
Gene Variants Implicated in Steatotic Liver Disease: Opportunities for Diagnostics and Therapeutics
 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
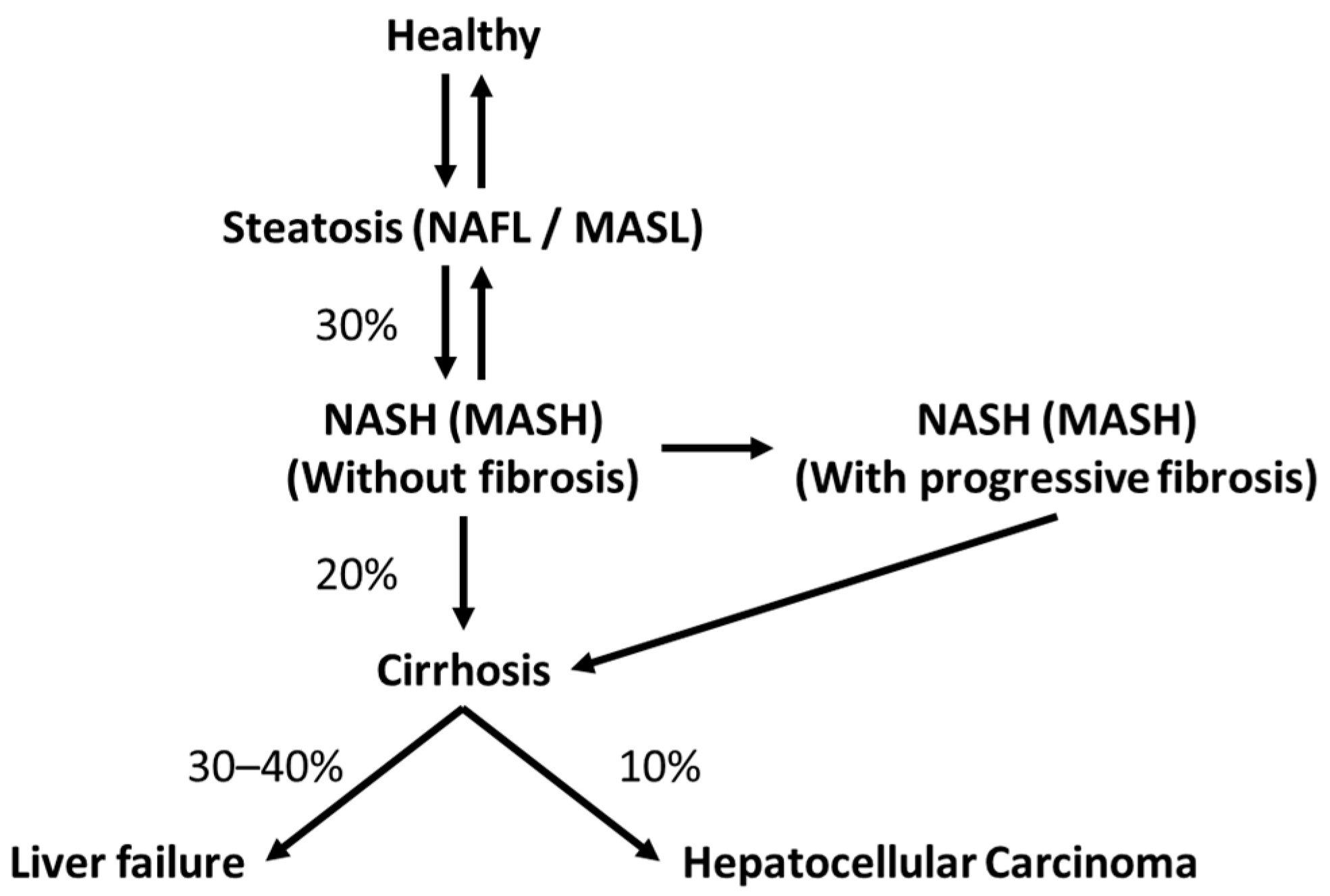
1.1. The Liver and Steatotic Liver Disease
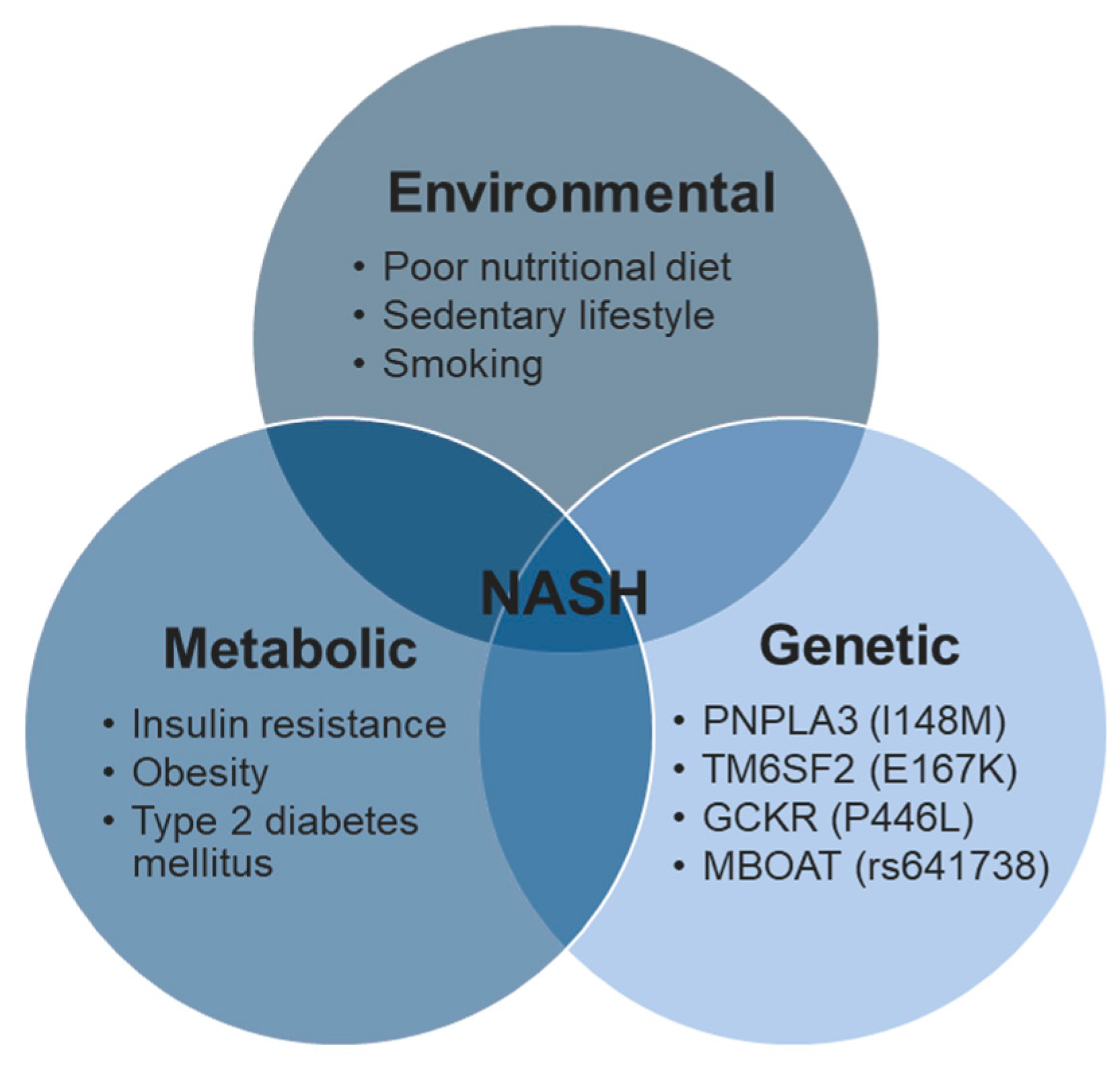
1.2. NAFLD Pathogenesis
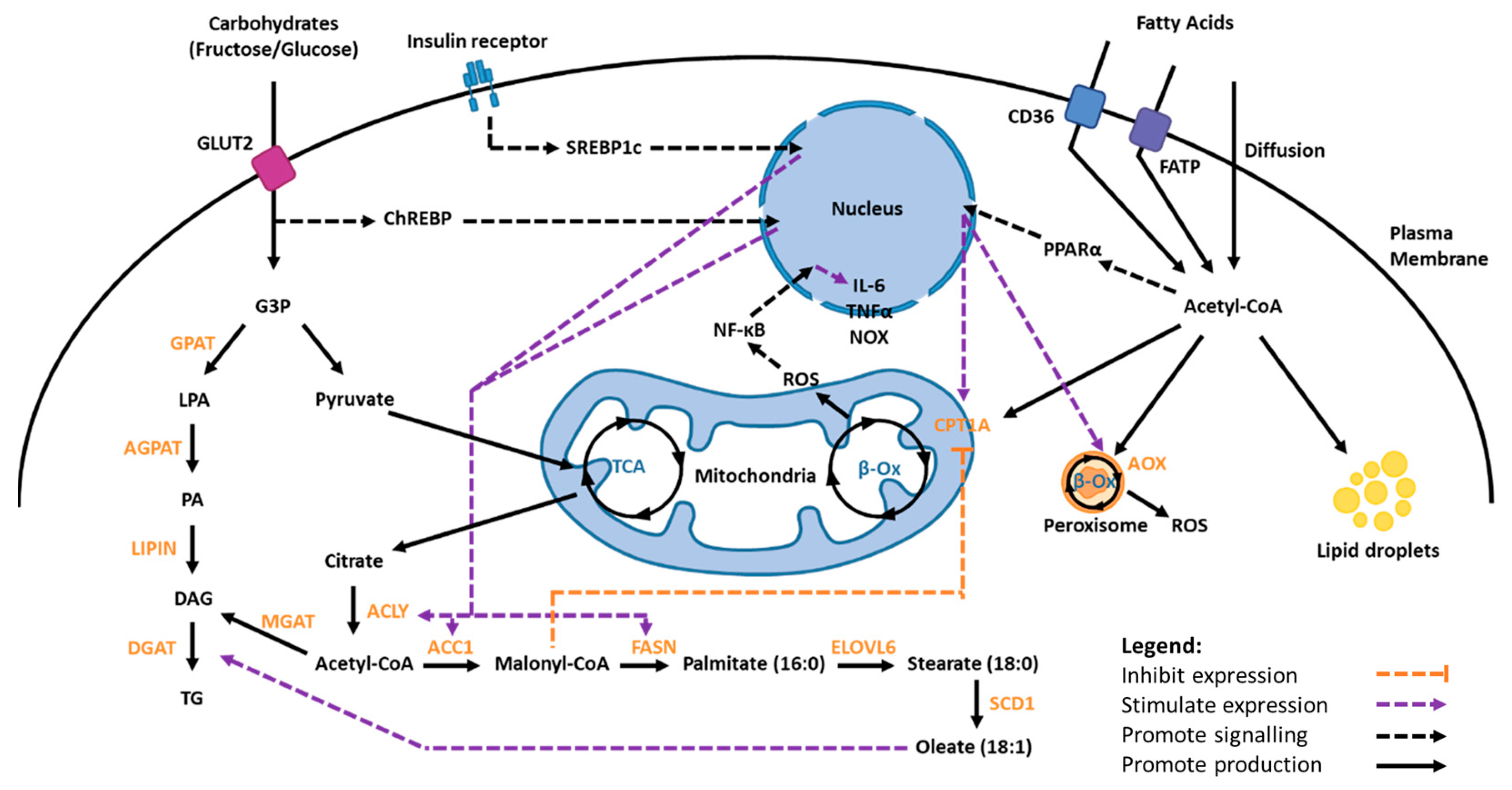
1.3. Lipid Metabolism
1.3.1. Fatty Acid Synthesis and Uptake
1.3.2. Fatty Acid Oxidation
1.4. NAFLD Risk Factors: Genetics
1.5. NAFLD Heritability
1.6. Gene Variants in NAFLD
1.6.1. PNPLA3
1.6.2. TM6SF2
1.6.3. MBOAT7
1.6.4. GCKR
1.6.5. HSD17B13
1.6.6. Other Gene Variants
1.7. Current and Potential Therapies
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Si-Tayeb, K.; Lemaigre, F.P.; Duncan, S.A. Organogenesis and development of the liver. Dev. Cell 2010, 18, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Remmer, H. The role of theliver in drug metabolism. Am. J. Med. 1970, 49, 617–629. [Google Scholar] [CrossRef]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol. Res. Health 2006, 29, 245–254. [Google Scholar] [PubMed]
- Boyer, J.L. Bile formation and secretion. Compr. Physiol. 2013, 3, 1035–1078. [Google Scholar] [CrossRef] [PubMed]
- Albahrani, A.A.; Greaves, R.F. Fat-Soluble Vitamins: Clinical Indications and Current Challenges for Chromatographic Measurement. Clin. Biochem. Rev. 2016, 37, 27–47. [Google Scholar]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Day, C.P. The genetics of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 645–655. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- American Association for the Study of Liver Diseases; Latin American Association for the Study of the Liver; European Association for the Study of the Liver. A call for unity: The path towards a more precise and patient-centric nomenclature for NAFLD. Hepatology 2023, 78, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Levene, A.P.; Goldin, R.D. The epidemiology, pathogenesis and histopathology of fatty liver disease. Histopathology 2012, 61, 141–152. [Google Scholar] [CrossRef] [PubMed]
- McCullough, A.J. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin. Liver Dis. 2004, 8, 521–533, viii. [Google Scholar] [CrossRef] [PubMed]
- Gomez, O.; Amatya, S.; Kamberov, L.; Dhaibar, H.; Khanna, P.; Rom, O.; Yurdagul, A., Jr.; Orr, W.A.; Nunez, K.; Thevenot, P.; et al. SLAMF1 is expressed and secreted by Hepatocytes and the Liver in Non-Alcoholic Fatty Liver Disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 323, G177–G187. [Google Scholar] [CrossRef]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef]
- Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Vairetti, M.; Ferrigno, A. Nonalcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: Current Issues and Future Perspectives in Preclinical and Clinical Research. Int. J. Mol. Sci. 2020, 21, 9646. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.L.; Chen, H.; Wang, C.L.; Liang, L. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model”. World J. Gastroenterol. 2018, 24, 2974–2983. [Google Scholar] [CrossRef] [PubMed]
- Juanola, O.; Martinez-Lopez, S.; Frances, R.; Gomez-Hurtado, I. Non-Alcoholic Fatty Liver Disease: Metabolic, Genetic, Epigenetic and Environmental Risk Factors. Int. J. Environ. Res. Public Health 2021, 18, 5227. [Google Scholar] [CrossRef] [PubMed]
- Petaja, E.M.; Yki-Jarvinen, H. Definitions of Normal Liver Fat and the Association of Insulin Sensitivity with Acquired and Genetic NAFLD-A Systematic Review. Int. J. Mol. Sci. 2016, 17, 633. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.P.F.; Schrauwen, P.; Verhoef, P.; Byrne, C.D.; Mela, D.J.; Pfeiffer, A.F.H.; Riserus, U.; Rosendaal, F.R.; Schrauwen-Hinderling, V. Liver fat: A relevant target for dietary intervention? Summary of a Unilever workshop. J. Nutr. Sci. 2017, 6, e15. [Google Scholar] [CrossRef]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Le Bloc’h, J.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Coleman, R.A.; Lewin, T.M.; Muoio, D.M. Physiological and nutritional regulation of enzymes of triacylglycerol synthesis. Annu. Rev. Nutr. 2000, 20, 77–103. [Google Scholar] [CrossRef]
- Grabner, G.F.; Xie, H.; Schweiger, M.; Zechner, R. Lipolysis: Cellular mechanisms for lipid mobilization from fat stores. Nat. Metab. 2021, 3, 1445–1465. [Google Scholar] [CrossRef]
- Ericsson, J.; Jackson, S.M.; Kim, J.B.; Spiegelman, B.M.; Edwards, P.A. Identification of glycerol-3-phosphate acyltransferase as an adipocyte determination and differentiation factor 1- and sterol regulatory element-binding protein-responsive gene. J. Biol. Chem. 1997, 272, 7298–7305. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Shimomura, I. Sterol regulatory element-binding proteins: Activators of cholesterol and fatty acid biosynthesis. Curr. Opin. Lipidol. 1999, 10, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef]
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–25662. [Google Scholar] [CrossRef]
- Goto, T.; Itoh, M.; Suganami, T.; Kanai, S.; Shirakawa, I.; Sakai, T.; Asakawa, M.; Yoneyama, T.; Kai, T.; Ogawa, Y. Obeticholic acid protects against hepatocyte death and liver fibrosis in a murine model of nonalcoholic steatohepatitis. Sci. Rep. 2018, 8, 8157. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Sanyal, A.J.; Loomba, R.; Rinella, M.; Harrison, S.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; MacConell, L.; Shringarpure, R.; et al. REGENERATE: Design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemp. Clin. Trials 2019, 84, 105803. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef]
- Cipriani, S.; Mencarelli, A.; Palladino, G.; Fiorucci, S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J. Lipid Res. 2010, 51, 771–784. [Google Scholar] [CrossRef]
- Romero-Gomez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef]
- Hamabe, A.; Uto, H.; Imamura, Y.; Kusano, K.; Mawatari, S.; Kumagai, K.; Kure, T.; Tamai, T.; Moriuchi, A.; Sakiyama, T.; et al. Impact of cigarette smoking on onset of nonalcoholic fatty liver disease over a 10-year period. J. Gastroenterol. 2011, 46, 769–778. [Google Scholar] [CrossRef]
- Targher, G.; Alberiche, M.; Zenere, M.B.; Bonadonna, R.C.; Muggeo, M.; Bonora, E. Cigarette smoking and insulin resistance in patients with noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1997, 82, 3619–3624. [Google Scholar] [CrossRef]
- Weitzman, M.; Cook, S.; Auinger, P.; Florin, T.A.; Daniels, S.; Nguyen, M.; Winickoff, J.P. Tobacco smoke exposure is associated with the metabolic syndrome in adolescents. Circulation 2005, 112, 862–869. [Google Scholar] [CrossRef]
- Yuan, H.; Shyy, J.Y.; Martins-Green, M. Second-hand smoke stimulates lipid accumulation in the liver by modulating AMPK and SREBP-1. J. Hepatol. 2009, 51, 535–547. [Google Scholar] [CrossRef]
- Masuoka, H.C.; Chalasani, N. Nonalcoholic fatty liver disease: An emerging threat to obese and diabetic individuals. Ann. N. Y. Acad. Sci. 2013, 1281, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipokines in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1062–1079. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.M.T.; Won, J.; Khan, M.; Chavin, K.D.; Singh, I. Peroxisomal footprint in the pathogenesis of nonalcoholic steatohepatitis. Ann. Hepatol. 2020, 19, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Dharmalingam, M.; Yamasandhi, P.G. Nonalcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus. Indian. J. Endocrinol. Metab. 2018, 22, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Non-Alcoholic Fatty Liver Disease Study Group; Lonardo, A.; Bellentani, S.; Argo, C.K.; Ballestri, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V.; et al. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig. Liver Dis. 2015, 47, 997–1006. [Google Scholar] [CrossRef]
- Rich, N.E.; Oji, S.; Mufti, A.R.; Browning, J.D.; Parikh, N.D.; Odewole, M.; Mayo, H.; Singal, A.G. Racial and Ethnic Disparities in Nonalcoholic Fatty Liver Disease Prevalence, Severity, and Outcomes in the United States: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 198–210.e2. [Google Scholar] [CrossRef]
- Struben, V.M.; Hespenheide, E.E.; Caldwell, S.H. Nonalcoholic steatohepatitis and cryptogenic cirrhosis within kindreds. Am. J. Med. 2000, 108, 9–13. [Google Scholar] [CrossRef]
- Willner, I.R.; Waters, B.; Patil, S.R.; Reuben, A.; Morelli, J.; Riely, C.A. Ninety patients with nonalcoholic steatohepatitis: Insulin resistance, familial tendency, and severity of disease. Am. J. Gastroenterol. 2001, 96, 2957–2961. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Celedon, M.A.; Lavine, J.E.; Salem, R.; Campbell, N.; Schork, N.J.; Shiehmorteza, M.; Yokoo, T.; Chavez, A.; Middleton, M.S.; et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009, 136, 1585–1592. [Google Scholar] [CrossRef]
- Bathum, L.; Petersen, H.C.; Rosholm, J.U.; Hyltoft Petersen, P.; Vaupel, J.; Christensen, K. Evidence for a substantial genetic influence on biochemical liver function tests: Results from a population-based Danish twin study. Clin. Chem. 2001, 47, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Makkonen, J.; Pietilainen, K.H.; Rissanen, A.; Kaprio, J.; Yki-Jarvinen, H. Genetic factors contribute to variation in serum alanine aminotransferase activity independent of obesity and alcohol: A study in monozygotic and dizygotic twins. J. Hepatol. 2009, 50, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Pelusi, S.; Baselli, G.; Pietrelli, A.; Dongiovanni, P.; Donati, B.; McCain, M.V.; Meroni, M.; Fracanzani, A.L.; Romagnoli, R.; Petta, S.; et al. Rare Pathogenic Variants Predispose to Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 3682. [Google Scholar] [CrossRef] [PubMed]
- Momozawa, Y.; Mizukami, K. Unique roles of rare variants in the genetics of complex diseases in humans. J. Hum. Genet. 2021, 66, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kim, Y.; Tsang, E.K.; Davis, J.R.; Damani, F.N.; Chiang, C.; Hess, G.T.; Zappala, Z.; Strober, B.J.; Scott, A.J.; et al. The impact of rare variation on gene expression across tissues. Nature 2017, 550, 239–243. [Google Scholar] [CrossRef]
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L.; Petta, S.; Maier, S.; Rosso, C.; De Luca, L.; et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 4492. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Darlay, R.; Cockell, S.; Meroni, M.; Govaere, O.; Tiniakos, D.; Burt, A.D.; Bedossa, P.; Palmer, J.; Liu, Y.L.; et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort(). J. Hepatol. 2020, 73, 505–515. [Google Scholar] [CrossRef]
- Namjou, B.; Lingren, T.; Huang, Y.; Parameswaran, S.; Cobb, B.L.; Stanaway, I.B.; Connolly, J.J.; Mentch, F.D.; Benoit, B.; Niu, X.; et al. GWAS and enrichment analyses of non-alcoholic fatty liver disease identify new trait-associated genes and pathways across eMERGE Network. BMC Med. 2019, 17, 135. [Google Scholar] [CrossRef]
- Kleinstein, S.E.; Rein, M.; Abdelmalek, M.F.; Guy, C.D.; Goldstein, D.B.; Mae Diehl, A.; Moylan, C.A. Whole-Exome Sequencing Study of Extreme Phenotypes of NAFLD. Hepatol. Commun. 2018, 2, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Rakela, J.; Rule, J.; Ganger, D.; Lau, J.; Cunningham, J.; Dehankar, M.; Baheti, S.; Lee, W.M.; Acute Liver Failure Study, G. Whole Exome Sequencing among 26 Patients with Indeterminate Acute Liver Failure: A Pilot Study. Clin. Transl. Gastroenterol. 2019, 10, e00087. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjaerg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.; Allison, M.E.; Alexander, G.J.; Piguet, A.C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Wagenknecht, L.E.; Palmer, N.D.; Bowden, D.W.; Rotter, J.I.; Norris, J.M.; Ziegler, J.; Chen, Y.D.; Haffner, S.; Scherzinger, A.; Langefeld, C.D. Association of PNPLA3 with non-alcoholic fatty liver disease in a minority cohort: The Insulin Resistance Atherosclerosis Family Study. Liver Int. 2011, 31, 412–416. [Google Scholar] [CrossRef]
- Pingitore, P.; Dongiovanni, P.; Motta, B.M.; Meroni, M.; Lepore, S.M.; Mancina, R.M.; Pelusi, S.; Russo, C.; Caddeo, A.; Rossi, G.; et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 2016, 25, 5212–5222. [Google Scholar] [CrossRef]
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 2011, 286, 37085–37093. [Google Scholar] [CrossRef]
- Pan, J.J.; Fallon, M.B. Gender and racial differences in nonalcoholic fatty liver disease. World J. Hepatol. 2014, 6, 274–283. [Google Scholar] [CrossRef]
- Pingitore, P.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Indiveri, C.; Pujia, A.; Montalcini, T.; Hedfalk, K.; Romeo, S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta 2014, 1841, 574–580. [Google Scholar] [CrossRef]
- Kumari, M.; Schoiswohl, G.; Chitraju, C.; Paar, M.; Cornaciu, I.; Rangrez, A.Y.; Wongsiriroj, N.; Nagy, H.M.; Ivanova, P.T.; Scott, S.A.; et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012, 15, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Caligiuri, A.; Stulnig, T.M.; Marra, F.; Trauner, M. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890. [Google Scholar] [CrossRef] [PubMed]
- Basu Ray, S. PNPLA3-I148M: A problem of plenty in non-alcoholic fatty liver disease. Adipocyte 2019, 8, 201–208. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef]
- Goffredo, M.; Caprio, S.; Feldstein, A.E.; D’Adamo, E.; Shaw, M.M.; Pierpont, B.; Savoye, M.; Zhao, H.; Bale, A.E.; Santoro, N. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: A multiethnic study. Hepatology 2016, 63, 117–125. [Google Scholar] [CrossRef]
- Trepo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Boren, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, B.; Linden, D.; Brolen, G.; Liljeblad, M.; Bjursell, M.; Romeo, S.; Loomba, R. Review article: The emerging role of genetics in precision medicine for patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2020, 51, 1305–1320. [Google Scholar] [CrossRef]
- Thangapandi, V.R.; Knittelfelder, O.; Brosch, M.; Patsenker, E.; Vvedenskaya, O.; Buch, S.; Hinz, S.; Hendricks, A.; Nati, M.; Herrmann, A.; et al. Loss of hepatic Mboat7 leads to liver fibrosis. Gut 2021, 70, 940–950. [Google Scholar] [CrossRef]
- Beer, N.L.; Tribble, N.D.; McCulloch, L.J.; Roos, C.; Johnson, P.R.; Orho-Melander, M.; Gloyn, A.L. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum. Mol. Genet. 2009, 18, 4081–4088. [Google Scholar] [CrossRef]
- Sliz, E.; Sebert, S.; Wurtz, P.; Kangas, A.J.; Soininen, P.; Lehtimaki, T.; Kahonen, M.; Viikari, J.; Mannikko, M.; Ala-Korpela, M.; et al. NAFLD risk alleles in PNPLA3, TM6SF2, GCKR and LYPLAL1 show divergent metabolic effects. Hum. Mol. Genet. 2018, 27, 2214–2223. [Google Scholar] [CrossRef] [PubMed]
- Fernandes Silva, L.; Vangipurapu, J.; Kuulasmaa, T.; Laakso, M. An intronic variant in the GCKR gene is associated with multiple lipids. Sci. Rep. 2019, 9, 10240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.B.; Su, W.; Xu, H.; Zhang, X.Y.; Guan, Y.F. HSD17B13: A Potential Therapeutic Target for NAFLD. Front. Mol. Biosci. 2021, 8, 824776. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Belyaeva, O.V.; Brown, P.M.; Fujita, K.; Valles, K.; Karki, S.; de Boer, Y.S.; Koh, C.; Chen, Y.; Du, X.; et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated with Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 1504–1519. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Y.; Guo, T.; Yang, F.; Mao, Y.; Li, L.; Liu, C.; Gao, H.; Jin, Y.; Che, Y.; et al. Genetic variant rs72613567 of HSD17B13 gene reduces alcohol-related liver disease risk in Chinese Han population. Liver Int. 2020, 40, 2194–2202. [Google Scholar] [CrossRef]
- Fairfield, C.J.; Drake, T.M.; Pius, R.; Bretherick, A.D.; Campbell, A.; Clark, D.W.; Fallowfield, J.A.; Hayward, C.; Henderson, N.C.; Joshi, P.K.; et al. Genome-Wide Association Study of NAFLD Using Electronic Health Records. Hepatol. Commun. 2022, 6, 297–308. [Google Scholar] [CrossRef]
- Sveinbjornsson, G.; Ulfarsson, M.O.; Thorolfsdottir, R.B.; Jonsson, B.A.; Einarsson, E.; Gunnlaugsson, G.; Rognvaldsson, S.; Arnar, D.O.; Baldvinsson, M.; Bjarnason, R.G.; et al. Multiomics study of nonalcoholic fatty liver disease. Nat. Genet. 2022, 54, 1652–1663. [Google Scholar] [CrossRef]
- Lu, W.; Mei, J.; Yang, J.; Wu, Z.; Liu, J.; Miao, P.; Chen, Y.; Wen, Z.; Zhao, Z.; Kong, H.; et al. ApoE deficiency promotes non-alcoholic fatty liver disease in mice via impeding AMPK/mTOR mediated autophagy. Life Sci. 2020, 252, 117601. [Google Scholar] [CrossRef]
- van den Berg, E.H.; Corsetti, J.P.; Bakker, S.J.L.; Dullaart, R.P.F. Plasma ApoE elevations are associated with NAFLD: The PREVEND Study. PLoS ONE 2019, 14, e0220659. [Google Scholar] [CrossRef]
- Ellis, J.M.; Paul, D.S.; Depetrillo, M.A.; Singh, B.P.; Malarkey, D.E.; Coleman, R.A. Mice deficient in glycerol-3-phosphate acyltransferase-1 have a reduced susceptibility to liver cancer. Toxicol. Pathol. 2012, 40, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Harley, I.T.; Stankiewicz, T.E.; Giles, D.A.; Softic, S.; Flick, L.M.; Cappelletti, M.; Sheridan, R.; Xanthakos, S.A.; Steinbrecher, K.A.; Sartor, R.B.; et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 2014, 59, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.C.; Sasaki, M.; Cohen, D.M.; Cui, J.; Smith, M.A.; Yenilmez, B.O.; Steger, D.J.; Rader, D.J. Tribbles-1 regulates hepatic lipogenesis through posttranscriptional regulation of C/EBPalpha. J. Clin. Investig. 2015, 125, 3809–3818. [Google Scholar] [CrossRef]
- Ishizuka, Y.; Nakayama, K.; Ogawa, A.; Makishima, S.; Boonvisut, S.; Hirao, A.; Iwasaki, Y.; Yada, T.; Yanagisawa, Y.; Miyashita, H.; et al. TRIB1 downregulates hepatic lipogenesis and glycogenesis via multiple molecular interactions. J. Mol. Endocrinol. 2014, 52, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Hikiba, Y.; Fujiwara, H.; Ikenoue, T.; Sue, S.; Sugimori, M.; Matsubayashi, M.; Kaneko, H.; Irie, K.; Sasaki, T.; et al. NAFLD exacerbates cholangitis and promotes cholangiocellular carcinoma in mice. Cancer Sci. 2021, 112, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Cefalu, A.B.; Pirruccello, J.P.; Noto, D.; Gabriel, S.; Valenti, V.; Gupta, N.; Spina, R.; Tarugi, P.; Kathiresan, S.; Averna, M.R. A novel APOB mutation identified by exome sequencing cosegregates with steatosis, liver cancer, and hypocholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2021–2025. [Google Scholar] [CrossRef]
- Baselli, G.A.; Jamialahmadi, O.; Pelusi, S.; Ciociola, E.; Malvestiti, F.; Saracino, M.; Santoro, L.; Cherubini, A.; Dongiovanni, P.; Maggioni, M.; et al. Rare ATG7 genetic variants predispose patients to severe fatty liver disease. J. Hepatol. 2022, 77, 596–606. [Google Scholar] [CrossRef]
- Botstein, D.; Risch, N. Discovering genotypes underlying human phenotypes: Past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 2003, 33, 228–237. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, W.; Huang, S.; Xu, L.; Miao, M.; Wu, C.; Yu, C.; Li, Y.; Xu, C. Serum apoB levels independently predict the development of non-alcoholic fatty liver disease: A 7-year prospective study. Liver Int. 2017, 37, 1202–1208. [Google Scholar] [CrossRef]
- Hennig, P.; Fenini, G.; Di Filippo, M.; Karakaya, T.; Beer, H.D. The Pathways Underlying the Multiple Roles of p62 in Inflammation and Cancer. Biomedicines 2021, 9, 707. [Google Scholar] [CrossRef]
- Takahashi, Y.; Sugimoto, K.; Inui, H.; Fukusato, T. Current pharmacological therapies for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2015, 21, 3777–3785. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Kaimal, R.; Song, X.; Yan, B.; King, R.; Deng, R. Differential modulation of farnesoid X receptor signaling pathway by the thiazolidinediones. J. Pharmacol. Exp. Ther. 2009, 330, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Nevola, R.; Epifani, R.; Imbriani, S.; Tortorella, G.; Aprea, C.; Galiero, R.; Rinaldi, L.; Marfella, R.; Sasso, F.C. GLP-1 Receptor Agonists in Non-Alcoholic Fatty Liver Disease: Current Evidence and Future Perspectives. Int. J. Mol. Sci. 2023, 24, 1703. [Google Scholar] [CrossRef]
- Jouihan, H.; Will, S.; Guionaud, S.; Boland, M.L.; Oldham, S.; Ravn, P.; Celeste, A.; Trevaskis, J.L. Superior reductions in hepatic steatosis and fibrosis with co-administration of a glucagon-like peptide-1 receptor agonist and obeticholic acid in mice. Mol. Metab. 2017, 6, 1360–1370. [Google Scholar] [CrossRef]
- Dong, X.C. PNPLA3-A Potential Therapeutic Target for Personalized Treatment of Chronic Liver Disease. Front. Med. 2019, 6, 304. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, G.; Wallace, D.F.; Powell, E.E.; Rahman, T.; Clark, P.J.; Subramaniam, V.N. Gene Variants Implicated in Steatotic Liver Disease: Opportunities for Diagnostics and Therapeutics. Biomedicines 2023, 11, 2809. https://doi.org/10.3390/biomedicines11102809
Huang G, Wallace DF, Powell EE, Rahman T, Clark PJ, Subramaniam VN. Gene Variants Implicated in Steatotic Liver Disease: Opportunities for Diagnostics and Therapeutics. Biomedicines. 2023; 11(10):2809. https://doi.org/10.3390/biomedicines11102809
Chicago/Turabian StyleHuang, Gary, Daniel F. Wallace, Elizabeth E. Powell, Tony Rahman, Paul J. Clark, and V. Nathan Subramaniam. 2023. "Gene Variants Implicated in Steatotic Liver Disease: Opportunities for Diagnostics and Therapeutics" Biomedicines 11, no. 10: 2809. https://doi.org/10.3390/biomedicines11102809