11(11):2983. https://doi.org/10.3390/biomedicines11112983
Chicago/Turabian StyleAlmatrafi, Ahmad M., Majed M. Alluqmani, and Sulman Basit. 2023. "Homozygous Duplication in the CHRNE in a Family with Congenital Myasthenic Syndrome 4C: 18-Year Follow Up" Biomedicines 11, no. 11: 2983. https://doi.org/10.3390/biomedicines11112983