

SLC31A1 Identifying a Novel Biomarker with Potential Prognostic and Immunotherapeutic Potential in Pan-Cancer
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Gene Expression Analysis
2.1.1. GEPIA Database
2.1.2. HPA Database
2.2. Gene Enrichment Analysis
2.2.1. GeneMANIA Database
2.2.2. STRING Database
2.3. Genetic Alteration Analysis and DNA Methylation Analysis
2.3.1. The cBioPortal Database
2.3.2. UALCAN Database
2.4. Survival Prognosis Analysis
2.5. Immune Infiltration Analysis
3. Results
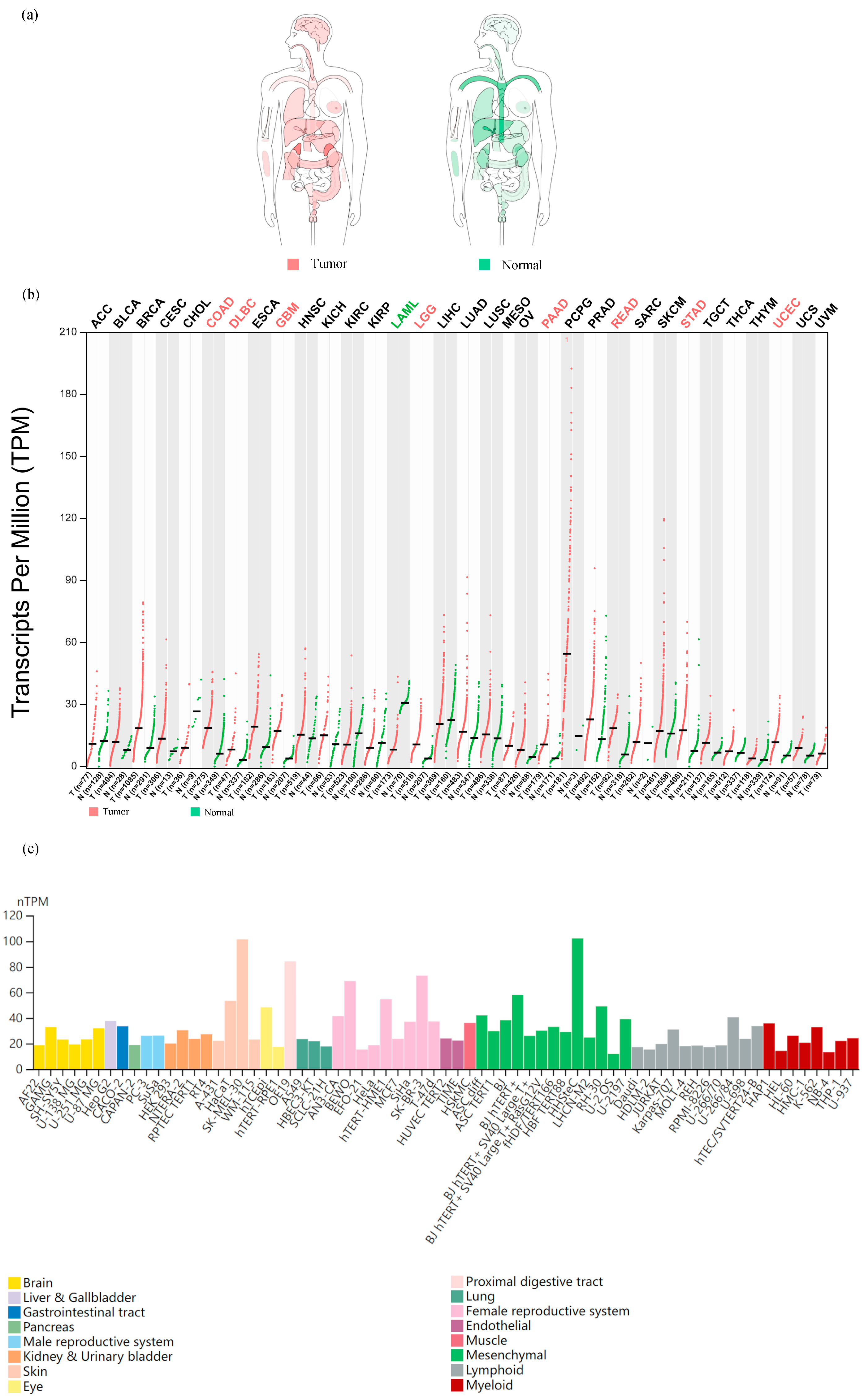
- A pan-cancer landscape of mRNA expression: we used the GEPIA dataset to analyze the mRNA levels of SLC31A1 in the interactive body map to learn more about its role in human pan-cancer. SLC31A1 expression was shown to be altered throughout many human tumor tissues compared to their corresponding normal tissues. This was notably true for the central nervous system, circulatory system, gastrointestinal system, urinary system, parathyroid glands, and thyroid (Figure 1a). Considering these results, we next examined the mRNA expression levels in 33 malignancies and adjacent normal tissues. Astonishingly, only eight tumor tissues (COAD, DLBC, GBM, LGG, PAAD, READ, STAD, and UCEC) showed higher median mRNA levels of SLC31A1 than normal tissues (Figure 1b). Finally, we examined SLC31A1’s cellular mRNA expression levels using data from the HPA database. The skin, the proximal gastrointestinal tract, the female reproductive system, the eye, and mesenchyme were among the tissue organ cell lines with higher SLC31A1 mRNA expression levels (Figure 1c).
- SLC31A1 expression and the pathological staging of cancers have been shown to have a substantial relationship. The pathological staging of malignancies is one of the key indications of patient prospects. As a result, our research investigated the connection between the SLC31A1 expression levels in cancers and their pathological stages using GEPIA, and it included 17 different malignancies. It is interesting to note that the level of SLC31A1 expression was not found to relate to the pathological stage of any other tumors, apart from ACC (p = 0.0152), KIRC (p = 0.000562), OV (p = 0.0405), and THCA (p = 0.00861); SLC31A1 exhibited an upward trend in relation to the pathological stage in ACC, while displaying a contrasting pattern in KIRC, OV, and THCA (Figure 2). The level of expression of SLC31A1 was shown to be linked with the pathological staging of ACC, KIRC, OV, and THCA, which suggests that it may be of importance in guiding the pathological staging of these malignancies. Interestingly, additional analyses conducted on the identical open-source database produced congruent experimental outcomes to ours, thereby providing further validation of the dependability of our results [17].
- 3.
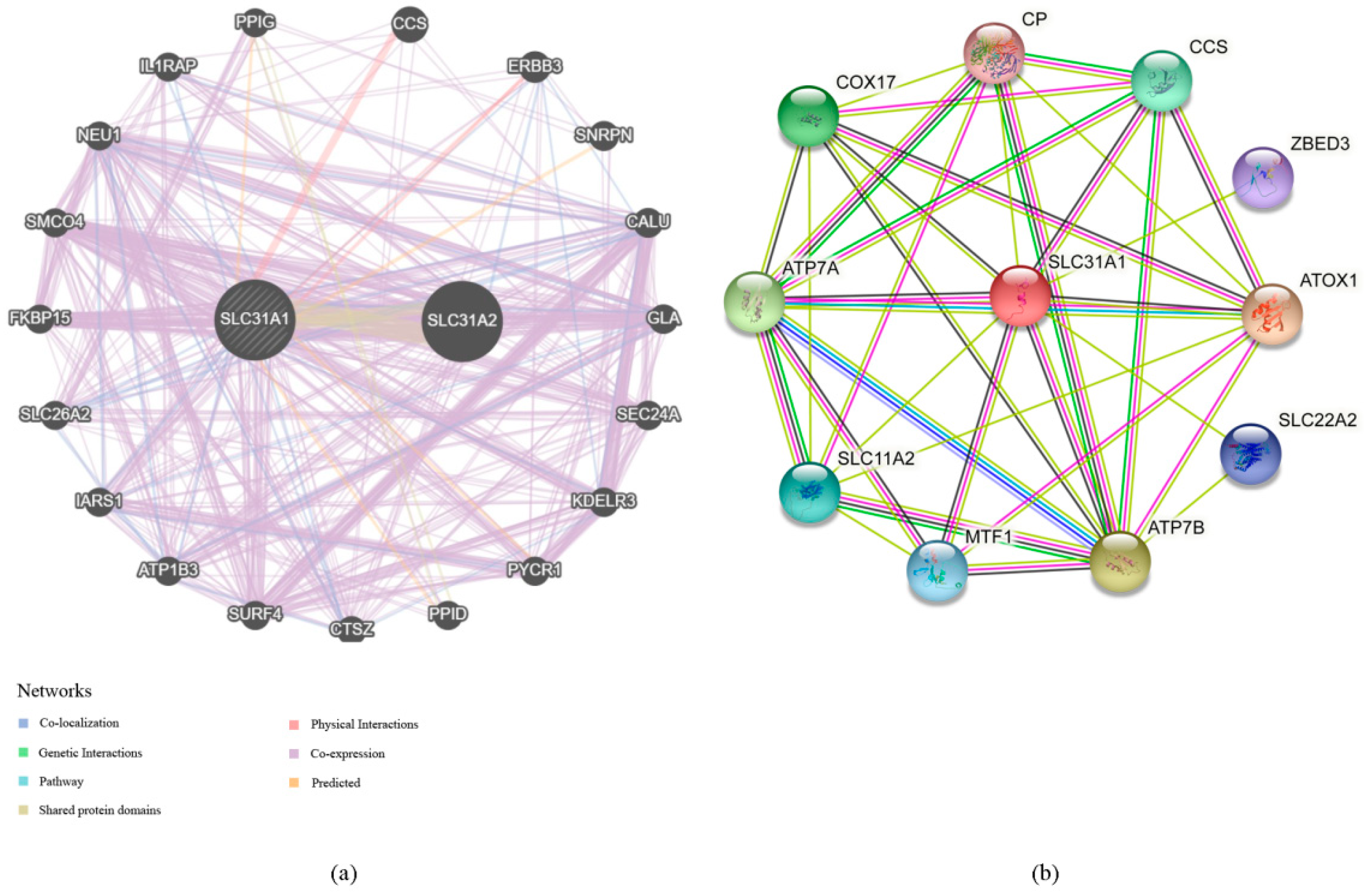
- Our investigation into the GeneMANIA databases led us to the discovery of 20 genes that are linked with the protein–protein interactions of SLC31A1 (Figure 4a). The small molecule route and protein–protein interaction network of SLC31A1 are shown below. According to the information found in the STRING database, there are a total of 10 nodes connected to the SLC31A1 gene (Figure 4b).
- 4.
- An investigation into the mutations of the SLC31A1 gene and the methylation levels of pan-cancerous tumors: the cBioPortal database was analyzed, and the results showed that 2.1% (54 out of 2584) of pan-cancer patients had mutations in the SLC31A1 gene (Figure 5a). In addition, we investigated the prevalence of mutations in the SLC31A1 gene among the various tumor types. The results showed that the disease with the highest frequency of aberrations was pancreatic cancer, followed by esophageal and gastric cancer and bone cancer (Figure 5b). Notably, mutations are the most common SLC31A1 aberrations. Our research found a total of two mutation sites, both of which were situated between numbers 0 and 200 (Figure 5c). This was done so that we could learn more about the SLC31A mutation sites found throughout the protein domains involved in cancer.
- 5.
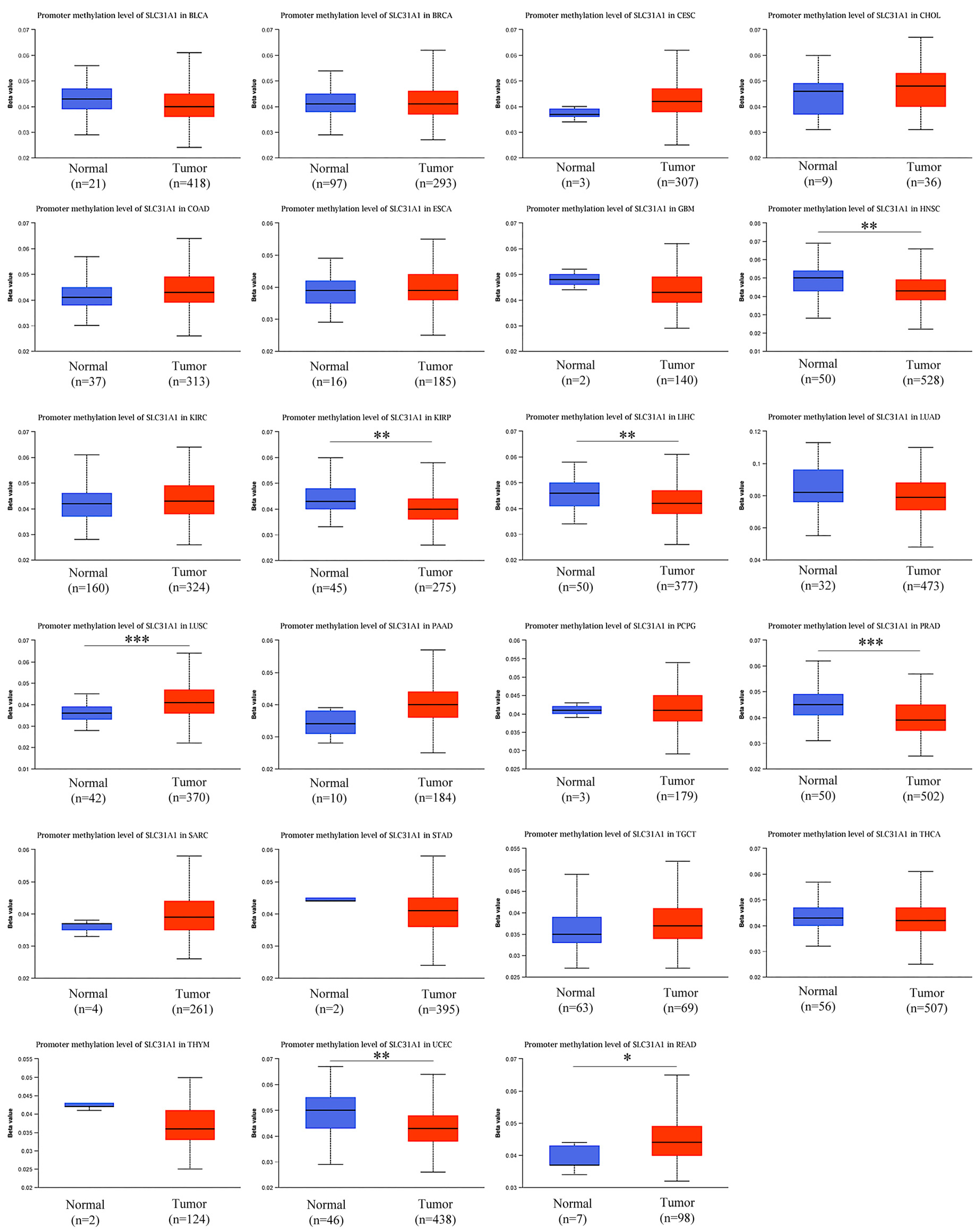
- DNA that has been methylated incorrectly is a substantial contributor to the development of cancer. Therefore, in the next step, we analyzed SLC31A1 methylation across cancers and the tissues that correlate with it using the UALCAN database. Compared to normal tissues, the levels of SLC31A1 methylation in HNSC, KIIRP, LIHC, LUSC, PRAD, READ, and UCEC tissues were found to be very different (Figure 6).
- 6.
- The expression and permeation of immunocytes in pan-cancers: in terms of the reality that there is a connection between SLC31A1 and the immune response, we decided to carry out pan-cancer research to investigate the link between SLC31A1 and the degree to which immune cells infiltrated the cancerous tissue. According to the data available here, 20 tumors were related to CD8+ T cells, 14 tumors were related to CD4+ T cells, 20 tumors were related to neutrophils, 21 tumors were related to medullary dendritic cells, 23 tumors were related to macrophages, and 13 tumors were related to B cells (Figure 7a).
- 7.
- We performed an analysis of the expression of SLC31A1 across many cancer types, together with the immune regulators TMB and MSI, and the immunological checkpoints. We assessed the link between SLC31A1 expression and two important immune regulators to quantify the relationship between SLC31A1 expression and the TME in the pan-cancer dataset. This allowed us to better understand the nature of this interaction. Positive associations were found between immune checkpoint genes and most different types of cancer, including UVM, UCEC, STAD, READ, OV, PAAD, LGG, LUSC, LAML, LUAD, DLBC, COAD, and BLCA. Only a small percentage of cancers, including THCA and CHOL tumors, were shown to have a negative association with immune checkpoint genes (Figure 8a).
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SLC31A1 | Solute carrier family 31 member 1 |
| ACC | Adrenocortical carcinoma |
| BLCA | Bladder urothelial carcinoma |
| BRCA | Breast invasive carcinoma |
| CESC | Cervical squamous cell carcinoma |
| CHOL | Cholangiocarcinoma |
| COAD | Colon adenocarcinoma |
| DLBC | Lymphoid neoplasm diffuse large B cell lymphoma |
| ESCA | Esophageal carcinoma |
| GBM | Glioblastoma |
| LGG | Brain lower grade glioma |
| HNSC | Head and neck squamous cell carcinoma |
| KICH | Kidney chromophobe |
| KIRC | Kidney renal clear cell carcinoma |
| KIRP | Kidney renal papillary cell carcinoma |
| LAML | Acute myeloid leukemia |
| LIHC | Liver hepatocellular carcinoma |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| MESO | Mesothelioma |
| OV | Ovarian serous cystadenocarcinoma |
| PAAD | Pancreatic adenocarcinoma |
| PCPG | Pheochromocytoma and paraganglioma |
| PRAD | Prostate adenocarcinoma |
| READ | Rectum adenocarcinoma |
| SARC | Sarcoma |
| SKCM | Skin cutaneous melanoma |
| STAD | Stomach adenocarcinoma |
| TGCT | Testicular germ cell tumors |
| THCA | Thyroid carcinoma |
| THYM | Thymoma |
| UCEC | Uterine corpus endometrial carcinoma |
| UCS | Uterine carcinosarcoma |
| UVM | Uveal melanoma |
| COX17 | Cytochrome c oxidase copper chaperone |
| ATOX1 | Antioxidant 1 |
References
- Hoffmann, R.; Große, J.; Nagl, M.; Niederwieser, D.; Mehnert, A.; Kersting, A. Internet-based grief therapy for bereaved individuals after loss due to Haematological cancer: Study protocol of a randomized controlled trial. BMC Psychiatry 2018, 18, 52. [Google Scholar] [CrossRef]
- Galván Morales, M.A.; Rodríguez, R.B.; Cruz, J.R.S.; Teran, L.M. Overview of New Treatments with Immunotherapy for Breast Cancer and a Proposal of a Combination Therapy. Molecules 2020, 25, 5686. [Google Scholar] [CrossRef]
- Li, F.; Gao, L.; Wang, P.; Hu, Y. Identifying Cancer Specific Driver Modules Using a Network-Based Method. Molecules 2018, 23, 1114. [Google Scholar] [CrossRef] [PubMed]
- Eilenberger, C.; Rothbauer, M.; Ehmoser, E.-K.; Ertl, P.; Küpcü, S. Effect of Spheroidal Age on Sorafenib Diffusivity and Toxicity in a 3D HepG2 Spheroid Model. Sci. Rep. 2019, 9, 4863. [Google Scholar] [CrossRef]
- Wakai, T.; Prasoon, P.; Hirose, Y.; Shimada, Y.; Ichikawa, H.; Nagahashi, M. Next-generation sequencing-based clinical sequencing: Toward precision medicine in solid tumors. Int. J. Clin. Oncol. 2019, 24, 115–122. [Google Scholar] [CrossRef]
- Wang, H. Genomic-guided individualized precision therapy in refractory metastatic solid tumor patients with extensively poor performance status: A Chinese single institutional prospective observational real-world study. Ann. Oncol. 2019, 30, v765. [Google Scholar] [CrossRef]
- Matsudera, S.; Kano, Y.; Aoyagi, Y.; Tohyama, K.; Takahashi, K.; Kumaki, Y.; Mitsumura, T.; Kimura, K.; Onishi, I.; Takemoto, A.; et al. A Pilot Study Analyzing the Clinical Utility of Comprehensive Genomic Profiling Using Plasma Cell-Free DNA for Solid Tumor Patients in Japan (PROFILE Study). Ann. Surg. Oncol. 2021, 28, 8497–8505. [Google Scholar] [CrossRef] [PubMed]
- Bayle, A.; Belcaid, L.; Aldea, M.; Peyraud, F.; Romano, P.M.; Blanc-Durand, F.; Clodion, R.; Ponce, S.; Nicotra, C.; Hollebecque, A.; et al. Clinical utility of circulating tumor DNA sequencing with a large panel: The experience of Gustave Roussy/National Center for Precision Medicine (PRISM). Cancer Res. 2022, 82 (Suppl. 12), 3413. [Google Scholar] [CrossRef]
- Taghizadeh, H.; Unseld, M.; Spalt, M.; Mader, R.M.; Müllauer, L.; Fuereder, T.; Raderer, M.; Sibilia, M.; Hoda, M.A.; Aust, S.; et al. Targeted Therapy Recommendations for Therapy Refractory Solid Tumors-Data from the Real-World Precision Medicine Platform MONDTI. J. Pers. Med. 2020, 10, 188. [Google Scholar] [CrossRef]
- Coombs, C.C.; Gillis, N.K.; Tan, X.; Berg, J.S.; Ball, M.; Balasis, M.E.; Montgomery, N.D.; Bolton, K.L.; Parker, J.S.; Mesa, T.E.; et al. Identification of Clonal Hematopoiesis Mutations in Solid Tumor Patients Undergoing Unpaired Next-Generation Sequencing Assays. Clin. Cancer Res. 2018, 24, 5918–5924. [Google Scholar] [CrossRef] [PubMed]
- Zimnicka, A.M.; Ivy, K.; Kaplan, J.H.; Fukai, T.; Ushio-Fukai, M.; Horn, C.C.; Meyers, K.; Lim, A.; Dye, M.; Pak, D.; et al. Acquisition of dietary copper: A role for anion transporters in intestinal apical copper uptake. Am. J. Physiol. Cell Physiol. 2011, 300, C588–C599. [Google Scholar] [CrossRef] [PubMed]
- Maryon, E.B.; Zhang, J.; Jellison, J.W.; Kaplan, J.H. Human copper transporter 1 lacking O-linked glycosylation is proteolytically cleaved in a Rab9-positive endosomal compartment. J. Biol. Chem. 2009, 284, 28104–28114. [Google Scholar] [CrossRef] [PubMed]
- Maryon, E.B.; Molloy, S.A.; Ivy, K.; Yu, H.; Kaplan, J.H. Rate and regulation of copper transport by human copper transporter 1 (hCTR1). J. Biol. Chem. 2013, 288, 18035–18046. [Google Scholar] [CrossRef] [PubMed]
- Voli, F.; Valli, E.; Lerra, L.; Kimpton, K.; Saletta, F.; Giorgi, F.M.; Mercatelli, D.; Rouaen, J.R.C.; Shen, S.; Murray, J.E.; et al. Intratumoral Copper Modulates PD-L1 Expression and Influences Tumor Immune Evasion. Cancer Res. 2020, 80, 4129–4144. [Google Scholar] [CrossRef]
- Li, L.; Li, L.; Sun, Q. High expression of cuproptosis-related SLC31A1 gene in relation to unfavorable outcome and deregulated immune cell infiltration in breast cancer: An analysis based on public databases. BMC Bioinform. 2022, 23, 350. [Google Scholar] [CrossRef]
- Wang, J.; Li, S.; Guo, Y.; Zhao, C.; Chen, Y.; Ning, W.; Yang, J.; Zhang, H. Cuproptosis-related gene SLC31A1 expression correlates with the prognosis and tumor immune microenvironment in glioma. Funct. Integr. Genom. 2023, 23, 279. [Google Scholar] [CrossRef]
- Kong, F.S.; Ren, C.-Y.; Jia, R.; Zhou, Y.; Chen, J.-H.; Ma, Y. Systematic pan-cancer analysis identifies SLC31A1 as a biomarker in multiple tumor types. BMC Med. Genom. 2023, 16, 61. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Digre, A.; Lindskog, C. The Human Protein Atlas-Spatial localization of the human proteome in health and disease. Protein Sci. 2021, 30, 218–233. [Google Scholar] [CrossRef]
- Franz, M.; Rodriguez, H.; Lopes, C.; Zuberi, K.; Montojo, J.; Bader, G.D.; Morris, Q. GeneMANIA update 2018. Nucleic Acids Res. 2018, 46, W60–W64. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.-Y.; Andre, F. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Turski, M.L.; Brady, D.C.; Kim, H.J.; Kim, B.-E.; Nose, Y.; Counter, C.M.; Winge, D.R.; Thiele, D.J. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol. Cell Biol. 2012, 32, 1284–1295. [Google Scholar] [CrossRef]
- Deo, A.; Chaudhury, S.; Kannan, S.; Rekhi, B.; Maheshwari, A.; Gupta, S.; Ray, P. IGF1R predicts better survival in high-grade serous epithelial ovarian cancer patients and correlates with hCtr1 levels. Biomark. Med. 2019, 13, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhou, R.; Zhao, Y.; Pan, Y.; Liang, H.; Zhang, J.; Tai, S.; Jin, L.; Teng, C. Blockage of SLC31A1-dependent copper absorption increases pancreatic cancer cell autophagy to resist cell death. Cell Prolif. 2019, 52, e12568. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yin, Y.-L.; Liu, X.-Z.; Shen, P.; Zheng, Y.-G.; Lan, X.-R.; Lu, C.-B.; Wang, J.-Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef]
- Uriu-Adams, J.Y.; Scherr, R.E.; Lanoue, L.; Keen, C.L. Influence of copper on early development: Prenatal and postnatal considerations. Biofactors 2010, 36, 136–152. [Google Scholar] [CrossRef]
- Nose, Y.; Kim, B.E.; Thiele, D.J. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 2006, 4, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; McCormick, F.; Smith-McCune, K.; Hanahan, D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell 2010, 17, 574–583. [Google Scholar] [CrossRef]
- Tsang, T.; Posimo, J.M.; Gudiel, A.A.; Cicchini, M.; Feldser, D.M.; Brady, D.C. Copper is an essential regulator of the autophagic kinases ULK1/2 to drive lung adenocarcinoma. Nat. Cell Biol. 2020, 22, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Brady, D.C.; Crowe, M.S.; Turski, M.L.; Hobbs, G.A.; Yao, X.; Chaikuad, A.; Knapp, S.; Xiao, K.; Campbell, S.L.; Thiele, D.J.; et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 2014, 509, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Clatworthy, S.A.; Gamell, C.; Smith, Z.M.; Francis, P.S.; Denoyer, D.; Meggyesy, P.M.; La Fontaine, S.; Cater, M.A. Copper accumulation in senescent cells: Interplay between copper transporters and impaired autophagy. Redox Biol. 2018, 16, 322–331. [Google Scholar] [CrossRef]
- Suzuki, C.; Daigo, Y.; Kikuchi, T.; Katagiri, T.; Nakamura, Y. Identification of COX17 as a therapeutic target for non-small cell lung cancer. Cancer Res. 2003, 63, 7038–7041. [Google Scholar]
- Blockhuys, S.; Zhang, X.; Wittung-Stafshede, P. Single-cell tracking demonstrates copper chaperone Atox1 to be required for breast cancer cell migration. Proc. Natl. Acad. Sci. USA 2020, 117, 2014–2019. [Google Scholar] [CrossRef]
- Blockhuys, S.; Wittung-Stafshede, P. Copper chaperone Atox1 plays role in breast cancer cell migration. Biochem. Biophys. Res. Commun. 2017, 483, 301–304. [Google Scholar] [CrossRef]
- Chanock, S.J. The paradox of mutations and cancer. Science 2018, 362, 893–894. [Google Scholar] [CrossRef]
- Soma, S.; Latimer, A.J.; Chun, H.; Vicary, A.C.; Timbalia, S.A.; Boulet, A.; Rahn, J.J.; Chan, S.S.L.; Leary, S.C.; Kim, B.-E.; et al. Elesclomol restores mitochondrial function in genetic models of copper deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, 8161–8166. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar]
- Luo, H.; Zhao, Q.; Wei, W.; Zheng, L.; Yi, S.; Li, G.; Wang, W.; Sheng, H.; Pu, H.; Mo, H.; et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci. Transl. Med. 2020, 12, eaax7533. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Zhao, Y.; Huang, W.; Gao, Y.; Xu, W.; Tao, J.; Yang, M.; Li, L.; Ping, W.; Shen, H.; et al. Non-invasive diagnosis of early-stage lung cancer using high-throughput targeted DNA methylation sequencing of circulating tumor DNA (ctDNA). Theranostics 2019, 9, 2056–2070. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Gu, Y.; Zhu, J.; Ci, C.; Guo, Z.; Chen, C.; Wei, Y.; Lv, W.; Liu, H.; et al. Specific breast cancer prognosis-subtype distinctions based on DNA methylation patterns. Mol. Oncol. 2018, 12, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA methylation, its mediators and genome integrity. Int. J. Biol. Sci. 2015, 11, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.K.; Yang, J.-Y.; Xu, Z.-Z.; Yu, W.-H. DNA Methylation and Uveal Melanoma. Chin. Med. J. 2018, 131, 845–851. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.e14. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Yang, C.-M.; Cheng, J.-T.; Tsai, K.-W.; Fu, T.-Y.; Liou, H.-H.; Tseng, H.-H.; Lee, J.-H.; Li, G.-C.; Wang, J.-S.; et al. Global DNA hypomethylation is associated with the development and poor prognosis of tongue squamous cell carcinoma. J. Oral Pathol. Med. 2016, 45, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Venza, M.; Visalli, M.; Catalano, T.; Beninati, C.; Teti, D.; Venza, I. DSS1 promoter hypomethylation and overexpression predict poor prognosis in melanoma and squamous cell carcinoma patients. Hum. Pathol. 2017, 60, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef] [PubMed]
- De Andrade, M.; Daw, E.W.; Kraja, A.T.; Fisher, V.; Wang, L.; Hu, K.; Li, J.; Romanescu, R.; Veenstra, J.; Sun, R.; et al. The challenge of detecting genotype-by-methylation interaction: GAW20. BMC Genet. 2018, 19, 81. [Google Scholar] [CrossRef]
- O’ Leary, P.C.; Penny, S.A.; Dolan, R.T.; Kelly, C.M.; Madden, S.F.; Rexhepaj, E.; Brennan, D.J.; McCann, A.H.; Pontén, F.; Uhlén, M.; et al. Systematic antibody generation and validation via tissue microarray technology leading to identification of a novel protein prognostic panel in breast cancer. BMC Cancer 2013, 13, 175. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Herman, J.G. Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2002, 196, 1–7. [Google Scholar] [CrossRef]
- Fukushige, S.; Horii, A. DNA methylation in cancer: A gene silencing mechanism and the clinical potential of its biomarkers. Tohoku J. Exp. Med. 2013, 229, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, S.; Anupriya. Drugs targeting epigenetic modifications and plausible therapeutic strategies against colorectal cancer. Front. Pharmacol. 2019, 10, 588. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wang, M.; Liu, Q.; Liu, Y.; Zhu, K.; Chen, L.; Guo, H.; Li, Y.; Shi, B. Identification of gene expression profiles and immune cell infiltration signatures between low and high tumor mutation burden groups in bladder cancer. Int. J. Med. Sci. 2020, 17, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yu, W.; Xiao, H.; Lin, K. BIRC5 is a prognostic biomarker associated with tumor immune cell infiltration. Sci. Rep. 2021, 11, 390. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Chen, W.; Xu, H.; Yang, J.; Jiang, J.; Jiang, Y.; Xu, G. Correlation of CCL8 expression with immune cell infiltration of skin cutaneous melanoma: Potential as a prognostic indicator and therapeutic pathway. Cancer Cell Int. 2021, 21, 635. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Yang, H.; Zhu, K.; Chang, C.; Lv, W.; Li, R.; Li, X.; Ye, T.; Cao, D. SLC31A1 Identifying a Novel Biomarker with Potential Prognostic and Immunotherapeutic Potential in Pan-Cancer. Biomedicines 2023, 11, 2884. https://doi.org/10.3390/biomedicines11112884
Zhang P, Yang H, Zhu K, Chang C, Lv W, Li R, Li X, Ye T, Cao D. SLC31A1 Identifying a Novel Biomarker with Potential Prognostic and Immunotherapeutic Potential in Pan-Cancer. Biomedicines. 2023; 11(11):2884. https://doi.org/10.3390/biomedicines11112884
Chicago/Turabian StyleZhang, Pei, Heqi Yang, Kaiguo Zhu, Chen Chang, Wanrui Lv, Ruizhen Li, Xiaoying Li, Tinghong Ye, and Dan Cao. 2023. "SLC31A1 Identifying a Novel Biomarker with Potential Prognostic and Immunotherapeutic Potential in Pan-Cancer" Biomedicines 11, no. 11: 2884. https://doi.org/10.3390/biomedicines11112884