
Implications for Combination Therapy of Selective Monoamine Reuptake Inhibitors on Dopamine Transporters
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Preparation
2.2. Neurotransmitter Uptake Assay
2.3. Computational Method
2.4. Data Analysis
3. Results and Discussion
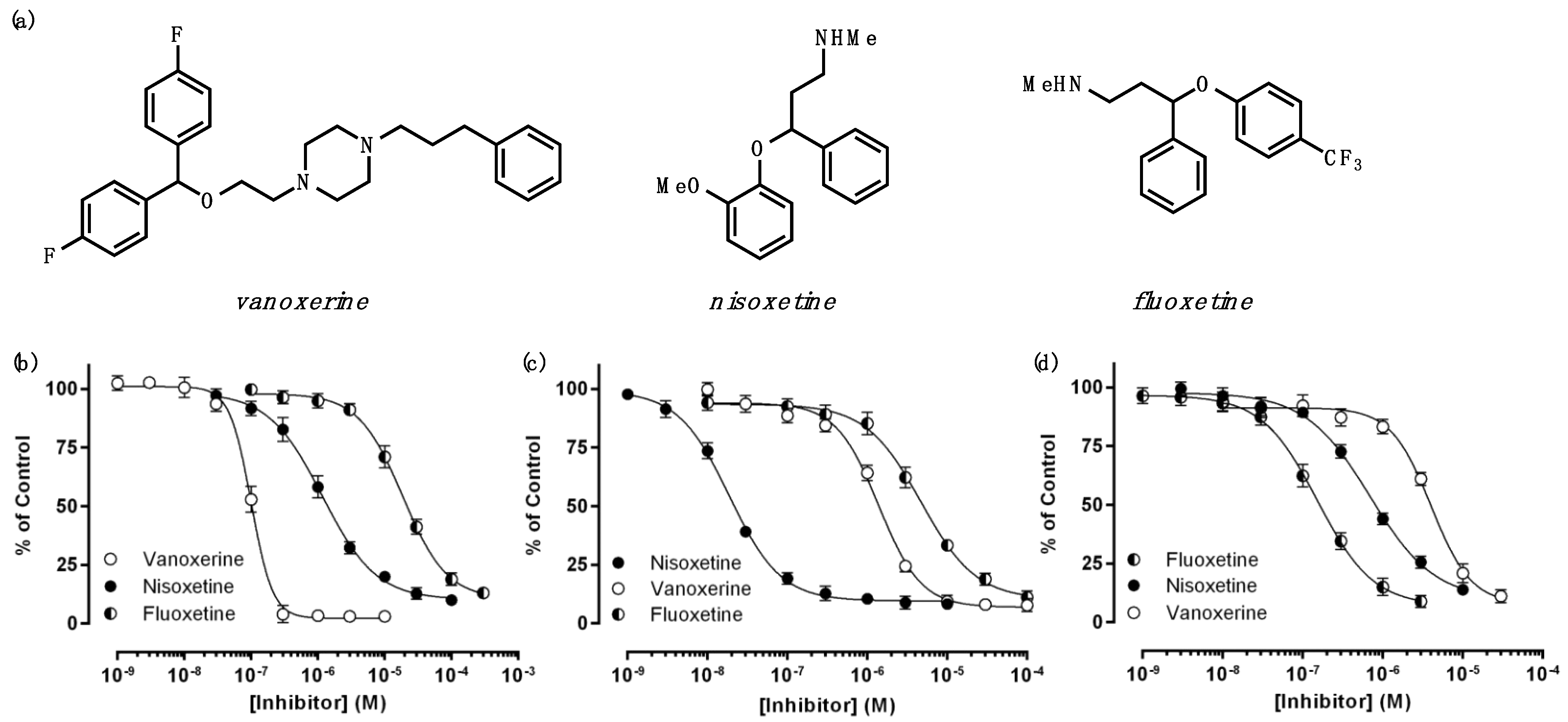
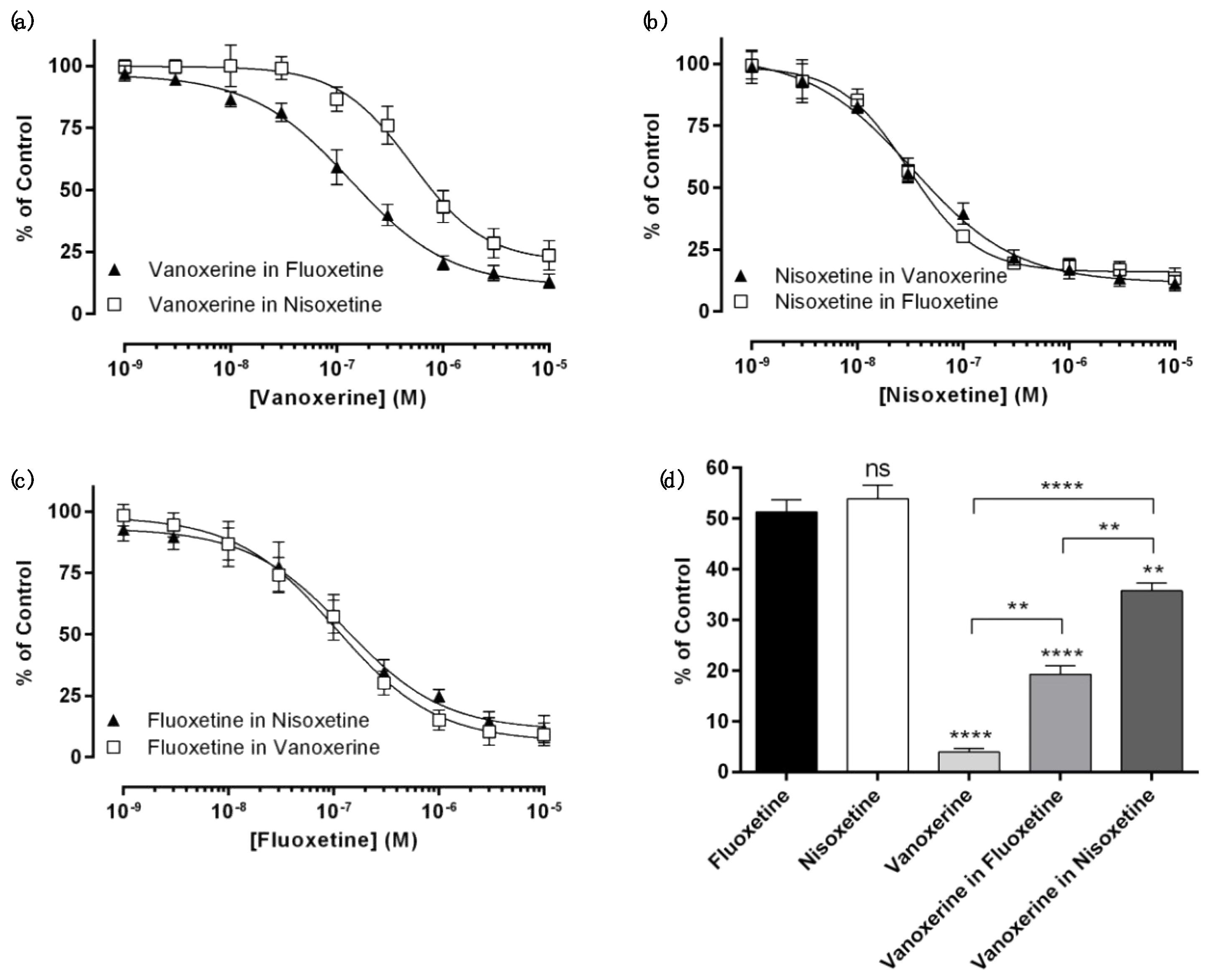
3.1. Antagonistic Effects of Multiple Drugs on DAT Inhibition
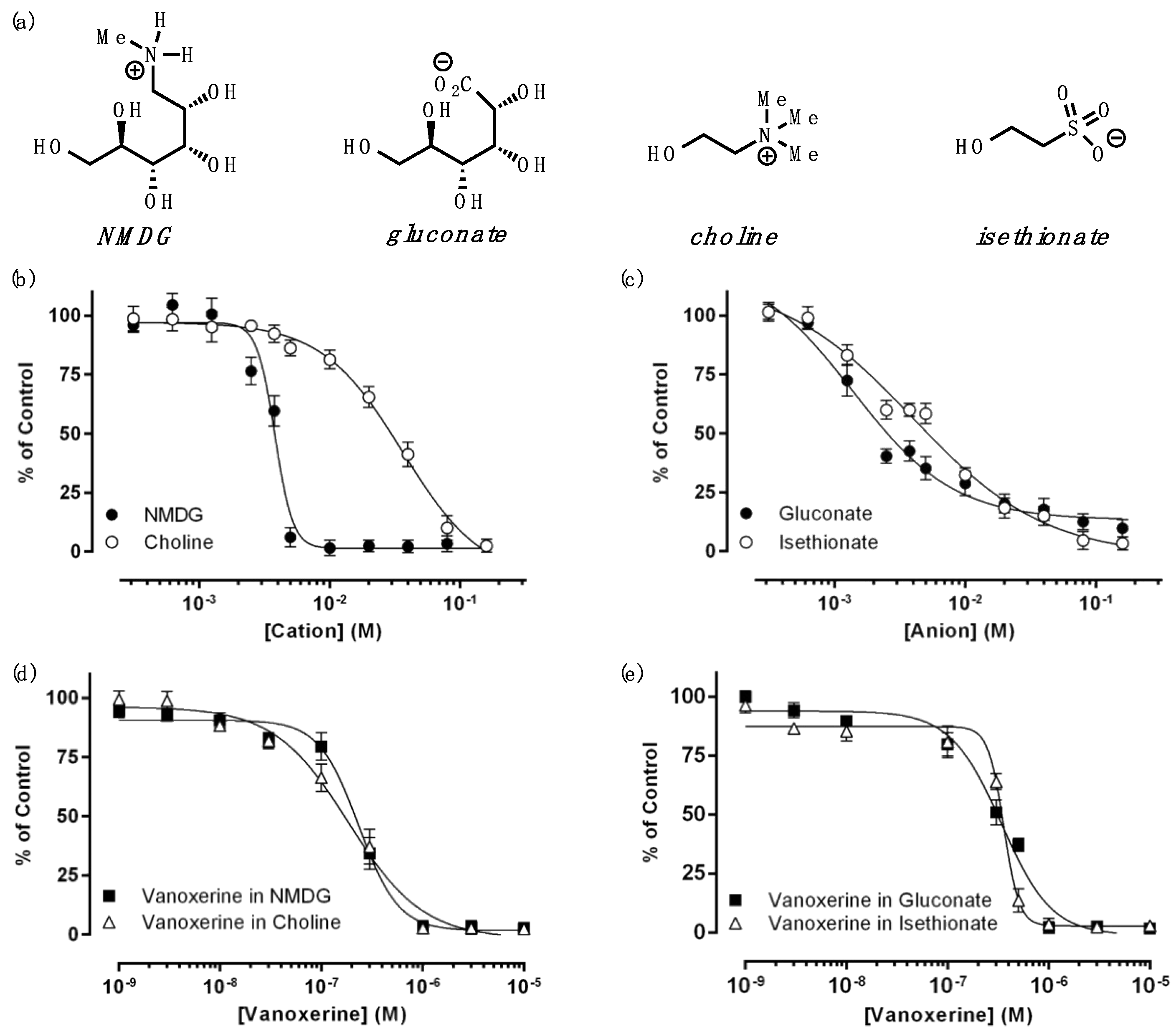
3.2. Effects of Monovalent Ions on DAT Inhibition by Vanoxerine
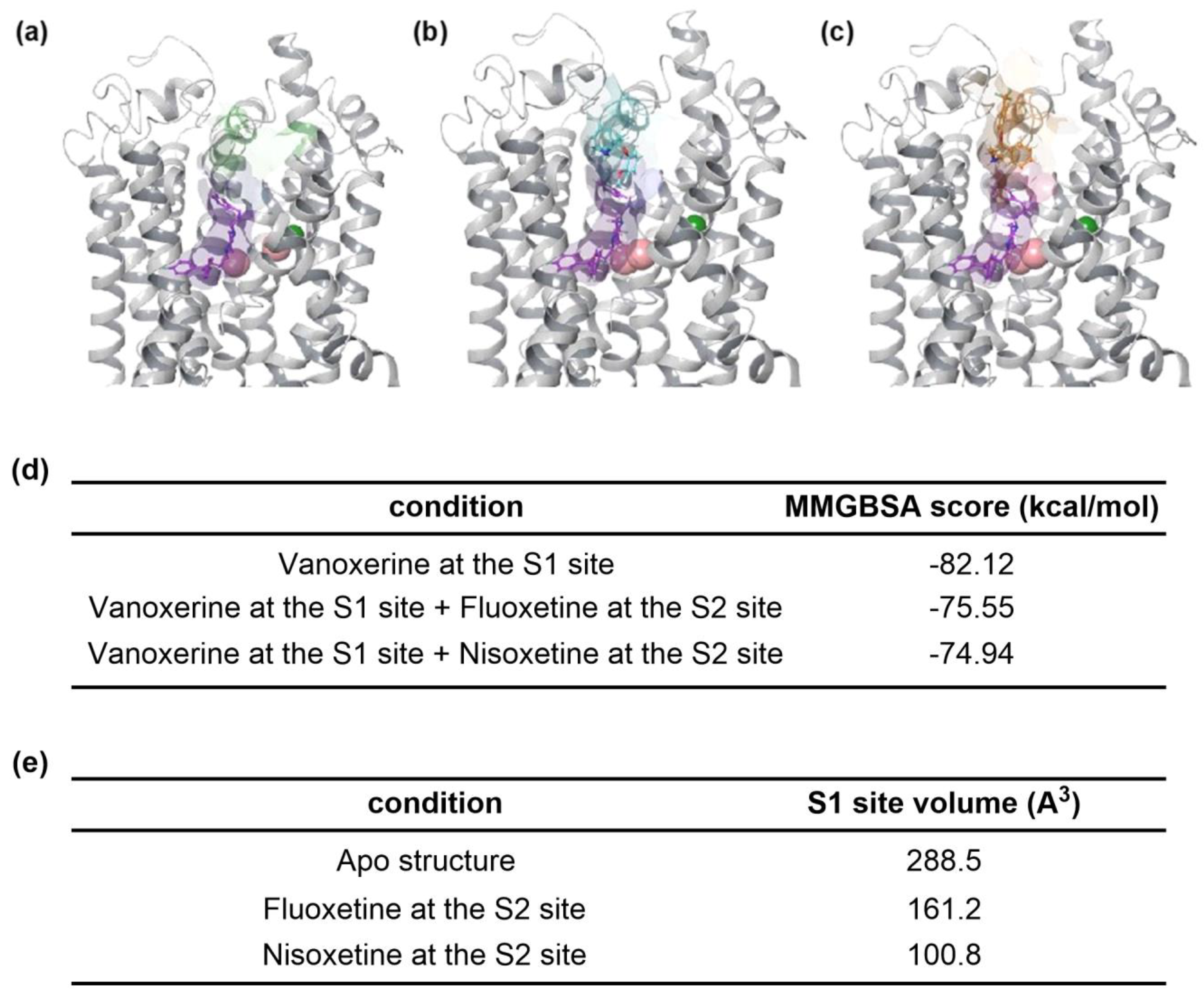
3.3. Structural Insights of DAT Inhibition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torres, G.E.; Gainetdinov, R.R.; Caron, M.G. Plasma membrane monoamine transporters: Structure, regulation and function. Nat. Rev. Neurosci. 2003, 4, 13–25. [Google Scholar]
- Weinglass, A.B.; Garcia, M.L.; Kaczorowski, G.L. Technologies for transporter drug discovery. Channels 2008, 2, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Kanner, B.I.; Zomot, E. Sodium-coupled neurotransmitter transporters. Chem. Rev. 2008, 108, 1654–1668. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J. Dual- and triple-acting agents for treating core and co-morbid symptoms of major depression: Novel concepts, new drugs. Neurotherapeutics 2009, 6, 53–77. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, S.; Nielsen, E.O.; Peters, D.; Dyhring, T. Validation of a fluorescence-based high-throughput assay for the meas-urement of neurotransmitter transporter uptake activity. J. Neurosci. Methods 2008, 169, 168–176. [Google Scholar] [CrossRef]
- Eshleman, A.J.; Carmolli, M.; Cumbay, M.; Martens, C.R.; Neve, K.A.; Janowsky, A. Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. J. Pharmacol. Exp. Ther. 1999, 289, 877–885. [Google Scholar]
- Frnka, J.V.; Chang, A.S.; Lam, D.M. Pharmacological characteristics of high-affinity serotonin uptake systems established through gene transfer. J. Pharmacol. Exp. Ther. 1991, 256, 734–740. [Google Scholar]
- Hansard, M.J.; Smith, L.A.; Jackson, M.J.; Cheetham, S.C.; Jenner, P. Dopamine, but not norepinephrine or serotonin, reuptake inhibition reverses motor deficits in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primates. J. Pharmacol. Exp. Ther. 2002, 303, 952–958. [Google Scholar] [CrossRef]
- Bonnet, J.J. Interactions of cations and anions with the binding of uptake blockers to the dopamine transporter. Eur. J. Phar-macol. 2003, 479, 199–212. [Google Scholar] [CrossRef]
- Jope, R.S.; Gu, X. Seizures increase acetylcholine and choline concentrations in rat brain regions. Neurochem. Res. 1991, 16, 1219–1226. [Google Scholar] [CrossRef]
- Klein, J.; Koppen, A.; Loffelholz, K.; Schmitthenner, J. Uptake and metabolism of choline by rat brain after acute choline ad-ministration. J. Neurochem. 1992, 58, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Corera, A.T.; Costentin, J.; Bonnet, J.J. Binding of uptake blockers to the neuronal dopamine transporter: Further investigation about cationic and anionic requirements. Naunyn. Schmiedebergs. Arch. Pharmacol. 2000, 362, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Amejdki-Chab, N.; Benmansour, S.; Costentin, J.; Bonnet, J.J. Effects of several cations on the neuronal uptake of dopamine and the specific binding of [3H]GBR 12783: Attempts to characterize the Na+ dependence of the neuronal transport of dopamine. J. Neurochem. 1992, 59, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Sun, L.; Reith, M.E. Cationic interactions at the human dopamine transporter reveal binding conformations for dopamine distinguishable from those for the cocaine analog 2 alpha-carbomethoxy-3 alpha-(4-fluorophenyl)tropane. J. Neurochem. 2002, 81, 1383–1393. [Google Scholar] [CrossRef]
- Coffey, L.L.; Reith, M.E. [3H]WIN 35,428 binding to the dopamine uptake carrier. I. Effect of tonicity and buffer composition. J. Neurosci. Methods 1994, 51, 23–30. [Google Scholar] [CrossRef]
- Milner, H.E.; Jarvis, S.M. Ionic requirements of [3H]GBR-12935 binding to the dopamine transporter in canine striatal membranes. Biochem. Soc. Trans. 1992, 20, 243S. [Google Scholar] [CrossRef]
- Shi, L.; Quick, M.; Zhao, Y.; Weinstein, H.; Javitch, J.A. The mechanism of a neurotransmitter: Sodium symporter--inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol. Cell 2008, 30, 667–677. [Google Scholar] [CrossRef]
- Singh, S.K.; Yamashita, A.; Gouaux, E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature 2007, 448, 952–956. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhen, J.; Karpowich, N.K.; Goetz, R.M.; Law, C.J.; Reith, M.E.; Wang, D.N. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science 2007, 317, 1390–1393. [Google Scholar] [CrossRef]
- Schmitt, K.C.; Reith, M.E. The atypical stimulant and nootropic modafinil interacts with the dopamine transporter in a dif-ferent manner than classical cocaine-like inhibitors. PLoS ONE 2011, 6, e25790. [Google Scholar] [CrossRef]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 2005, 437, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Beuming, T.; Kniazeff, J.; Bergmann, M.L.; Shi, L.; Gracia, L.; Raniszewska, K.; Raniszewska, K.; Newman, A.H.; Javitch, J.A.; Weinstein, H.; et al. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat. Neurosci. 2008, 11, 780–789. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vanoxerine | Nisoxetine | Fluoxetine | ||
|---|---|---|---|---|
| DAT | IC50 (μM) | 0.09 ± 0.01 | 1.15 ± 0.17 | 18.4 ± 2.7 |
| %Amin | 0.0 ± 0.8 | 9.2 ± 2.4 | 9.4 ± 2.8 | |
| NET | IC50 (μM) | 1.46 ± 0.13 | 0.019 ± 0.001 | 4.41 ± 0.33 |
| SERT | IC50 (μM) | 3.84 ± 0.22 | 0.70 ± 0.07 | 0.13 ± 0.03 |
| DAT | Vanoxerine | Vanoxerine in Fluoxetine | Vanoxerine in Nisoxetine | |
| IC50 (μM) | 0.09 ± 0.01 | 0.14 ± 0.03 | 0.55 ± 0.09 | |
| %Amin | 0.0 ± 0.8 | 10.5 ± 3.5 | 21.7 ± 5.6 | |
| Significance | * | **** | ||
| NET | Nisoxetine | Nisoxetine in Vanoxerine | Nisoxetine in Fluoxetine | |
| IC50 (μM) | 0.019 ± 0.001 | 0.032 ± 0.002 | 0.030 ± 0.02 | |
| Significance | NS | NS | ||
| SERT | Fluoxetine | Fluoxetine in Nisoxetine | Fluoxetine in Vanoxerine | |
| IC50 (μM) | 0.13 ± 0.03 | 0.13 ± 0.04 | 0.11 ± 0.03 | |
| Significance | NS | NS |
| Cation | Anion | |||
|---|---|---|---|---|
| NMDG | Choline | Gluconate | Isethionate | |
| IC50 (mM) | 3.7 ± 0.2 | 36.6 ± 2.0 | 1.4 ± 0.2 | 3.3 ± 0.6 |
| h | 10.6 ± 3.8 | 1.4 ± 0.2 | 1.2 ± 0.3 | 0.8 ± 0.2 |
| Control | Cation | Anion | |||
|---|---|---|---|---|---|
| NMDG | Choline | Gluconate | Isethionate | ||
| IC50 (μM) | 0.09 ± 0.01 | 0.17 ± 0.01 | 0.14 ± 0.01 | 0.20 ± 0.02 | 0.23 ± 0.01 |
| Significance | *** | ** | ** | **** | |
| NS | NS | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, H.; Park, K.; Kim, D.; Chi, S.-G.; Choi, K.-H.; Han, S.-J.; Song, C. Implications for Combination Therapy of Selective Monoamine Reuptake Inhibitors on Dopamine Transporters. Biomedicines 2023, 11, 2846. https://doi.org/10.3390/biomedicines11102846
Ahn H, Park K, Kim D, Chi S-G, Choi K-H, Han S-J, Song C. Implications for Combination Therapy of Selective Monoamine Reuptake Inhibitors on Dopamine Transporters. Biomedicines. 2023; 11(10):2846. https://doi.org/10.3390/biomedicines11102846
Chicago/Turabian StyleAhn, Hyomin, Kichul Park, Dongyoung Kim, Sung-Gil Chi, Kee-Hyun Choi, Seo-Jung Han, and Chiman Song. 2023. "Implications for Combination Therapy of Selective Monoamine Reuptake Inhibitors on Dopamine Transporters" Biomedicines 11, no. 10: 2846. https://doi.org/10.3390/biomedicines11102846