
Therapeutic Strategies in Huntington’s Disease: From Genetic Defect to Gene Therapy


Abstract
:1. Introduction
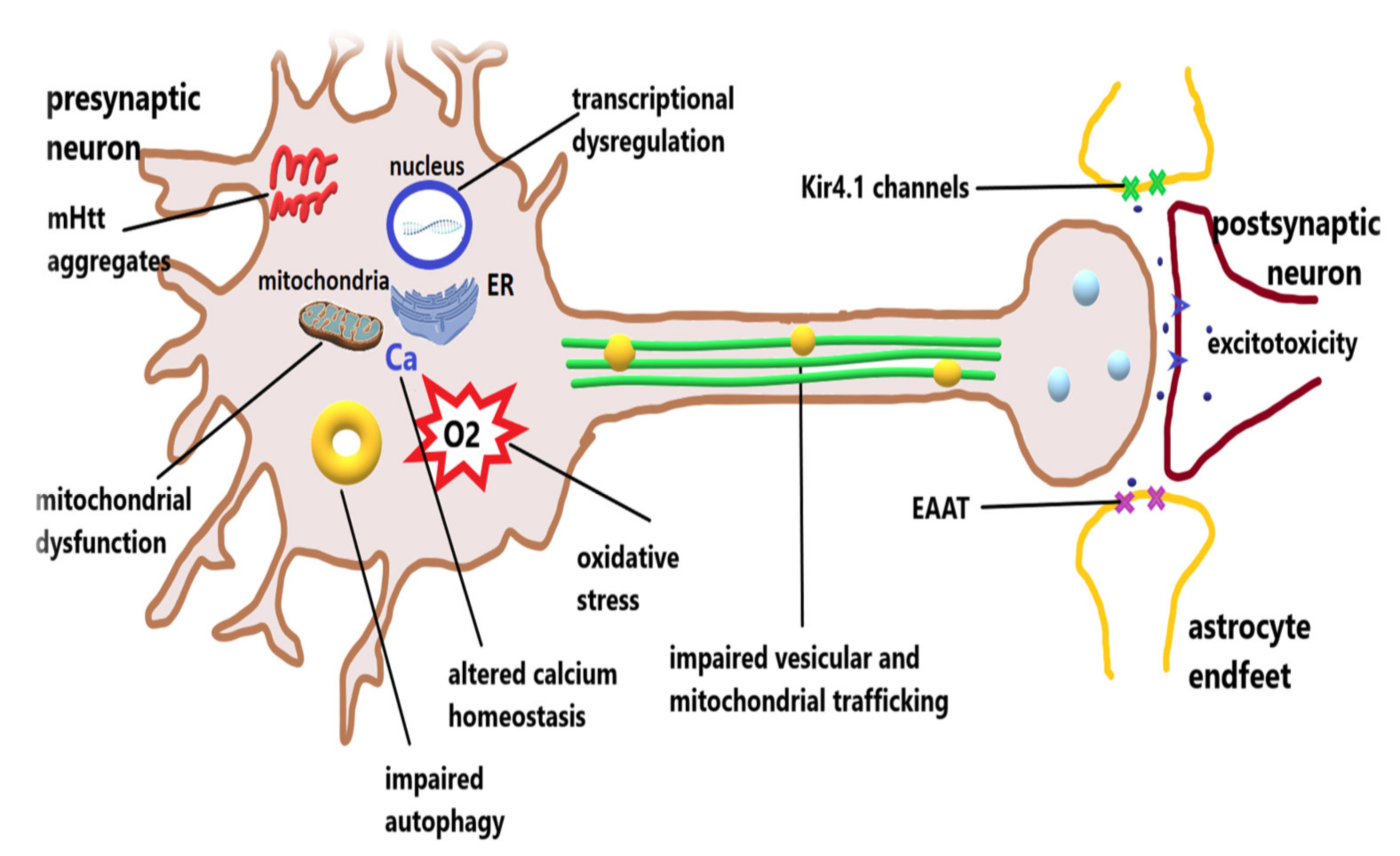
2. Pathophysiology of Huntington’s Disease
2.1. Excitotoxicity
2.2. Impairments of the Proteostasis
2.3. Mitochondrial Dysfunction
2.4. Oxidative Stress
2.5. Transcriptional Dysregulation
2.6. Impaired BDNF Synthesis and Transport
2.7. Dysfunctions of Glial Cells
2.8. Neuroinflammation in HD
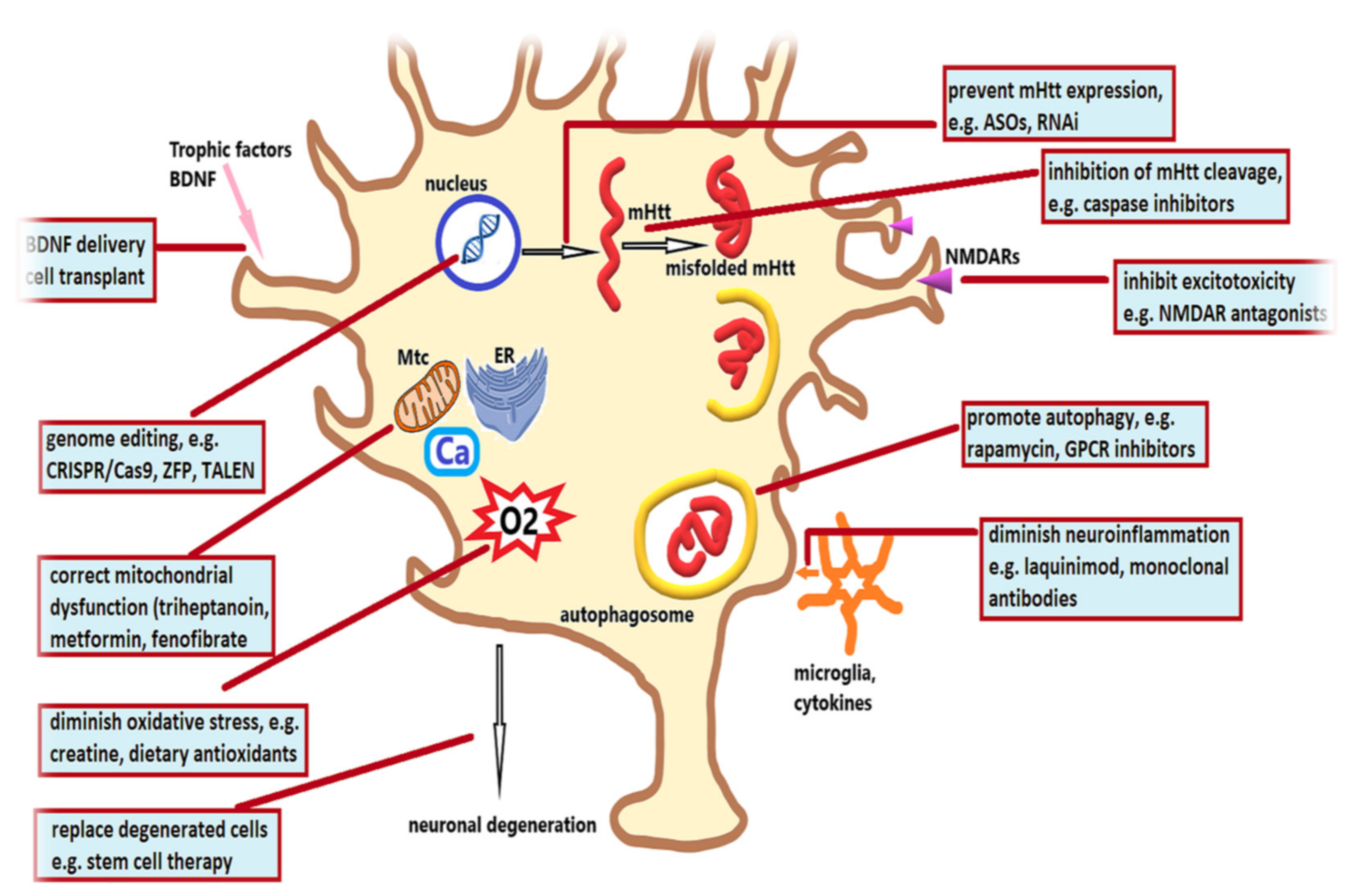
3. Therapeutic Strategies in Huntington’s Disease
3.1. Therapeutic Strategies Targeting Pathogenic Cascades
3.2. Therapeutic Strategies Targeting Mutant Huntingtin
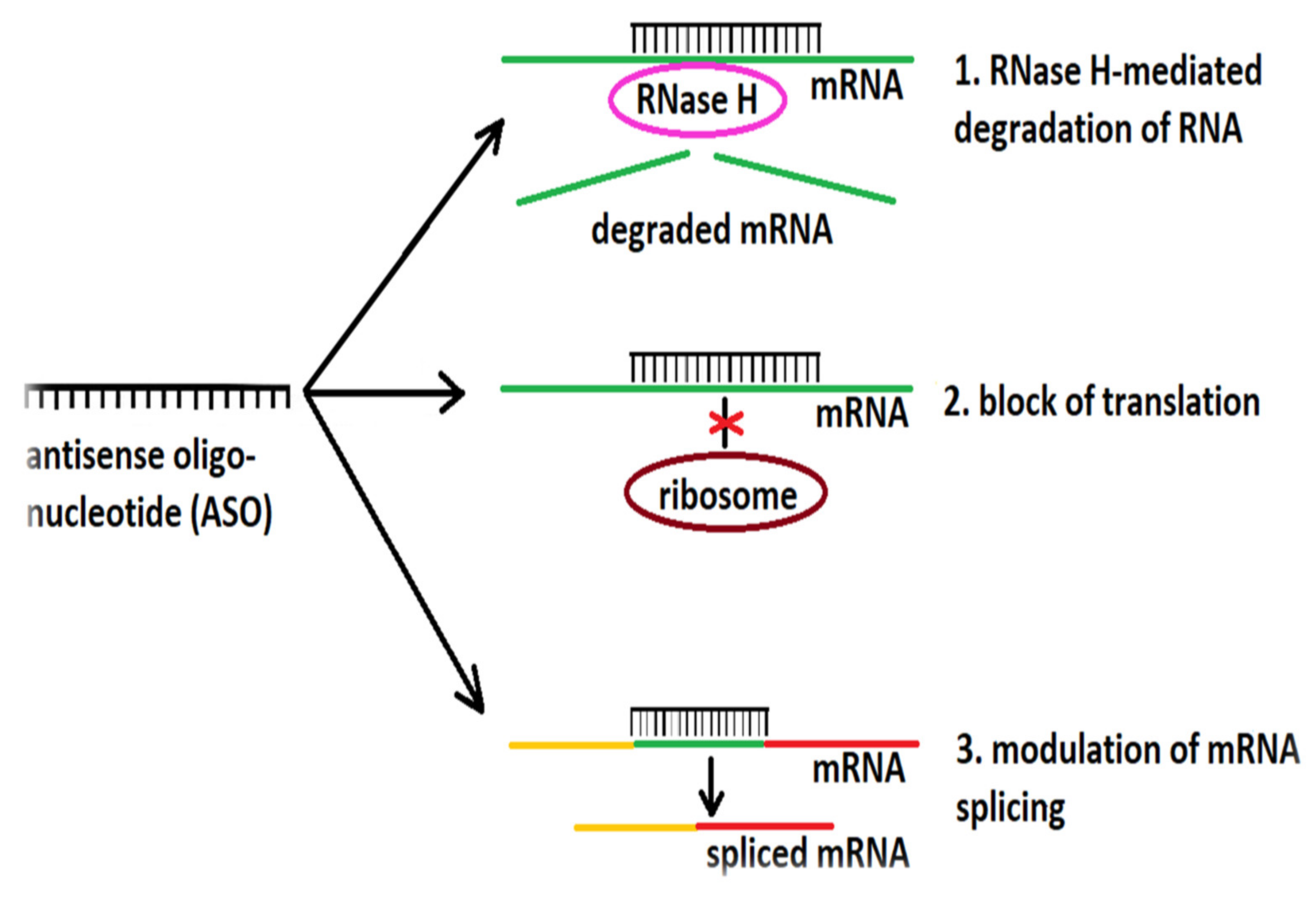
3.2.1. Targeting the RNA
3.2.2. Targeting the DNA
3.3. Therapeutic Strategies Targeting Cell Loss
3.4. Strategies Targeting Neuroinflammation
3.4.1. Laquinimod
3.4.2. Drugs Targeting TNF-α
3.4.3. Antibody-Based Therapies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutic targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef] [PubMed]
- HD Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Jansen, A.H.P.; van Hall, M.; Op den Kelder, I.C.; Meier, R.T.; de Ruiter, A.A.; Schut, M.H.; Smith, D.L.; Grit, C.; Brouwer, N.; Kamphuis, W.; et al. Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell-type-specific. Glia 2017, 65, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Kojer, K.; Hering, T.; Bazenet, C.; Weiss, A.; Herrmann, F.; Taanman, J.W.; Orth, M. Huntingtin aggregates and mitochondrial pathology in skeletal muscle but not heart of late-stage R6/2 mice. J. Huntingt. Dis. 2019, 8, 145–159. [Google Scholar] [CrossRef]
- Jurcau, A. Molecular pathophysiological mechanisms in Huntington’s disease. Biomedicines 2022, 10, 1432. [Google Scholar] [CrossRef]
- Beal, M.F.; Ferrante, R.J.; Swartz, K.J.; Kowall, N.W. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J. Neurosci. 1991, 11, 1649–1659. [Google Scholar] [CrossRef]
- Shenoy, S.A.; Zheng, S.; Liu, W.; Dai, Y.; Liu, Y.; Hou, Z.; Mori, S.; Tang, Y.; Cheng, J.; Duan, W.; et al. A novel and accurate full-length HTT mouse model for Huntington’s disease. eLife 2022, 11, e70217. [Google Scholar] [CrossRef]
- Eremeev, A.V.; Lebedeva, O.S.; Bogomiakova, M.E.; Lagarkova, M.A.; Bogomazova, A.N. Cerebral organoids—Challenges to establish a brain prototype. Cells 2021, 10, 1790. [Google Scholar] [CrossRef]
- Reiner, A.; Deng, Y.-P. Disrupted striatal neuron inputs and outputs in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 250–280. [Google Scholar] [CrossRef]
- Cowan, C.M.; Fan, M.M.; Fan, J.; Shehadeh, J.; Zhang, L.Y.; Graham, R.K.; Hayden, M.R.; Raymond, L.A. Polyglutamine-modulated striatal calpain activity in YAC transgenic Huntington disease mouse model: Impact on NMDA receptor function and toxicity. J. Neurosci. 2008, 28, 12725–12735. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Zhang, Y.; Parsons, C.G.; Liu, Y.F. Expression of polyglutamine-expanded huntingtin induces tyrosine phosphorylation of N-methyl-D-aspartate receptors. J. Biol. Chem. 2003, 278, 33364–33369. [Google Scholar] [CrossRef] [Green Version]
- Jurcau, A.; Ardelean, I.A. Molecular pathophysiological mechanisms of ischemia/reperfusion injuries after recanalization therapy for acute ischemic stroke. J. Integr. Neurosci. 2021, 20, 727–744. [Google Scholar]
- Raymond, L.A. Striatal synaptic dysfunction and altered calcium regulation in Huntington disease. Biochem. Biophys. Res. Commun. 2017, 483, 1051–1062. [Google Scholar] [CrossRef]
- Jurcau, A.; Ardelean, A.I. Oxidative stress in ischemia/reperfusion injuries following acute ischemic stroke. Biomedicines 2022, 10, 574. [Google Scholar] [CrossRef]
- Gladding, C.M.; Sepers, M.D.; Xu, J.; Zhang, L.Y.; Milnerwood, A.J.; Lombroso, P.J.; Raymond, L.A. Calpain and STriatal-Enriched protein tyrosine phosphatase (STEP) activation contribute to extrasynaptic NMDA receptor localization in a Huntington’s disease mouse model. Hum. Mol. Genet. 2012, 21, 3739–3752. [Google Scholar] [CrossRef]
- Xifró, X.; García-Martínez, J.M.; Del Toro, D.; Alberch, J.; Pérez-Navarro, E. Calcineurin is involved in the early activation of NMDA-mediated cell death in mutant huntingtin knock-in striatal cells. J. Neurochem. 2008, 105, 1596–1612. [Google Scholar] [CrossRef]
- Harding, R.J.; Tong, Y.F. Proteostasis in Huntington’s disease: Disease mechanisms and therapeutic opportunities. Acta Pharmacol. Sin. 2018, 39, 754–769. [Google Scholar] [CrossRef] [Green Version]
- Jurcau, A.; Nunkoo, V.S. Tau-targeted therapy in Alzheimer’s disease: History and current state. In Frontiers in Clinical Drug Research; Ibarra Arias, J.J.J., Ed.; Bentham Science Publishers: Singapore, 2021; Volume 2, pp. 56–138. [Google Scholar]
- Moily, N.S.; Ormsby, A.R.; Stojilovic, A.; Ramdzan, Y.M.; Diesch, J.; Hannan, R.D.; Zajac, M.S.; Hannan, A.J.; Oshlack, A.; Hatters, D.M. Transcriptional profiles for distinct aggregation states of mutant huntingtin exon 1 protein unmask new Huntington’s disease pathways. Mol. Cell. Neurosci. 2017, 83, 103–112. [Google Scholar] [CrossRef]
- Nath, S.R.; Lieberman, A.P. The ubiquitination, disaggregation and proteasomal degradation machineries in polyglutamine disease. Front. Mol. Neurosci. 2017, 10, 78. [Google Scholar] [CrossRef]
- Li, X.J.; Li, S. Proteasomal dysfunction in aging and Huntington’s disease. Neurobiol. Dis. 2011, 43, 4–8. [Google Scholar] [CrossRef] [Green Version]
- Jurcau, A. Insights into the pathogenesis of neurodegenerative diseases: Focus on mitochondrial dysfunction and oxidative stress. Int. J. Mol. Sci. 2021, 22, 11847. [Google Scholar] [CrossRef]
- Soares, T.R.; Reis, S.D.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. Targeting the proteostasis network in Huntington’s disease. Ageing Res. Rev. 2019, 49, 92–103. [Google Scholar] [CrossRef]
- Mealer, R.G.; Murray, A.J.; Shahani, M.; Subramaniam, S.; Snyder, S.H. Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J. Biol. Chem. 2014, 289, 3547–3554. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Wu, P.; Pan, Y.; Sun, X.; DiFiglia, M.; Lu, B. A toxic mutant huntingtin species is resistant to selective autophagy. Nat. Chem. Biol. 2017, 13, 1152. [Google Scholar] [CrossRef]
- Kraus, F.; Ryan, M.T. The constriction and scission machineries involved in mitochondrial fission. J. Cell Sci. 2017, 30, 2953–2960. [Google Scholar] [CrossRef] [Green Version]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef]
- Sawant, N.; Morton, H.; Kshirsagar, S.; Reddy, A.P.; Reddy, P.H. Mitochondrial abnormalities and synaptic damage in Huntington’s disease: A focus on defective mitophagy and mitochondria-targeted therapeutics. Mol. Neurobiol. 2021, 58, 6350–6377. [Google Scholar] [CrossRef]
- Woo, J.H.; Cho, H.; SEol, Y.H.; Kim, S.H.; Park, C.; Yousefian-Yazi, A.; Hyeon, S.J.; Lee, J.; Ryu, H. Power failure of mitochondria and oxidative stress in neurodegeneration and its computational model. Antioxidants 2021, 10, 229. [Google Scholar] [CrossRef]
- López-Doménech, G.; Coville-Cooke, C.; Ivankovic, D.; Halff, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 2018, 37, 321–336. [Google Scholar] [CrossRef]
- Bossy-Wetzel, E.; Petrilli, A.; Knott, A.B. Mutant Huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008, 31, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Mackay, J.P.; Nassrallah, W.B.; Raymond, L.A. Cause or compensation?—Altered neuronal Ca2+ handling in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 301–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czeredys, M.; Maciag, F.; Methner, A.; Kuznicki, J. Tetrahydrocarbazoes decrease elevated SOCE in medium spiny neurons from transgenic YAC128 mice, a model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1194–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurcau, A.; Simion, A. Oxidative stress in the pathogenesis of Alzheimer’s disease and cerebrovascular disease with therapeutic implications. CNS Neurol. Disord. Drug Targets 2020, 19, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Fão, L.; Rego, C. Mitochondrial and redox-based therapeutic strategies in Huntington’s disease. Antioxid. Redox Signal. 2021, 34, 650–673. [Google Scholar] [CrossRef]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Ratan, R.R. Oxidative stress and Huntington’s disease: The good, the bad, and the ugly. J. Huntingt. Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef] [Green Version]
- Valadão, P.A.C.; Santos, K.B.S.; Ferreira, E.; Vieira, T.H.; Macedo, E.; Cordeiro, T.; Teixeira, A.L.; Guatimosim, C.; de Miranda, A.S. Inflammation in Huntington’s disease: A few new twists on an old tale. J. Neuroimmunol. 2020, 348, 577380. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F. Role of neuroinflammation in amyotrophic lateral sclerosis: Cellular mechanisms and therapeutic implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [Green Version]
- Jurcau, A.; Simion, A. Neuroinflammation in cerebral ischemia and ischemia/reperfusion injuries: From pathophysiology to therapeutic strategies. Int. J. Mol. Sci. 2021, 23, 14. [Google Scholar] [CrossRef]
- Ganner, A.; Pfeiffer, Z.-C.; Wingendorf, L.; Kreis, S.; Klein, M.; Walz, G.; Neumann-Haefelin, E. The acetyltransferase p300 regulates NRF2 stability and localization. Biochem. Biophys. Res. Commun. 2020, 524, 895–902. [Google Scholar] [CrossRef]
- Choi, Y.S.; Lee, B.; Cho, H.Y.; Reyes, I.B.; Pu, X.A.; Saido, T.C.; Hoyt, K.R.; Obrietan, K. CREB is a key regulator of striatal vulnerability in chemical and genetic models of Huntington’s disease. Neurobiol. Dis. 2009, 36, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Zuccato, C.; Belyaev, N.; Conforti, P.; Ooi, L.; Tartari, M.; Papadimou, E.; MacDonald, M.; Fossale, E.; Zeitlin, S.; Buckley, N.; et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J. Neurosci. 2007, 27, 6972–6983. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Zukin, R.S. REST, master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 2018, 48, 193–200. [Google Scholar] [CrossRef]
- Bae, B.I.; Xu, H.; Igarashi, S.; Fujimoro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 2004, 47, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Altar, C.A.; Cai, N.; Bliven, T.; Juhasz, M.; Conner, J.M.; Acheson, A.L.; Lindsay, R.M.; Wiegand, S.J. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 1997, 389, 856–860. [Google Scholar] [CrossRef]
- Pla, P.; Orvoen, S.; Saudou, F.; David, D.J.; Humbert, S. Mood disorders in Huntington’s disease: From behavior to cellular and molecular mechanisms. Front. Behav. Neurosci. 2014, 8, 135. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Yousefian-Jazi, A.; Choi, S.H.; Chang, I.; Lee, J.; Ryu, H. Non-cell autonomous and epigenetic mechanisms of Huntington’s disease. Int. J. Mol. Sci. 2021, 22, 12499. [Google Scholar] [CrossRef]
- Qin, Z.-H.; Wang, Y.; Sapp, E.; Cuiffo, B.; Wanker, E.; Hayden, M.R.; Kegel, K.B.; Aronin, N.; DiFiglia, M. Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J. Neurosci. 2004, 24, 269–281. [Google Scholar] [CrossRef]
- Khakh, B.S.; Beaumont, V.; Cachope, R.; Munoz-Sanjuan, I.; Goldman, S.A.; Grantyn, R. Unravelling and exploiting astrocyte dysfunction in Huntington’s disease. Trends Neurosci. 2017, 40, 422–437. [Google Scholar] [CrossRef]
- Ohno, Y.; Kinboshi, M.; Shimizu, S. Inwardly rectifying potassium channel Kir4. 1 as a novel modulator of BDNF expression in astrocytes. Int. J. Mol. Sci. 2018, 19, 3313. [Google Scholar] [CrossRef] [Green Version]
- Dvorzhak, A.; Helassa, N.; Török, K.; Schmitz, D.; Grantyn, R. Single synapse indicators of impaired glutamate clearance derived from fast iGluu imaging of cortical afferents in the striatum of normal and Huntington (Q175) mice. J. Neurosci. 2019, 39, 3970–3982. [Google Scholar] [CrossRef] [Green Version]
- Bardile, C.F.; Garcia-Miralles, M.; Caron, N.S.; Rayan, N.A.; Langley, S.R.; Harmston, N.; Rondelli, A.M.; Teo, R.T.Y.; Waltl, S.; Anderson, L.M.; et al. Intrinsic mutant HTT-mediated defects in oligodendroglia cause myelination deficits and behavioral abnormalities in Huntington disease. Proc. Natl. Acad. Sci. USA 2019, 116, 9622–9627. [Google Scholar] [CrossRef] [Green Version]
- O’Regan, G.C.; Farag, S.H.; Casey, C.S.; Wood-Kaczmar, A.; Pocock, J.M.; Tabrizi, S.J.; Andre, R. Human Huntington’s disease pluripotent stem cell-derived microglia develop normally but are abnormally hyper-reactive and release elevated levels of reactive oxygen species. J. Neuroinflamm. 2021, 18, 94. [Google Scholar] [CrossRef]
- Rocha, N.P.; Charron, O.; Latham, L.B.; Colpo, G.D.; Zanotti-Fregonara, P.; Yu, M.; Freeman, L.; Furr Stimming, E.; Teixeira, A.L. Microglia activation in basal ganglia is a late event in Huntington disease pathophysiology. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e984. [Google Scholar] [CrossRef]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The role of microglia and astrocytes in Huntington’s disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [Green Version]
- Bachoud-Lévi, A.C.; Ferreira, J.; Massart, R.; Youssov, K.; Rosser, A.; Busse, M.; Craufurd, D.; Reilmann, R.; De Michele, G.; Rae, D.; et al. International guidelines for the treatment of Huntington’s disease. Front. Neurol. 2019, 10, 710. [Google Scholar] [CrossRef] [Green Version]
- Priller, J. Effects of EGCG (Epigallocatechin Gallate) in Huntington’s Disease. The ETON-Study—A Randomized, Double-Blind, Stratified, Placebo-Controlled Prospective Investigator Initiated Multicenter Trial-Charite University, Berlin, Germany. Identifier: NCT01357681. Available online: https://clinicaltrials.gov/ct2/show/NCT01357681 (accessed on 4 June 2022).
- Pasinetti, G.M.; Wang, J.; Marambaud, P.; Ferruzzi, M.; Gregor, P.; Knable, L.A.; Ho, L. Neuroprotective and metabolic effects of resveratrol: Therapeutic implications for Huntington’s disease and other neurodegenerative disorders. Exp. Neurol. 2011, 232, 1–6. [Google Scholar] [CrossRef]
- Ho, D.J.; Calingasan, N.Y.; Wille, E.; Dumont, M.; Beal, M.F. Resveratrol protects against peripheral deficits in a mouse model of Huntington’s disease. Exp. Neurol. 2010, 225, 74–84. [Google Scholar] [CrossRef]
- Available online: www.clinicaltrials.gov (accessed on 7 May 2022).
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravillai, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington’s disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Ona, V.O.; Li, M.; Ferrante, R.J.; Fink, K.B.; Zhu, S.; Bian, J.; Guo, L.; Farrell, L.A.; Hersch, S.M.; et al. Minocycline inhibits caspase -1 1nd caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 2000, 6, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study Group DOMINO Investigators. A futility study of minocycline in Huntington’s disease. Mov. Disord. 2010, 25, 2219–2224. [Google Scholar] [CrossRef] [PubMed]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies. J. Parkinsons Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Schiefer, J.; Landwehrmeyer, G.B.; Luesse, H.G.; Sprunken, A.; Puls, C.; Milkereit, A.; Milkereit, E.; Kosinski, C.M. Riluzole prolongs survival time and alters nuclear inclusion formation in a transgenic mouse model of Huntington’s disease. Mov. Disord. 2002, 17, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Landwehrmeyer, G.B.; Dubois, B.; Garcia de Jébenes, J.; Kremer, B.; Gaus, W.; Kraus, P.H.; Przuntek, H.; Dib, M.; Doble, A.; Fischer, W.; et al. Riluzole in Huntington’s disease: A 3-year, randomized controlled study. Ann. Neurol. 2007, 62, 262–272. [Google Scholar] [CrossRef]
- Rosas, H.D.; Koroshetz, W.J.; Jenkins, B.G.; Chen, Y.I.; Hayden, D.L.; Beal, M.F.; Cudkowicz, M.E. Riluzole therapy in Huntington’s disease (HD). Mov. Disord. 1999, 14, 326–330. [Google Scholar] [CrossRef]
- Seppi, K.; Mueller, J.; Bodner, T.; Brandauer, E.; Benke, T.; Weirich-Schwaiger, H.; Poewe, W.; Wenning, G.K. Riluzole in Huntington’s disease (HD): An open label study with one year follow up. J. Neurol. 2001, 248, 866–869. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Park, J.E.; Kang, L.; Ko, S.Y.; Jung, K.H.; Kim, M. Memantine reduces striatal cell death with decreasing calpain level in 3-nitropropionic model of Huntington’s disease. Brain Res. 2006, 118, 199–207. [Google Scholar] [CrossRef]
- Dau, A.; Gladding, C.M.; Sepers, M.D.; Raymond, L.A. Chronic blockade of extrasynaptic NMDA receptors ameliorates synaptic dysfunction and pro-death signaling in Huntington disease transgenic mice. Neurobiol. Dis. 2014, 62, 533–542. [Google Scholar] [CrossRef]
- Cankurtan, E.S.; Ozalp, E.; Soygur, H.; Cakir, A. Clinical experience with risperidone and memantine in the treatment of Huntington’s disease. J. Natl. Med. Assoc. 2006, 98, 1353–1355. [Google Scholar]
- Kremer, B.; Clark, C.M.; Almqvist, E.W.; Raymond, L.A.; Graf, P.; Jacova, C.; Mezei, M.; Hardy, M.A.; Snow, B.; Martin, W.; et al. Influence of lamotrigine on progression of early Huntington disease: A randomized clinical trial. Neurology 1999, 53, 1000–1011. [Google Scholar] [CrossRef]
- Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: A randomized controlled trial. Neurology 2006, 66, 366–372. [Google Scholar] [CrossRef]
- Frank, S. Tetrabenazine as anti-chorea therapy in Huntington disease: An open-label continuation study. Huntington Study Group/TETRA-HD Investigators. BMC Neurol. 2009, 9, 62. [Google Scholar] [CrossRef] [Green Version]
- Huntington Study Group. Effect of deutetrabenazine on chorea among patients with Huntington disease. JAMA 2016, 316, 40. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, F.B.; Wild, E.J. Huntington’s Disease Clinical Trials Corner: April 2020. J. Huntingt. Dis. 2020, 9, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Andreassen, O.A.; Dedeoglu, A.; Ferrante, R.J.; Jenkins, B.G.; Ferrante, K.L.; Thomas, M.; Friedlich, A.; Browne, S.E.; Schilling, G.; Borchelt, D.R.; et al. Creatine increases survival and delays motor symptoms in a transgenic animal model of Huntington’s disease. Neurobiol. Dis. 2001, 8, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Hersch, S.M.; Gevorkian, S.; Marder, K.; Moskowitz, C.; Feigin, A.; Cox, M.; Como, P.; Zimmerman, C.; Lin, M.; Zhang, L.; et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2′dG. Neurology 2006, 66, 250–252. [Google Scholar] [CrossRef]
- Forbes, S.C.; Cordingley, D.M.; Cornish, S.M.; Gualano, B.; Roschel, H.; Ostojic, S.M.; Rawson, E.S.; Roy, B.D.; Prokopidis, K.; Giannos, P.; et al. Effects of creatine supplementation on brain function and health. Nutrients 2022, 14, 921. [Google Scholar] [CrossRef]
- Hersch, S.M.; Schifitto, G.; Oakes, D.; Bredlau, A.L.; Meyers, C.M.; Nahin, R.; Rosas, H.D. Huntington Study Group CREST-E Investigators and Coordinators The CREST-E study of creatine for Huntington disease: A randomized controlled trial. Neurology 2017, 89, 594–601. [Google Scholar] [CrossRef] [Green Version]
- McGarry, A.; McDermott, M.; Kieburtz, K.; de Blieck, E.A.; Beal, F.; Marder, K.; Ross, C.; Shoulson, I.; Gilbert, P.; Mallonee, W.M.; et al. A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology 2017, 88, 152–159. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.S.; Lonergan, P.E.; Boland, B.; Fogarty, M.P.; Brady, M.; Horrobin, D.F.; Campbell, V.A.; Lynch, M.A. Apoptotic changes in the aged brain are triggered by interleukin-1beta-induced activation of p38 and reversed by treatment with eicosapentaenoic acid. J. Biol. Chem. 2002, 277, 34239–34246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, B.K.; Leavitt, B.R.; Hayden, M.R.; Ross, C.A.; Rosenblatt, A.; Greenmayre, J.T.; Hersch, S.; Vaddadi, K.S.; Sword, A.; Horrobin, D.F.; et al. Ethyl-EPA in Huntington disease: A double-blind, randomized, placebo-controlled trial. Neurology 2005, 65, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study Group TREND-HD Investigators. Randomized controlled trial of ethyl-eicosapentaenoic acid in Huntington disease: The TREND-HD study. Arch. Neurol. 2008, 65, 1582–1589. [Google Scholar]
- Vázquez-Manrique, R.P.; Farina, F.; Cambon, K.; Dolores Sequedo, M.; Parker, A.J.; Millán, J.M.; Weiss, A.; Déglon, N.; Neri, C. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum. Mol. Genet. 2015, 25, 1043–1058. [Google Scholar] [CrossRef]
- Ma, T.C.; Buescher, J.L.; Oatis, B.; Funk, J.A.; Nash, A.J.; Carrier, R.L.; Hoyt, K.R. Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci. Lett. 2007, 411, 98–103. [Google Scholar] [CrossRef]
- Kim, A.; Lalonde, K.; Truesdell, A.; Gomes Welter, P.; Brocardo, P.S.; Rosenstock, T.R.; Gil-Mohapel, J. New avenues for the treatment of Huntington’s disease. Int. J. Mol. Sci. 2021, 22, 8363. [Google Scholar] [CrossRef]
- Adanyeguh, I.M.; Rinaldi, D.; Henry, P.G.; Caillet, S.; Valabregue, R.; Durr, A.; Mochel, F. Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 2015, 84, 490–495. [Google Scholar] [CrossRef]
- Duan, W.; Guo, Z.; Jiang, H.; Ladenheim, B.; Xu, X.; Cadet, J.L.; Mattson, M.P. Paroxetine Retards Disease Onset and Progression in Huntingtin Mutant Mice. Ann. Neurol. 2004, 55, 590–594. [Google Scholar] [CrossRef]
- Grote, H.E.; Bull, N.D.; Howard, M.L.; Van Dellen, A.; Blakemore, C.; Bartlett, P.F.; Hannan, A.J. Cognitive disorders and neurogenesis deficits in Huntington’s disease mice are rescued by fluoxetine. Eur. J. Neurosci. 2005, 22, 2081–2088. [Google Scholar] [CrossRef]
- Peng, Q.; Masuda, N.; Jiang, M.; Li, Q.; Zhao, M.; Ross, C.A.; Duan, W. The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/2 Huntington’s disease mouse model. Exp. Neurol. 2008, 210, 154–163. [Google Scholar] [CrossRef] [Green Version]
- Cong, W.N.; Chadwick, W.; Wang, R.; Daimon, C.M.; Cai, H.; Amma, J.; Wood, W.H.; Becker, K.G.; Martin, B.; Maudsley, S. Amitriptyline improves motor function via enhanced neurotrophin signaling and mitochondrial functions in the murine N171-82Q Huntington disease model. J. Biol. Chem. 2015, 290, 2728–2743. [Google Scholar] [CrossRef] [Green Version]
- Ellrichmann, G.; Blusch, A.; Fatoba, O.; Brunner, J.; Hayardeny, L.; Hayden, M.; Sehr, D.; Winklhofer, K.F.; Saft, C.; Gold, R. Laquinimod treatment in the R6/2 mouse model. Sci. Rep. 2017, 7, 4947. [Google Scholar] [CrossRef]
- Corey-Bloom, J.; Aikin, A.M.; Gutierrez, A.M.; Nadhem, J.S.; Howell, T.L.; Thomas, E.A. Beneficial effects of glatiramer acetate in Huntington’s disease mouse models: Evidence for BDNF-elevating and immunomodulatory mechanisms. Brain Res. 2017, 1673, 102–110. [Google Scholar] [CrossRef]
- Da Fonsêca, V.S.; da Silva Colla, A.R.; de Paula Nascimento-Castro, C.; Plácido, E.; Rosa, J.M.; Farina, M.; Gil-Mohapel, J.; Rodrigues, A.L.S.; Brocardo, P.S. Brain-derived neurotrophic factor prevents depressive-like behaviors in aarly-symptomatic YAC128 Huntington’s disease mice. Mol. Neurobiol. 2018, 55, 7201–7215. [Google Scholar] [CrossRef]
- Fatoba, O.; Kloster, E.; Reick, C.; Saft, C.; Gold, R.; Epplen, J.T.; Arning, L.; Ellrichmann, G. Activation of NPY-Y2 receptors ameliorates disease pathology in the R6/2 mouse and PC12 cell models of Huntington’s disease. Exp. Neurol. 2018, 302, 112–128. [Google Scholar] [CrossRef]
- Cabezas-Llobet, N.; Vidal-Sancho, L.; Masana, M.; Fournier, A.; Alberch, J.; Vaudry, D.; Xifró, X. Pituitary adenylate cyclase-activating polypeptide (PACAP) enhances hippocampal synaptic plasticity and improves memory performance in Huntington’s disease. Mol. Neurobiol. 2018, 55, 8263–8277. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kubilus, J.K.; Lee, J.; Ryu, H.; Beesen, A.; Zucker, B.; Smith, K.; Kowall, N.W.; Ratan, R.R.; Luthi-Carter, R.; et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 2003, 23, 9418–9427. [Google Scholar] [CrossRef]
- Oliveira, J.M.A.; Chen, S.; Almeida, S.; Riley, R.; Gonçalves, J.; Oliveira, C.R.; Hayden, M.R.; Nicholls, D.G.; Ellerby, L.M.; Rego, A.C. Mitochondrial-dependent Ca2+ handling in Huntington’s disease striatal cells: Effect of histone deacetylase inhibitors. J. Neurosci. 2006, 26, 11174–11186. [Google Scholar] [CrossRef] [Green Version]
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D.; et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Wang, Y.; Morris, C.D.; Jacques, V.; Gottesfeld, J.M.; Rusche, J.R.; Thomas, E.A. The effects of pharmacological inhibition of histone deacetylase 3 (HDAC3) in Huntington’s disease mice. PLoS ONE 2016, 11, e0152498. [Google Scholar] [CrossRef]
- Chopra, V.; Quinti, L.; Khanna, P.; Paganetti, P.; Kuhn, R.; Young, A.B.; Kazantsev, A.G.; Hersch, S. LBH589, A hydroxamic acid-derived HDAC inhibitor, is neuroprotective in mouse models of Huntington’s disease. J. Huntingt. Dis. 2016, 5, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Syed, A.; Lukacsovich, T.; Purcell, J.; Barbaro, B.A.; Worthge, S.A.; Wei, S.R.; Pollio, G.; Magnoni, L.; Scali, C.; et al. A potent and selective sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of huntington’s disease. Hum. Mol. Genet. 2014, 23, 2995–3007. [Google Scholar] [CrossRef] [Green Version]
- Hathorn, T.; Snyder-Keller, A.; Messer, A. Nicotinamide improves motor deficits and upregulates PGC-1α and BDNF gene expression in a mouse model of Huntington’s disease. Neurobiol. Dis. 2011, 41, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Naia, L.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira-Sousa, S.I.; Caldeira, G.L.; Carmo, C.; Laço, M.N.; Hayden, M.R.; Oliveira, C.R.; Rego, A.C. Comparative mitochondrial-based protective effects of resveratrol and nicotinamide in Huntington’s disease models. Mol. Neurobiol. 2017, 54, 5385–5399. [Google Scholar] [CrossRef]
- Claasen, D.O.; Carroll, B.; De Boer, L.M.; Wu, E.; Ayyagari, R.; Gandhi, S.; Stamler, D. Indirect tolerability comparison of deutetrabenazine and tetrabenazine for Huntington disease. J. Clin. Mov. Disord. 2017, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyant, K.J.; Ridder, A.J.; Dayalu, P. Huntington’s Disease—Update on Treatments. Curr. Neurol. Neurosci. Rep. 2017, 17, 33. [Google Scholar] [CrossRef]
- Uhlyar, S.; Rey, J.A. Valbenazine (Ingrezza): The first FDA-approved treatment for tardive dyskinesia. Pharm. Ther. 2018, 43, 328–331. [Google Scholar]
- Jurcau, A. The role of natural antioxidants in the prevention of dementia—Where do we stand and future perspectives. Nutrients 2021, 13, 282. [Google Scholar] [CrossRef] [PubMed]
- Khoury, N.; Koronowski, K.B.; Young, J.I.; Perez-Pinzon, M.A. The NAD+-dependent family of sirtuins in cerebral ischemia and preconditioning. Antioxid. Redox Signal. 2018, 28, 691–710. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Sandhir, R.; Mehrotra, A. Quercetin supplementation is effective in mitochondrial dysfunctions induced by 3-nitropropionic acid: Implications in Huntington’s disease. Biochim. Biophys. Acta 2013, 1832, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Padi, S.S.V.; Naidu, P.S.; Kumar, A. Possible neuroprotective mechanisms of curcumin in attenuating 3-nitropropionic acid-induced neurotoxicity. Methods Find. Exp. Clin. Pharmacol. 2007, 29, 19–25. [Google Scholar] [CrossRef]
- Sumathi, T.; Vedagiri, A.; Ramachandran, S.; Purushothaman, B. Quinolinic acid-induced Huntington disease-like symptoms mitigated by potent free radical scavenger edaravone—A pilot study on neurobehavioral, biochemical, and histological approach in male wistar rats. J. Mol. Neurosci. 2018, 66, 322–341. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Ferrante, R.J.; Dedeoglu, A.; Beal, M.F. Lipoic acid improves survival in transgenic mouse models of Huntington’s disease. Neuroreport 2001, 12, 3371–3373. [Google Scholar] [CrossRef]
- Elifani, F.; Amico, E.; Pepe, G.; Capocci, L.; Castaldo, S.; Rosa, P.; Montano, E.; Pollice, A.; Madonna, M.; Filosa, S.; et al. Curcumin dietary supplementation ameliorates disease phenotype in an animal model of Huntington’s disease. Hum. Mol. Genet. 2019, 28, 4012–4021. [Google Scholar] [CrossRef]
- Zajac, M.S.; Pang, T.Y.C.; Wong, N.; Weinrich, B.; Leang, L.S.K.; Craig, J.M.; Saffery, R.; Hannan, A.J. Wheel running and environmental enrichment differentially modify exon-specific BDNF expression in the hippocampus of wild-type and pre-motor symptomatic male and female Huntington’s disease mice. Hippocampus 2010, 20, 621–636. [Google Scholar] [CrossRef]
- Hockly, E.; Tse, J.; Barker, A.L.; Moolman, D.L.; Beunard, J.L.; Revington, A.P.; Holt, K.; Sunshine, S.; Moffitt, H.; Sathasivam, K.; et al. Evaluation of the benzothiazole aggregation inhibitors riluzole and PGL-135 as therapeutics for Huntington’s disease. Neurobiol. Dis. 2006, 21, 228–236. [Google Scholar] [CrossRef]
- Squitieri, F.; Orobello, S.; Cannella, M.; Martino, T.; Romanelli, P.; Giovacchini, G.; Frati, L.; Mansi, L.; Ciarmiello, A. Riluzole protects Huntington disease patients from brain glucose hypometabolism and grey matter volume loss and increases production of neurotrophins. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1113–1120. [Google Scholar] [CrossRef]
- Cho, S.R.; Benraiss, A.; Chmielnicki, E.; Samdani, A.; Economides, A.; Goldman, S.A. Induction of neostriatal neurogenesis slows disease progression in a transgenic murine model of Huntington disease. J. Clin. Investig. 2007, 117, 2889–2902. [Google Scholar] [CrossRef]
- Arregui, L.; Benítez, J.A.; Razgado, L.F.; Vergara, P.; Segovia, J. Adenoviral astrocyte-specific expression of BDNF in the striata of mice transgenic for Huntington’s disease delays the onset of the motor phenotype. Cell. Mol. Neurobiol. 2011, 31, 1229–1243. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Canals, J.M.; Pineda, J.R.; Torres-Peraza, J.F.; Bosch, M.; Martín-Ibañez, R.; Muñoz, M.T.; Mengod, G.; Ernfors, P.; Alberch, J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J. Neurosci. 2004, 24, 7727–7739. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.K.; Kim, J.; Cho, S.J.; Hatori, K.; Nagai, A.; Choi, H.B.; Lee, M.C.; McLarnon, J.G.; Kim, S.U. Proactive transplantation of human neural stem cells prevents degeneration of striatal neurons in a rat model of Huntington disease. Neurobiol. Dis. 2004, 16, 68–77. [Google Scholar] [CrossRef]
- Conti, L.; Cattaneo, E. Neural stem cell systems: Physiological players or in vitro entities? Nat. Rev. Neurosci. 2010, 11, 176–187. [Google Scholar] [CrossRef]
- Hoy, S.M. Nusinersen: First Global Approval. Drugs 2017, 77, 473–479. [Google Scholar] [CrossRef]
- Wild, E.J.; Tabrizi, S. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Dragatsis, I.; Dietrich, P.; Ren, H.; Deng, Y.P.; Del Mar, N.; Wang, H.B.; Johnson, I.M.; Jones, K.R.; Reiner, A. Effect of early embryonic deletion of huntingtin from pyramidal neurons on the development and long-term survival of neurons in cerebral cortex and striatum. Neurobiol. Dis. 2018, 111, 102–117. [Google Scholar] [CrossRef]
- Rossor, A.M.; Reilly, M.M.; Sleigh, J.N. Antisense oligonucleotides and other genetic therapies made simple. Pract. Neurol. 2018, 18, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Pulecio, J.; Verma, N.; Mejía-Ramírez, E.; Huangfu, D.; Raya, A. CRISPR/Cas9-based engineering of the epigenome. Cell Stem Cell 2017, 21, 431–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiFusco, D.; Dinallo, V.; Marafini, I.; Figliuzzi, M.M.; Romano, B.; Monteleone, G. Antisense oligonucleotide: Basic concepts and therapeutic application in inflammatory bowel disease. Front. Pharmacol. 2019, 10, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [Green Version]
- Schoch, K.M.; Miller, T.M. Antisense oligonucleotides: Translation from mouse models to human neurodegenerative diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.C.; Lobo, D.D.; Martins, I.M.; Lopes, S.M.; Henriques, C.; Duarte, S.P.; Dodart, J.C.; Nobre, R.J.; Pereira de Almeida, I. Antisense oligonucleotide therapeutics in neurodegenerative diseases: The case of polyglutamine disorders. Brain 2020, 143, 407–429. [Google Scholar] [CrossRef]
- Karaki, S.; Paris, C.; Rocchi, P. Antisense oligonucleotides, a novel developing targeting therapy. In Antisense Therapy; Sharad, S., Kapur, S., Eds.; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Vickers, T.A.; Liang, X.H. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef]
- Quemener, A.M.; Centomo, M.L.; Sax, S.L.; Panella, R. Small drugs, huge impact: The extraordinary impact of antisense oligonucleotides in research and drug development. Molecules 2022, 27, 536. [Google Scholar] [CrossRef]
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef]
- Zhan, X.; Deng, L.; Chen, G. Mechanisms and applications of peptide nucleic acids selectively binding to double-stranded RNA. Biopolymers 2022, 113, e23476. [Google Scholar] [CrossRef]
- Vasquez, G.; Freestone, G.C.; Wan, W.B.; Low, A.; De Hoyos, C.L.; Yu, J.; Prakash, T.P.; Ǿstergaard, M.E.; Liang, X.H.; Crooke, S.T.; et al. Site-specific incorporation of 5′-methyl DNA enhances the therapeutic profile of gapmer ASOs. Nucleic Acids Res. 2021, 49, 1828–1839. [Google Scholar] [CrossRef]
- Partridge, W.; Xia, S.; Kwoh, T.J.; Bhanot, S.; Geary, R.S.; Baker, B.F. Improvements in the tolerability profile of 2′-O-methoxyethyl chimeric antisense oligonucleotides in parallel with advances in design, screening, and other methods. Nucleic Acid Ther. 2021, 31, 417–426. [Google Scholar] [CrossRef]
- Kuespert, S.; Heydn, R.; Peters, S.; Wirkert, E.; Meyer, A.L.; Siebörger, M.; Johannesen, S.; Aigner, L.; Bogdahn, U.; Bruun, T.H. Antisense oligonucleotide in LNA-gapmer design targeting TGFBR2-a key single gene target for safe and effective inhibition of TGFβ signaling. Int. J. Mol. Sci. 2020, 21, 1952. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X.H. Antisense technology: An overview and prospectus. Nat. Rev. Drug Discov. 2021, 20, 427–453. [Google Scholar] [CrossRef]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Aartsma-Rus, A.; Goemans, N. A sequel to the Eteplirsen saga: Eteplirsen is approved in the United States but was not approved in Europe. Nucleic Acid Ther. 2019, 29, 13–15. [Google Scholar] [CrossRef]
- Blonda, A.; Lacosta, T.B.; Toumi, M.; Simoens, S. Assessing the value of Nusinersen for spinal muscular atrophy: A comparative analysis of reimbursement submission and appraisal in European countries. Front. Pharmacol. 2022, 12, 750742. [Google Scholar] [CrossRef]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef] [Green Version]
- Chi, X.; Gatti, P.; Papoian, T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef]
- Lu, X.-H.; Yang, X.W. “Huntingtin holiday”: Progress toward an antisense therapy for Huntington’s disease. Neuron 2012, 74, 964–966. [Google Scholar] [CrossRef] [Green Version]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Phase 1–2a IONIS-HTTRx Study Site Teams. Targeting huntingtin expression in patients with Huntington’s disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef]
- Ferguson, M.W.; Kennedy, C.J.; Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Current and possible future therapeutic options for Huntington’s disease. J. Cent. Nerv. Syst. Dis. 2022, 14, 11795735221092517. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Ferreira, J.J.; Wild, E.J. Huntington’s disease clinical trials corner: June 2019. J. Huntingt. Dis. 2019, 8, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Kennington, L.A.; Rosas, H.D.; Hersch, S.; Cha, J.-H.; Zamore, P.D.; Aronin, N. Linking SNPs to CAG repeat length in Huntington’s disease patients. Nat. Meth. 2008, 5, 951–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, M.M.; Pepers, B.A.; van Deutekorn, J.C.T.; Muldres, S.A.M.; den Dunnen, J.T.; Aartsma-Rus, A.; van Ommen, G.-J.B.; van Roon-Mom, W.M.C. Targeting several CAG expansion diseases by a single antisense oligonucleotide. PLoS ONE 2011, 6, e24308. [Google Scholar] [CrossRef] [PubMed]
- Kay, C.; Collins, J.A.; Skotte, N.H.; Southwell, A.L.; Warby, S.C.; Caron, N.S.; Doty, C.N.; Nguyen, B.; Griguoli, A.; Ross, C.J.; et al. Huntingtin haplotypes provide prioritized target panels for allele-specific silencing in Huntington’s disease patients of European ancestry. Mol. Ther. 2015, 23, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Datson, N.A.; González-Barriga, A.; Kourkouta, E.; Weij, R.; van de Giessen, J.; Mulders, S.; Kontkanen, O.; Heikkinen, T.; Lehtimäki, K.; van Deutekorn, J.C.T.; et al. The expanded CAG repeats in the huntingtin gene as a target for therapeutic RNA modulation throughout the HD mouse brain. PLoS ONE 2017, 12, e0171127. [Google Scholar] [CrossRef] [PubMed]
- Maxreiter, F.; Stemick, J.; Kohl, Z. Huntingtin lowering strategies. Int. J. Mol. Sci. 2020, 21, 2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wave Life Sciences Provides Update on Phase 1b/2a PRECISION-HD Trials. Available online: https://www.globenewswire.com/news-release/2021/03/29/2201081/0/en/Wave-Life-Sciences-Provides-Update-on-Phase-1b-2a-PRECISION-HD-Trials.html (accessed on 5 June 2022).
- Dale, E.; Frank-Kamenetsky, M.; Taborn, K.; Aklilu, K.; Liu, Y.; Berkovitch, S.; Brown, J.; Kandasamy, P.; Kumarasamy, J.; Mathieu, S.; et al. Stereopure oligonucleotides for the selective silencing of mutant Huntingtin (4703). Neurology 2020, 94, 4703. [Google Scholar]
- Skotte, N.H.; Southwell, A.L.; Østergaard, M.E.; Carroll, J.B.; Warby, S.C.; Doty, C.N.; Petoukhov, E.; Vaid, K.; Kordasiewicz, H.; Watt, A.T.; et al. Allele-specific suppression of mutant huntingtin using antisense oligonucleotides: Providing a therapeutic option for all Huntington disease patients. PLoS ONE 2014, 9, e107434. [Google Scholar] [CrossRef] [Green Version]
- Evers, M.M.; Tran, H.-D.; Zalachoras, I.; Meijer, O.C.; den Dunnen, J.T.; van Ommen, G.-J.B.; Aartsma-Rus, A.; van Roon-Mom, W.M.C. Preventing formation of toxic N-terminal Huntingtin fragments through antisense oligonucleotide-mediated protein modification. Nucleic Acid Therapeut. 2014, 24, 4–12. [Google Scholar] [CrossRef]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Wood, M.J.A. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef] [Green Version]
- Askeland, G.; Dosoudilova, Z.; Rodinova, M.; Klempir, J.; Liskova, I.; Kuśnierczyk, A.; Bjørås, M.; Nesse, G.; Klungland, A.; Hansikova, H.; et al. Increased nuclear DNA damage precedes mitochondrial dysfunction in peripheral blood mononuclear cells from Huntington’s disease patients. Sci. Rep. 2018, 8, 9817. [Google Scholar] [CrossRef] [Green Version]
- Enokido, Y.; Tamura, T.; Ito, H.; Arumughan, A.; Komuro, A.; Shiwaku, H.; Sone, M.; Foulle, R.; Sawada, H.; Ishiguro, H.; et al. Mutant Huntingtin impairs Ku70-mediated DNA repair. J. Cell Biol. 2010, 189, 425–443. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, S.; Gao, R.; Bush, K.; Zhang, N.; Wairkar, Y.P.; Sarkar, P.S. Polyglutamine expansion in huntingtin and mechanism of DNA damage repair defects in Huntington’s disease. Front. Cell. Neurosci. 2022, 16, 837576. [Google Scholar] [CrossRef]
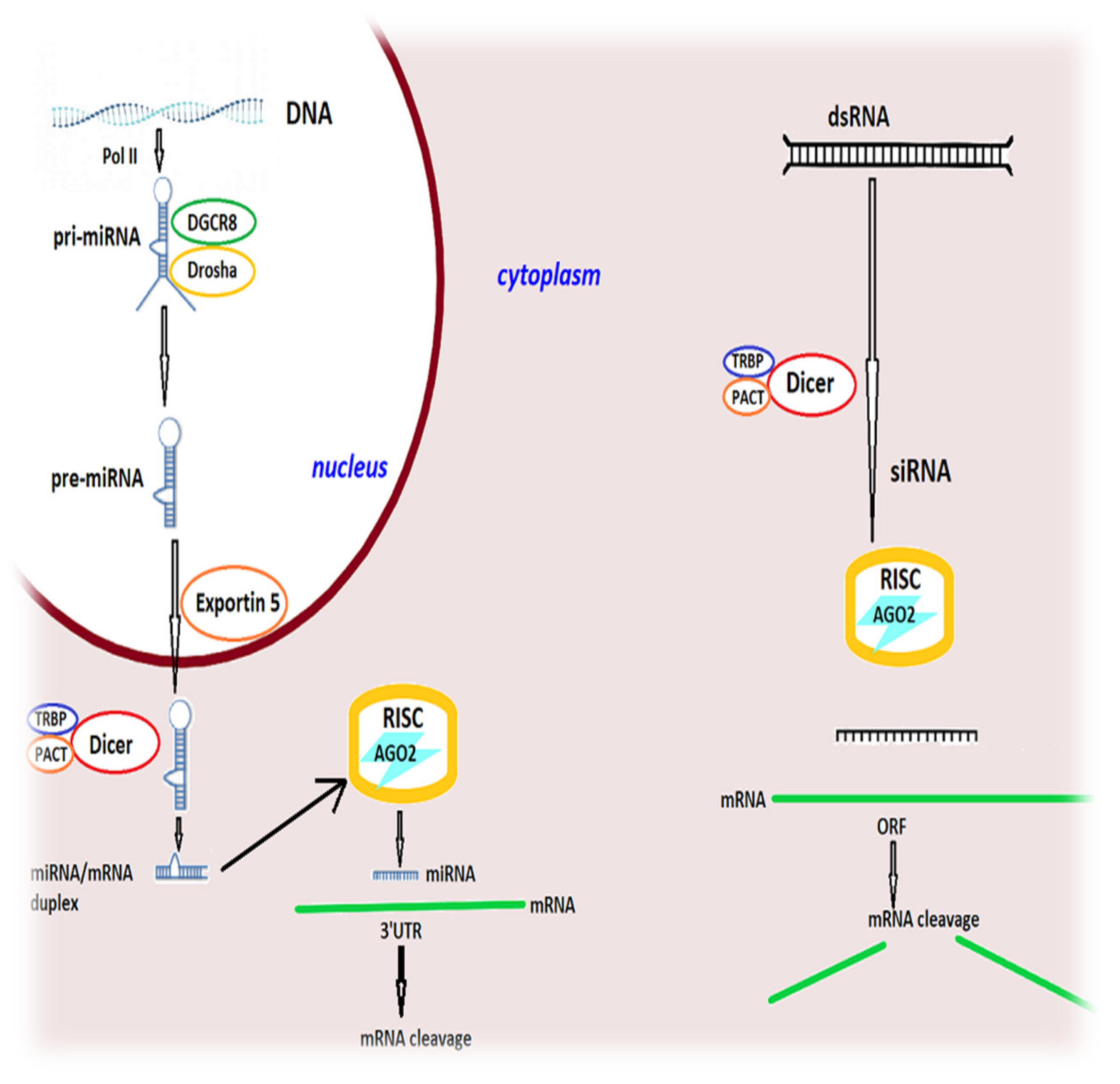
- Kim, D.H.; Rossi, J.J. Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 2007, 8, 173–184. [Google Scholar] [CrossRef]
- Orban, T.I.; Izaurralde, E. Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA 2005, 11, 459–469. [Google Scholar] [CrossRef] [Green Version]
- Preall, J.B.; Sontheimer, E.J. RNAi: RISC gets loaded. Cell 2005, 123, 543–545. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.; Fischer, N.; Grimm, D. Right on target: The next class of efficient, safe, and specific RNAi triggers. Mol. Ther. Nucleic Acids 2022, 28, 363–365. [Google Scholar] [CrossRef]
- Aguiar, S.; van der Gaag, B.; Bosco Cortese, F.A. RNAi mechanisms in Huntington’s disease therapy: siRNA versus shRNA. Transl. Neurodegener. 2017, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef]
- Herrera-Carillo, E.; Berkhout, B. Dicer-independent processing of small RNA duplexes: Mechanistic insights and applications. Nucleic Acids Res. 2017, 45, 10369–10379. [Google Scholar] [CrossRef] [Green Version]
- Askou, A.L.; Alsing, S.; Benckedorff, J.N.E.; Holmgaard, A.; Mikkelsen, J.G.; Aagaard, L.; Bek, T.; Corydon, T.J. Suppression of choroidal neovascularization by AAV-based dual-acting antiangiogenic gene therapy. Mol. Ther. Nucl. Acids 2019, 16, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Estevez-Fraga, C.; Rodrigues, F.B.; Tabrizi, S.J.; Wild, E.J. Huntington’s disease clinical trials corner: April 2022. J. Huntingt. Dis. 2022, 11, 105–118. [Google Scholar] [CrossRef]
- Evers, M.M.; Konstantinova, P. AAV5-miHTT gene therapy for Huntington disease: Lowering both huntingtins. Expert Opin. Biol. Ther. 2020, 20, 1121–1124. [Google Scholar] [CrossRef]
- Keskin, S.; Brouwers, C.C.; Sogorb-Gonzalez, M.; Martier, R.; Depla, J.A.; Vallès, A.; van Deventer, S.J.; Konstantinova, P.; Evers, M.M. AAV5-miHTT lowers Huntingtin mRNA and protein without off-target effects in patient-derived neuronal cultures and astrocytes. Mol. Ther. Methods Clin. Dev. 2019, 15, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Caron, N.S.; Southwell, A.L.; Brouwers, C.C.; Cengio, L.D.; Xie, Y.; Black, H.F.; Anderson, L.M.; Ko, S.; Zhu, X.; van Deventer, S.J.; et al. Potent and sustained huntingtin lowering via AAV5 encoding miRNA preserves striatal volume and cognitive function in a humanized mouse model of Huntington disease. Nucleic Acids Res. 2020, 48, 36–54. [Google Scholar] [CrossRef]
- Spronck, E.A.; Vallès, A.; Lampen, M.H.; Montenegro-Miranda, P.S.; Keskin, S.; Heijink, L.; Evers, M.M.; Petry, H.; van Deventer, S.J.; Konstantinova, P.; et al. Intrastriatal administration of AAV5-miHTT in non-human primates and rats Is well tolerated and results in miHTT transgene expression in key areas of Huntington disease pathology. Brain Sci. 2021, 11, 129. [Google Scholar] [CrossRef]
- Stanek, L.M.; Sardi, S.P.; Mastis, B.; Richards, A.R.; Treleaven, C.M.; Taksir, T.; Misra, K.; Cheng, S.H.; Shihabuddin, L.S. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum. Gene Ther. 2014, 25, 461–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabrizi, S.J.; Ghosh, R.; Leavitt, B.R. Huntingtin lowering strategies for disease modification in Huntington’s disease. Neuron 2019, 101, 801–819. [Google Scholar] [CrossRef] [Green Version]
- Vassena, R.; Heindrycks, B.; Peco, R.; Pennings, G.; Raya, A.; Sermon, K.; Veiga, A. Genome engineering through CRISPR/Cas9 technology in the human germline and pluripotent stem cells. Hum. Reprod. Update 2016, 22, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Klug, A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu. Rev. Biochem. 2010, 79, 213–231. [Google Scholar] [CrossRef] [Green Version]
- Garriga-Canut, M.; Agustin-Pavón, C.; Herrman, F.; Isalan, M. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc. Natl. Acad. Sci. USA 2012, 109, E3136–E3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butland, S.L.; Devon, R.S.; Huang, Y.; Mead, C.L.; Meynert, A.M.; Neal, S.J.; Lee, S.S.; Wilkinson, A.; Yang, G.S.; Yuen, M.M.S.; et al. CAG-encoded polyglutamine length polymorphism in the human genome. BMC Genom. 2007, 8, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agustín-Pavón, C.; Mielcarek, M.; Garriga-Canut, M.; Isalan, M. Deimmunization for gene therapy: Host matching of synthetic zinc finger constructs enables long-term mutant Huntingtin repression in mice. Mol. Neurodegener. 2016, 11, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitler, B.; Froelich, S.; Marlen, K.; Shivak, D.A.; Yu, Q.; Li, D.; Pearl, J.R.; Miller, J.C.; Zhang, L.; Paschon, D.E.; et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 2019, 25, 1131–1142. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Dabrowska, M.; Juzwa, W.; Krzyzosiak, W.J.; Olejniczak, M. Precise excision of the CAG tract from the Huntingtin gene by Cas9 nickases. Front. Neurosci. 2018, 12, 75. [Google Scholar] [CrossRef]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 editing of the mutant Huntingtin allele In vitro and in vivo. Mol. Ther. Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.W.; Kim, K.-H.; Chao, M.J.; Atwal, R.S.; Gillis, T.; MacDonald, M.E.; Gusella, J.F.; Lee, J.M. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016, 25, 4566–4576. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Tay, Y.; Sim, B.; Yoon, S.-I.; Huang, Y.; Ooi, J.; Utami, K.H.; Ziaei, A.; Ng, B.; Radulescu, C.; et al. Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in Huntington disease patient-derived induced pluripotent stem cells. Stem Cell Rep. 2017, 8, 619–633. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Quin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef] [Green Version]
- Ekman, F.; Ojala, D.; Adil, M.; Lopez, P.; Schaffer, D.; Gaj, T. CRISPR-Cas9-mediated genome editing increases lifespan and improves motor deficits in a Huntington’s disease mouse model. Mol. Ther. Nucleic Acids 2019, 17, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, K.A.; Wu, W.-H.; Colgan, D.F.; Tsang, S.H.; Bassuk, A.G.; Mahajan, V.B. Unexpected mutations after CRISPR-Cas9 editing in vivo. Nat. Methods 2017, 127, 547–548. [Google Scholar] [CrossRef]
- Fink, K.D.; Deng, P.; Gutierrez, J.; Anderson, J.S.; Torrest, A.; Komarla, A.; Kalomoiris, S.; Cary, W.; Anderson, J.D.; Gruenloh, W.; et al. Allele-specific reduction of the mutant huntingtin allele using transcription activator-like effectors in human Huntington’s disease fibroblasts. Cell Transplant. 2016, 25, 677–686. [Google Scholar] [CrossRef] [Green Version]
- Salado-Manzano, C.; Perpiña, U.; Straccia, M.; Molina-Ruiz, F.J.; Cozzi, E.; Rosser, A.E.; Canals, J.M. Is the immunological response a bottleneck for cell therapy in neurodegenerative diseases? Front. Cell Neurosci. 2020, 14, 250. [Google Scholar] [CrossRef]
- Macedo, J.; Pagani, E.; Wenceslau, C.V.; Ferrara, L.; Kerkis, I. A phase I clinical trial on intravenous administration of immature human dental pulp stem cells (Nestacell-HDTM) to Huntington’s disease patients. Cytotherapy 2021, 23, 1. [Google Scholar] [CrossRef]
- Fink, K.D.; Deng, P.; Torrest, A.; Stewart, H.; Pollock, K.; Gruenloh, W.; Annett, G.; Tempkin, T.; Wheelock, V.; Nolta, J.A. Developing stem cell therapies for juvenile and adult-onset Huntington’s disease. Regen. Med. 2015, 10, 623–646. [Google Scholar] [CrossRef]
- Fink, K.D.; Rossignol, J.; Crane, A.T.; Davis, K.K.; Bombard, M.C.; Bavar, A.M.; Clerc, S.; Lowrance, S.A.; Song, C.; Lescaudron, L.; et al. Transplantation of umbilical cord-derived mesenchymal stem cells into the striata of R6/2 mice: Behavioral and neuropathological analysis. Stem Cell Res. Ther. 2013, 4, 130. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, M.J.; Aliaghaei, A.; Boroujeni, M.E.; Khodagholi, F.; Meftahi, G.; Abdollahifar, M.A.; Ahmadi, H.; Danyali, S.; Daftari, M.; Sadeghi, Y. Human umbilical cord matrix stem cells reverse oxidative stress-induced cell death and ameliorate motor function and striatal atrophy in rat model of Huntington Disease. Neurotox. Res. 2018, 34, 273–284. [Google Scholar] [CrossRef]
- Liu, J. Induced pluripotent stem cell-derived neural stem cells: New hope for stroke? Stem Cell Res. Ther. 2013, 4, 115. [Google Scholar] [CrossRef] [Green Version]
- Mu, S.; Wang, J.; Zhou, G.; Peng, W.; He, Z.; Zhao, Z.; Mo, C.P.; Qu, J.; Zhang, J. Transplantation of induced pluripotent stem cells improves functional recovery in Huntington’s disease rat model. PLoS ONE 2014, 9, e101185. [Google Scholar] [CrossRef]
- Yoon, Y.; Kim, H.S.; Hong, C.P.; Li, E.; Jeon, I.; Park, H.J.; Lee, N.; Pei, Z.; Song, J. Neural transplants from human induced pluripotent stem cells rescue the pathology and behavioral defects in a rodent model of Huntington’s disease. Front. Neurosci. 2020, 14, 558204. [Google Scholar] [CrossRef]
- Al-Gharaibeh, A.; Culver, R.; Stewart, A.N.; Srinageshwar, B.; Spelde, K.; Frollo, L.; Kolli, N.; Story, D.; Paladugu, L.; Anwar, S.; et al. Induced pluripotent stem cell-derived neural stem cell transplantations reduced behavioral deficits and ameliorated neuropathological changes in YAC128 mouse model of Huntington’s disease. Front. Neurosci. 2017, 11, 628. [Google Scholar] [CrossRef]
- Barker, R.A.; Mason, S.L.; Harrower, T.P.; Swain, R.A.; Ho, A.K.; Sahakian, B.J.; Mathur, R.; Elneil, S.; Thornton, S.; Hurrelbrink, C.; et al. The long-term safety and efficacy of bilateral transplantation of human fetal striatal tissue in patients with mild to moderate Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2013, 84, 657–665. [Google Scholar] [CrossRef]
- Bachoud-Lévi, A.C.; Bourdet, C.; Brugières, P.; Nguyen, J.P.; Grandmougin, T.; Haddad, B.; Jény, R.; Bartolomeo, P.; Boissé, M.F.; Dalla Barba, G.; et al. Safety and tolerability assessment of intrastriatal neural allografts in five patients with Huntington’s disease. Exp. Neurol. 2000, 161, 194–202. [Google Scholar] [CrossRef]
- Krebs, S.S.; Trippel, M.; Prokop, T.; Omer, T.N.; Landwehrmeyer, B.; Weber, W.A.; Nikkhah, G. Immune response after striatal engraftment of fetal neuronal cells in patients with Huntington’s disease: Consequences for cerebral transplantation programs. Clinic. Exp. Neuroimmunol. 2011, 2, 25–32. [Google Scholar] [CrossRef]
- Drew, C.; Sharouf, F.; Randell, E.; Brookes-Howell, L.; Smallman, K.; Sewell, B.; Burrell, A.; Kirby, N.; Mills, L.; Precious, S.; et al. Protocol for an open label: Phase I trial within a cohort of foetal cell transplants in people with Huntington’s disease. Brain Commun. 2021, 3, fcaa230. [Google Scholar] [CrossRef]
- Bachoud-Lévi, A.C.; Gaura, V.; Brugières, P.; Lefaucheur, J.P.; Boissé, M.F.; Maison, P.; Baudic, S.; Ribeiro, M.J.; Bourdet, C.; Remy, P.; et al. Effect of fetal neural transplants in patients with Huntington’s disease 6 years after surgery: A long-term follow-up study. Lancet Neurol. 2006, 5, 303–309. [Google Scholar] [CrossRef]
- Dietrich, P.; Johnson, I.M.; Alli, S.; Dragatsis, I. Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. PLoS Genet. 2017, 13, e1006846. [Google Scholar] [CrossRef] [Green Version]
- Cho, I.K.; Hunter, C.E.; Ye, S.; Pongos, A.L.; Chan, A.W.S. Combination of stem cell and gene therapy ameliorates symptoms in Huntington’s disease mice. NPJ Regen. Med. 2019, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Onyango, I.G.; Jauregui, G.V.; Čarná, M.; Bennett, J.P., Jr.; Stokin, G.B. Neuroinflammation in Alzheimer’s disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, S.; Chang, S.C.; Lee, J. Significant roles of neuroinflammation in Parkinson’s disease: Therapeutic targets for PD prevention. Arch. Pharm. Res. 2019, 42, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Saba, J.; Couselo, F.L.; Bruno, J.; Carniglia, L.; Durand, D.; Lasaga, M.; Caruso, C. Neuroinflammation in Huntington’s disease: A starring role for astrocyte and microglia. Curr. Neuropharmacol. 2022, 20, 1116–1143. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-M.; Yang, S.; Huang, S.-S.; Tang, B.-S.; Guo, J.-F. Microglial activation in the pathogenesis of Huntington’s disease. Front. Aging Neurosci. 2017, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Brück, W.; Wegner, C. Insight into the mechanism of laquinimod action. J. Neurol. Sci. 2011, 306, 173–179. [Google Scholar] [CrossRef]
- Gurevich, M.; Gritzman, T.; Orbach, R.; Tuller, T.; Feldman, A.; Achiron, A. Laquinimod suppress antigen presentation in relapsing-remitting multiple sclerosis: In-vitro high-throughput gene expression study. J. Neuroimmunol. 2010, 221, 87–94. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Caron, N.S.; Deng, Y.; Qiu, X.; Tsang, M.; Hayden, M.R. Laquinimod decreases Bax expression and reduces caspase-6 activation in neurons. Exp. Neurol. 2016, 283, 121–128. [Google Scholar] [CrossRef]
- Fatoba, O.; Ohtake, Y.; Itokazu, T.; Yamashita, T. Immunotherapies in Huntington’s disease and α-synucleinopathies. Front. Immunol. 2020, 11, 337. [Google Scholar] [CrossRef]
- Politis, M.; Lahiri, N.; Niccolini, F.; Su, P.; Wu, K.; Giannetti, P.; Scahill, R.I.; Turkheimer, F.E.; Tabrizi, S.J.; Piccini, P. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol. Dis. 2015, 83, 115–121. [Google Scholar] [CrossRef]
- Hsiao, H.Y.; Chiu, F.L.; Chen, C.M.; Wu, Y.R.; Chen, H.M.; Chen, Y.C.; Kuo, H.C.; Chern, Y. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 2014, 23, 4328–4344. [Google Scholar] [CrossRef] [Green Version]
- Pido-Lopez, J.; Tanudjojo, B.; Farag, S.; Bondulich, M.K.; Andre, R.; Tabrizi, S.J.; Bates, G.P. Inhibition of tumour necrosis factor alpha in the R6/2 mouse model of Huntington’s disease by etanercept treatment. Sci. Rep. 2019, 9, 7202. [Google Scholar] [CrossRef]
- Lansita, J.A.; Mease, K.M.; Qiu, H.; Yednock, T.; Sankaranarayanan, S.; Kramer, S. Nonclinical development of ANX005: A humanized anti-C1q antibody for treatment of autoimmune and neurodegenerative diseases. Int. J. Toxicol. 2017, 36, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Leonard, J.E.; Fisher, T.L.; Winter, L.A.; Cornelius, C.A.; Reilly, C.; Smith, E.S.; Zauderer, M. Nonclinical safety evaluation of VX15/2503, a humanized IgG4 Anti-SEMA4D antibody. Mol. Cancer Therapeut. 2015, 14, 964. [Google Scholar] [CrossRef] [Green Version]
- Southwell, A.L.; Franciosi, S.; Villanueva, E.B.; Xie, Y.; Winter, L.A.; Veeraraghavan, J.; Jonason, A.; Felczak, B.; Zhang, W.; Kovalik, V.; et al. Anti-semaphorin 4D immunotherapy ameliorates neuropathology and some cognitive impairment in the YAC128 mouse model of Huntington disease. Neurobiol. Dis. 2015, 76, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Denis, H.L.; David, L.S.; Cicchetti, F. Antibody-based therapies for Huntington’s disease: Current status and future directions. Neurobiol. Dis. 2019, 132, 104569. [Google Scholar] [CrossRef]
- Amaro, I.A.; Henderson, L.A. An intrabody drug (rAAV6-INT41) reduces the binding of N-terminal Huntingtin fragment(s) to DNA to basal levels in PC12 cells and delays cognitive loss in the R6/2 animal model. J. Neurodegen. Dis. 2016, 2016, 7120753. [Google Scholar] [CrossRef] [Green Version]
- Denis, H.L.; Lauruol, F.; Cicchetti, F. Are immunotherapies for Huntington’s disease a realistic option? Mol. Psychiatr. 2019, 24, 364–377. [Google Scholar] [CrossRef]
- Bartl, S.; Oueslati, A.; Southwell, A.L.; Siddu, A.; Parth, M.; David, L.S.; Maxan, A.; Salhat, N.; Burkert, M.; Mairhofer, A.; et al. Inhibiting cellular uptake of mutant huntingtin using a monoclonal antibody: Implications for the treatment of Huntington’s disease. Neurobiol. Dis. 2020, 141, 104943. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Mestre, T.A.; Sampaio, C. Huntington disease: Linking pathogenesis to the development of experimental therapeutics. Curr. Neurol. Neurosci. Rep. 2017, 17, 18. [Google Scholar] [CrossRef]
- Du, G.; Dong, W.; Yang, Q.; Yu, X.; Ma, J.; Gu, W.; Huang, Y. Altered gut microbiota related to inflammatory responses in patients with Huntington’s disease. Front. Immunol. 2020, 11, 603594. [Google Scholar] [CrossRef]
- Kong, G.; Ellul, S.; Narayana, V.K.; Kanojia, K.; Ha, H.T.T.; Li, S.; Renoir, T.; Cao, K.A.L.; Hannan, A.J. An integrated metagenomics and metabolomics approach implicates the microbiota-gut-brain axis in the pathogenesis of Huntington’s disease. Neurobiol. Dis. 2021, 148, 105199. [Google Scholar] [CrossRef]
- Kaemmerer, W.F.; Grondin, R.C. The effects of huntingtin-lowering: What do we know so far? Degener. Neurol. Neuromuscul. Dis. 2019, 9, 3–17. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathogenic Mechanism | Action of mHtt | Resulting Abnormalities | References |
|---|---|---|---|
| Excitotoxicity |
|
| [10,11] [15,16] |
| Impaired proteostasis |
|
| [17,18] [24] |
| Mitochondrial dysfunction |
|
| [28] [28] [29] [12] [5] [32] |
| Oxidative stress |
|
| [35] [37] |
| Transcriptional dysregulation |
|
| [36] [44,45] [46] |
| Reduced BDNF |
|
| [44,45,48] [49] |
| Dysfunction of glial cells |
|
| [52] [54] |
| Neuroinflammation |
|
| [41] [57] |
| Targeted Mechanism | Molecule | Effect | Trials | Results | References |
|---|---|---|---|---|---|
| mHtt aggregation | Epigallocatechin 3 gallate | Suppresses Htt aggregation | Clinical trial (NCT01357681, ETON trial) | Not published | [59] |
| Resveratrol | Activates AMPK, SIRT1, increasing PGC-1α and Nrf2 mRNA expression | Mouse models Clinical trial (NCT02336633, REVHD) | Not posted | [60,61] [62] | |
| Rapamycin | Inhibits mTOR and activates autophagy, promoting mHtt aggregate clearance | Drosophila and mouse HD models, toxicity precludes human use | Decreased accumulation of mHtt aggregates, improved motor performance | [63] | |
| mHtt cleavage | Minocycline | Inhibits caspase 1 and caspase 3 | R6/2 mice Clinical trial (NCT00029874) | Improved phenotype Negative results | [64] [65] |
| Nilotinib | Inhibits a tyrosine kinase involved in apoptosis, autophagy modulator | Clinical trial (NCT03764215, Tasigna-HD) | No published results | [62,66] | |
| Excitotoxicity | Riluzole | Inhibits glutamate neurotransmission, enhances EAAT2 activity | Transgenic mice, primate HD model HD patients (NCT00277602) | Improved abnormal movements, reduced striatal atrophy, increasing survival Transient improvement of chorea | [67,68] [69,70] |
| Memantine | Antagonizes extrasynaptic NMDARs | Chemical animal HD models Transgenic mouse models Memantine + risperidone in single HD patient | Reduced striatal neuronal loss Worsened outcome Slowed down progression of motor symptoms and cognitive decline | [71] [72,73] | |
| Lamotrigine | Glutamate antagonist | Chemical mouse models Clinical trial | Restored antioxidative defense mechanisms, improved behavior Null results | [1] [74] | |
| Tetrabenazine, Deutetrabenazine, Valbenazine | Inhibits vesicular monoamine transporter type 2 (VMAT2), decreases dopamine in the striatum | HD patients (NCT01451463; NCT00219804) (NCT01795859) HD patients (NCT04102579, KINECT-HD), NCT04400331 | Diminished chorea No published results | [75,76] [62,77,78] | |
| Dextromethorphan | NMDAR antagonist | Clinical trial (NCT03854019), in combination with quinidine | No results released | [62] | |
| Mitochondrial dysfunction and oxidative stress | Creatine | Stimulates mitochondrial respiration | R6/2 mouse models HD patients (NCT00026988) NCT01412151, CREST-X NCT00712426, CREST-E | Neuroprotective, slowed down the development of neuropathology Reduced markers of oxidative DNA damage No clinical benefit No clinical benefit | [79] [80] [62,68] [81,82] |
| Coenzyme Q10 | Interacts with ROS, improves ATP production | HD patients, NCT00608881, 2CARE | No clinical benefit | [83] | |
| Eicosapentaenoic acid (EPA) | Binds to mitochondrial PPAR, inhibits caspases, downregulates the JNK pathway | HD patients, NCT00146211, TREND-HD | No clinical benefit | [84,85,86] | |
| Metformin | Activates AMPK | In vitro and fly models R6/2 mice HD patients (NCT048266920, Test-ing METformin) | Neuroprotective effect Increased lifespan Ongoing, assessing its effect on cognitive decline | [87] [88] [62,89] | |
| Fenofibrate | Activates PGC-1α, promotes mitochondrial biogenesis | HD patients (NCT03515213) | ongoing | [62] | |
| Triheptanoin | Increases acetyl-CoA, a substrate of the Krebs cycle | HD patients, NCT02453061, TRIHEP3 | No results released | [62,90] | |
| Increase in BDNF | Selective serotonin reuptake inhibition (paroxetine, fluoxetine, sertraline, amitriptyline) | Activates the MAPK/ERK signaling pathway, BDNF/tyrosine kinase B pathway | Mouse models | Improved HD symptoms | [91,92,93,94] |
| Immunomodulators: laquinimod, glatiramer acetate | Reduces NF-κB activation, upregulates BDNF | Mouse models | Improved mitochondrial function, reduced pro-inflammatory cytokines, increased BDNF levels | [95,96] | |
| Intranasal delivery of BDNF, PACAP38 | Enhances synaptic plasticity | mouse models | Improved behavior, attenuated memory deficits, reduced HD neuropathology | [97,98,99] | |
| Transcriptional dysregulation | Sodium butyrate | Modulates HDACs | R6/2 mice | Improved motor performance, extended survival, reduced HD pathology | [100,101] |
| Synthetic molecules, such as 4b, RGFP966 | Promotes pyruvate dehydrogenase activity, improves mitochondrial dysfunction | Transgenic mice | Improved motor performance, reduced striatal atrophy | [102,103,104] | |
| Selistat, nicotinamide | HDAC inhibition, SIRT1 inhibitor | Transgenic mice | Improved motor performances, increased BDNF levels | [105,106,107] |
| Modification | Resulted ASO | Main Properties | Mechanism of Action | References |
|---|---|---|---|---|
| Backbone modifications | Phosphorothioate (PS) |
| Degradation of mRNA by RNase H | [138] |
| Phosphoroamidate (PA) |
| Non-degrading RNA mechanisms | [139] | |
| Phosphorodiamidate morpholino oligomers (PMO) |
| Non-degrading RNA mechanisms | [140] | |
| Peptide nucleic acids (PNA) |
| Non-degrading RNA mechanisms | [141] | |
| Sugar modifications | 2′-O-methyl (2′-OMe) |
| Non-degrading RNA mechanisms and RNase activity with gapmer design | [142] |
| 2′-O-methoxyl-ethyl (2′-MOE) |
| Non-degrading RNA mechanisms and RNase activity with gapmer design | [143] | |
| Locked nucleic acids (LNA) |
| Non-degrading RNA mechanisms and RNase activity with gapmer design | [144] | |
| S-constrained-ethyl (cEt) |
| Non-degrading RNA mechanisms and RNase activity with gapmer design | [145] |
| Small Interfering RNA (siRNA) | Short Hairpin RNA (shRNA) | |
|---|---|---|
| Source | Exogenous source | Nuclear expression |
| Delivery methods | Via synthetic or natural polymers or lipids to the cytoplasm | Via viral or other vectors to the nucleus |
| Persistence | Short-lasted (rapid degradation) | Expressed for months to years |
| Required dosage | High | Low |
| Incidence of “off-target” effects | Higher than for shRNA approaches, higher immune activation, toxicity | Lower than for siRNA therapies, low toxicity and immune activation |
| Therapeutic applications | In acute diseases, where high doses can be tolerated without significant toxicity and which do not require lifelong frequent administrations | In chronic disorders, where low doses acting for a longer time are desirable |
| Target | Category | Molecule | Development | Strengths | Limitations |
|---|---|---|---|---|---|
| mHtt RNA | ASO | IONIS-HTTRx | Phase 3 clinical trial | Dose-dependent reduction of mHtt RNA and protein | Intrathecal delivery, requires multiple doses |
| WVE-120102/120101 | Phase 1b/2a trial | Allele-specific (rs362331/ 362307, do not lower wild-type Htt | Applicable only to the targeted SNP carrier, require multiple doses | ||
| (CUG)7 | Pre-clinical | Allele-specific, less reduction of wild-type Htt | Multiple dosing | ||
| RNAi | AMT-130 | Phase 1b/2a | Single dose | Allele-non-specific, requires intrastriatal viral vector-mediated delivery | |
| VY-HTT01 | Pre-clinical | Single dose | Viral vector-mediated intracranial injection | ||
| Small molecules | Branaplam | Pre-clinical | Oral administration | Multiple doses, lack of specificity, risk of off-target effects | |
| PTC518 | Phase 1 | Oral administration | Multiple doses, lack of specificity, risk of off-target effects | ||
| DNA | ZFP | TAK-686 ZF-KOX1 | Pre-clinical | Prevents mHtt transcription without altering the gene itself, single dose | Viral vector, intrastriatal injection; can trigger immune reactions |
| CRISPR/Cas9 | Unnamed CRISPR/Cas9 molecules | Pre-clinical | Single dose; corrects genetic defect | Viral vector-mediated intracranial delivery; risk of off-target mutations | |
| TALEN | Still in development | Pre-clinical | Not known | Not known | |
| Cell loss | Stem cells | Cellavita | Phase 2/3 | Intravenous delivery | Inconsistent effects |
| Fetal stem cell transplant | Phase 1 | Single dose | Intrastriatal injection, risk of graft rejection | ||
| Autologous stem cells | Clinical trial | Intravenous delivery | Uncertain cerebral penetration | ||
| Neuroinflammation | Monoclonal antibodies | ANX005 | Phase 2 | Intravenous delivery | Multiple doses required |
| VX15/2503 | Phase 2 | Intravenous delivery | Multiple doses required | ||
| Intrabodies | rAAV6-INT41 | Pre-clinical | Prevents protein misfolding, promotes aggregate clearance | Intrastriatal injection, viral vector-mediated delivery |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurcau, A.; Jurcau, M.C. Therapeutic Strategies in Huntington’s Disease: From Genetic Defect to Gene Therapy. Biomedicines 2022, 10, 1895. https://doi.org/10.3390/biomedicines10081895
Jurcau A, Jurcau MC. Therapeutic Strategies in Huntington’s Disease: From Genetic Defect to Gene Therapy. Biomedicines. 2022; 10(8):1895. https://doi.org/10.3390/biomedicines10081895
Chicago/Turabian StyleJurcau, Anamaria, and Maria Carolina Jurcau. 2022. "Therapeutic Strategies in Huntington’s Disease: From Genetic Defect to Gene Therapy" Biomedicines 10, no. 8: 1895. https://doi.org/10.3390/biomedicines10081895