
Brain Cancer Chemotherapy through a Delivery System across the Blood-Brain Barrier into the Brain Based on Receptor-Mediated Transcytosis Using Monoclonal Antibody Conjugates

Abstract
:1. Introduction
2. Discussion
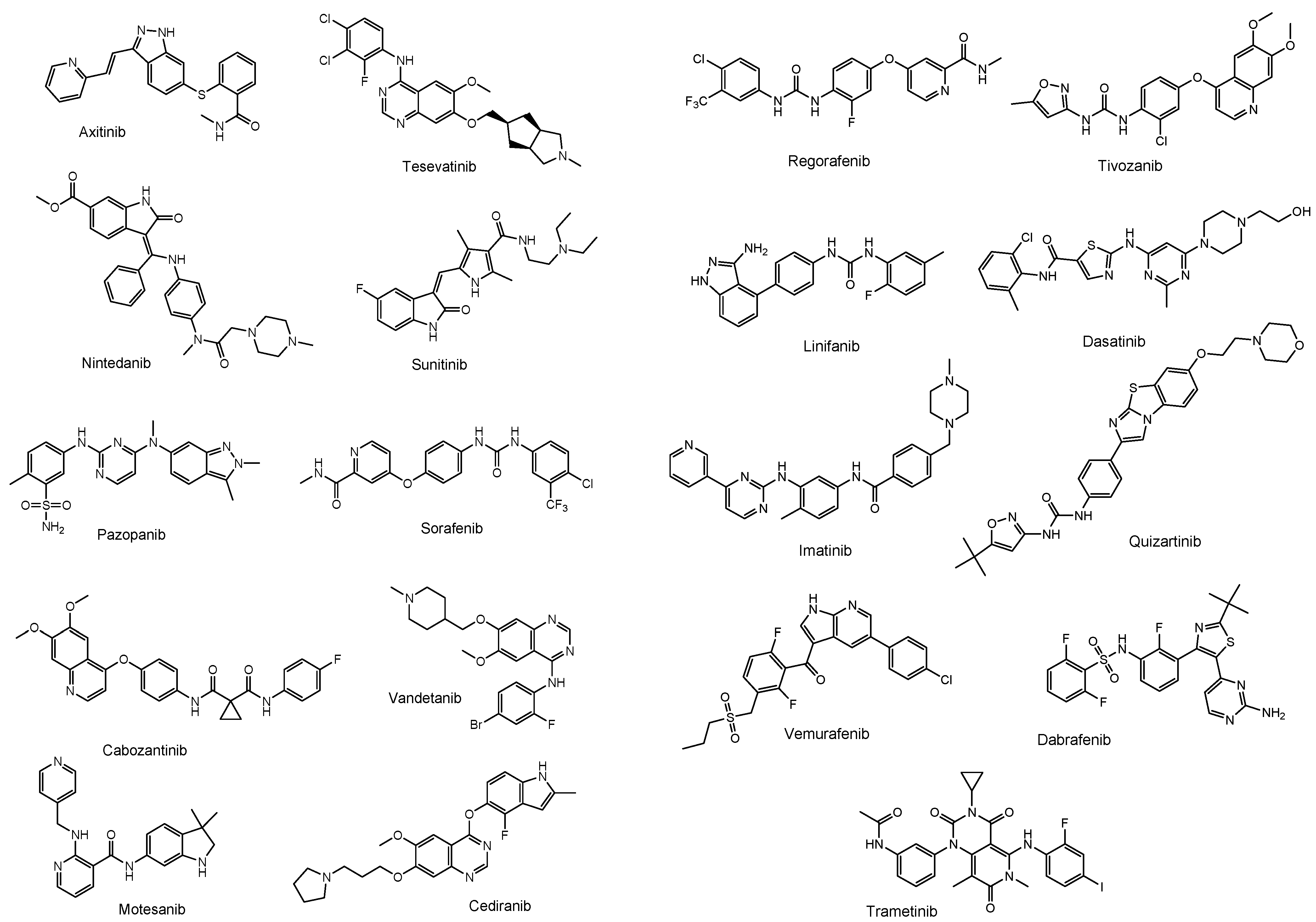
2.1. Brain Cancers and Their Chemotherapy
2.2. Possibility and Implement of ADCs
2.2.1. mAb Drugs
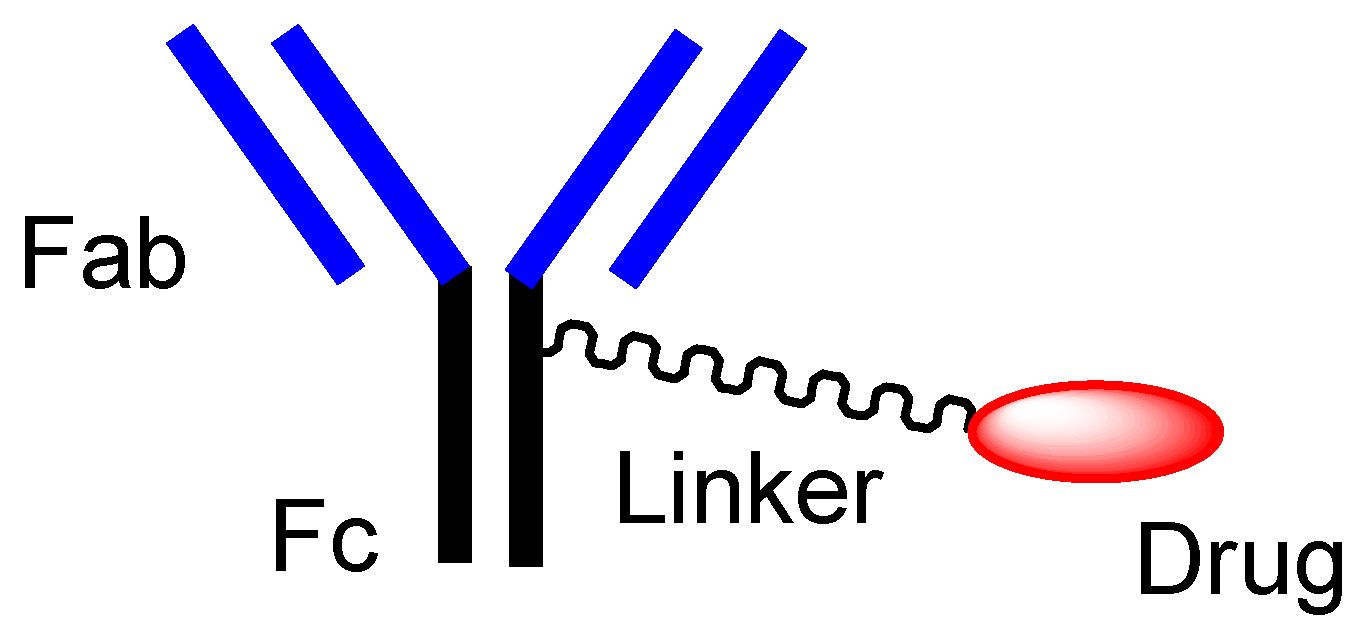
2.2.2. Orthodox ADCs
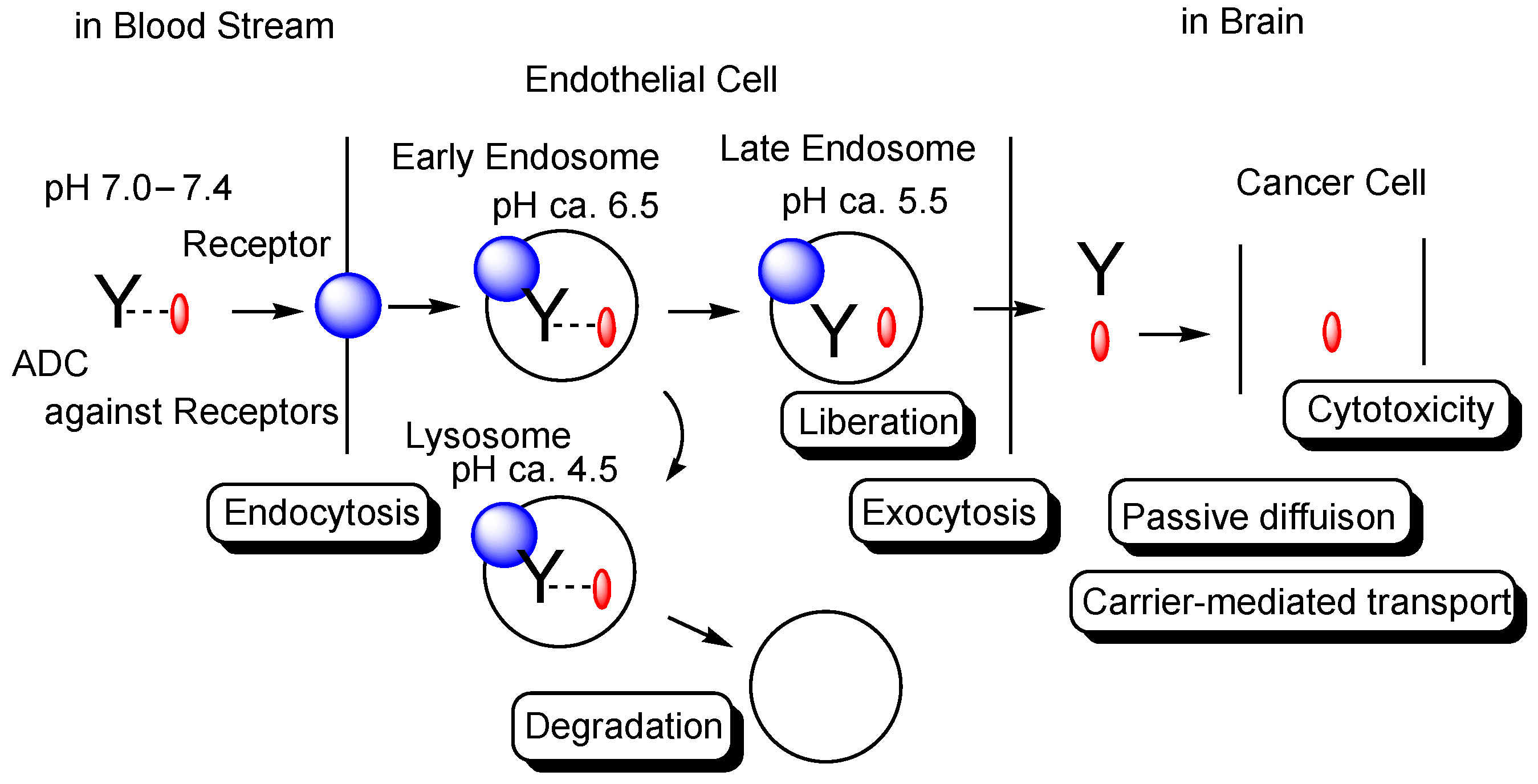
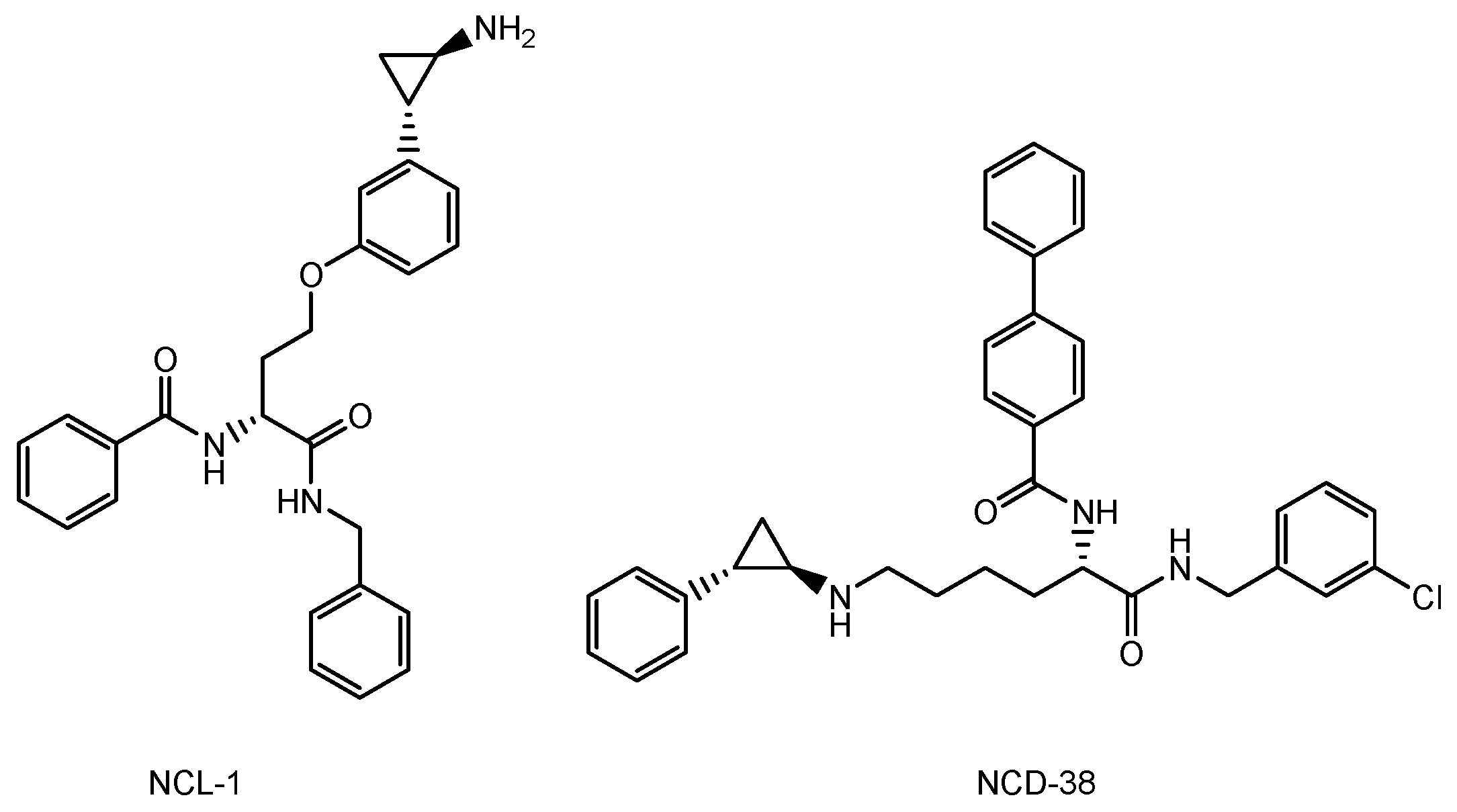
2.2.3. Anti-Receptor ADCs That Cross the Endothelium via RMT
2.2.4. Anti-Receptor ADCs including Bispecific ADCs
2.3. Activity Expression of ADCs
2.4. Promising Implement of ADCs for the Treatment of Brain Cancers
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stimulus package. Nat. Med. 2018, 24, 247. [CrossRef] [Green Version]
- Tashima, T. Smart Strategies for Therapeutic Agent Delivery into Brain across the Blood-Brain Barrier Using Receptor-Mediated Transcytosis. Chem. Pharm. Bull. 2020, 68, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The Blood-Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Tashima, T. Intriguing possibilities and beneficial aspects of transporter-conscious drug design. Bioorg. Med. Chem. 2015, 23, 4119–4131. [Google Scholar] [CrossRef]
- Tashima, T. Intelligent substance delivery into cells using cell-penetrating peptides. Bioorg. Med. Chem. Lett. 2017, 27, 121–130. [Google Scholar] [CrossRef]
- Tashima, T. Effective cancer therapy based on selective drug delivery into cells across their membrane using receptor-mediated endocytosis. Bioorg. Med. Chem. Lett. 2018, 28, 3015–3024. [Google Scholar] [CrossRef]
- Tashima, T. Shortcut Approaches to Substance Delivery into the Brain Based on Intranasal Administration Using Nanodelivery Strategies for Insulin. Molecules 2020, 25, 5188. [Google Scholar] [CrossRef]
- Tashima, T. Delivery of Orally Administered Digestible Antibodies Using Nanoparticles. Int. J. Mol. Sci. 2021, 22, 3349. [Google Scholar] [CrossRef]
- Tashima, T. Delivery of Intravenously Administered Antibodies Targeting Alzheimer’s Disease-Relevant Tau Species into the Brain Based on Receptor-Mediated Transcytosis. Pharmaceutics 2022, 14, 411. [Google Scholar] [CrossRef]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosentha, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Prim. 2015, 1, 15017. [Google Scholar] [CrossRef]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar]
- Norden, A.D.; Young, G.S.; Setayesh, K.; Muzikansky, A.; Klufas, R.; Ross, G.L.; Ciampa, A.S.; Ebbeling, L.G.; Levy, B.; Drappatz, J.; et al. Bevacizumab for recurrent malignant gliomas: Efficacy, toxicity, and patterns of recurrence. Neurology 2008, 70, 779–787. [Google Scholar] [CrossRef]
- Perry, J.; Chambers, A.; Spithoff, K.; Laperriere, N. Gliadel wafers in the treatment of malignant glioma: A systematic review. Curr. Oncol. 2007, 14, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Goulet, D.R.; Atkins, W.M. Considerations for the Design of Antibody-Based Therapeutics. J. Pharm. Sci. 2020, 109, 74–103. [Google Scholar] [CrossRef] [Green Version]
- Shitara, K. Potelligent antibodies as next generation therapeutic antibodies. Yakugaku Zasshi 2009, 129, 3–9. [Google Scholar] [CrossRef]
- Ohashi, Y. Safe Use of Recent New Drugs―Current Status and Challenges. Yakugaku Zasshi 2018, 138, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.J. Tocilizumab: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1865–1879. [Google Scholar] [CrossRef] [Green Version]
- Gunturi, A.; McDermott, D.F. Nivolumab for the treatment of cancer. Expert Opin. Investig. Drugs 2015, 24, 253–260. [Google Scholar] [CrossRef]
- Beck, A.; Reichert, J.M. Marketing approval of mogamulizumab: A triumph for glyco-engineering. MAbs 2012, 4, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Insogna, K.L.; Briot, K.; Imel, E.A.; Kamenický, P.; Ruppe, M.D.; Portale, A.A.; Weber, T.; Pitukcheewanont, P.; Cheong, H.I.; Beur, S.J.d.; et al. A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Evaluating the Efficacy of Burosumab, an Anti-FGF23 Antibody, in Adults With X-Linked Hypophosphatemia: Week 24 Primary Analysis. J. Bone Miner. Res. 2018, 33, 1383–1393. [Google Scholar] [CrossRef] [Green Version]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.D.; Bussel, J.B. Neonatal Fc receptor in human immunity: Function and role in therapeutic intervention. J. Allergy Clin. Immunol. 2020, 146, 467–478. [Google Scholar] [CrossRef] [PubMed]
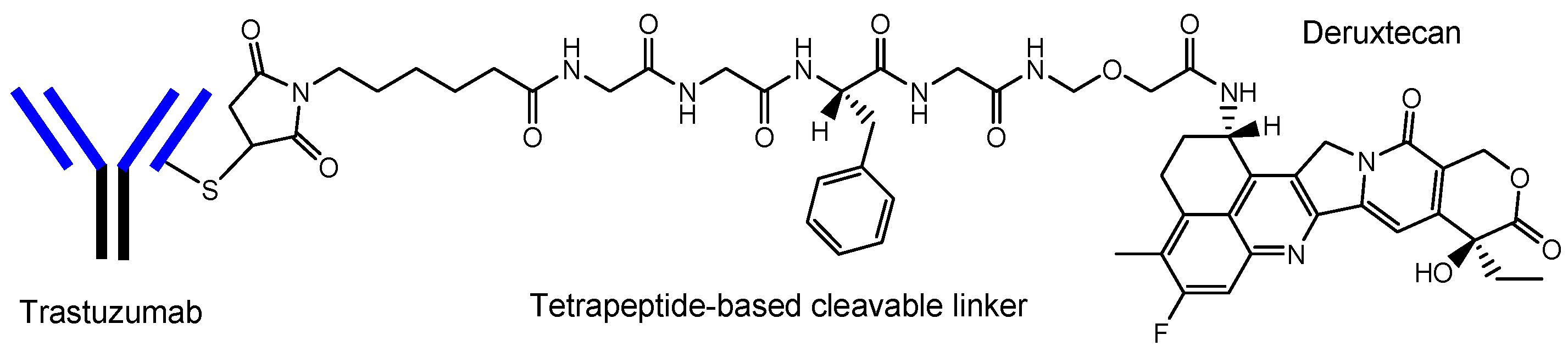
- Nakada, T.; Naito, H.; Morita, K.; Masuda, T.; Ogitani, Y. Discovery of Trastuzumab Deruxtecan (T-DXd, DS-8201a) as Next-Generation ADC. Medchem. News 2020, 30, 71–77. Available online: https://www.jstage.jst.go.jp/article/medchem/30/2/30_71/_pdf/-char/ja (accessed on 27 June 2022).
- Okajima, D.; Yasuda, S.; Maejima, T.; Karibe, T.; Sakurai, K.; Aida, T.; Toki, T.; Yamaguchi, J.; Kitamura, M.; Kamei, R.; et al. Datopotamab Deruxtecan, a Novel TROP2-directed Antibody-drug Conjugate, Demonstrates Potent Antitumor Activity by Efficient Drug Delivery to Tumor Cells. Mol. Cancer Ther. 2021, 20, 2329–2340. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Baik, C.; Su, W.C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated Non-Small Cell Lung Cancer. Cancer Discov. 2022, 12, 74–89. [Google Scholar] [CrossRef]
- Patel, M.R.; Johnson, M.L.; Falchook, G.S.; Doi, T.; Friedman, C.F.; Piha-Paul, S.A.; Gutierrez, M.; Shimizu, T.; Cheng, B.; Qian, M.; et al. DS-7300 (B7-H3 DXd-ADC) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): A subgroup analysis of a phase 1/2 multicenter study. J. Clin. Oncol. 2022, 40, 87. [Google Scholar] [CrossRef]
- Murakami, M.; Tokui, T.; Nakamaru, K.; Cogswell, J.; Ford, S.K.; Gallant, G. Expanded precision medicine capabilities for strengthening oncology drug development at Daiichi Sankyo. J. Precis. Med. 2021, 7, 14–19. Available online: https://www.thejournalofprecisionmedicine.com/wp-content/uploads/expanded-precision.pdf (accessed on 27 June 2022).
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Kida, S.; Kinoshita, M.; Tanaka, S.; Okumura, M.; Koshimura, Y.; Morimoto, H. Non-clinical evaluation of a blood-brain barrier-penetrating enzyme for the treatment of mucopolysaccharidosis type I. Mol. Genet. Metab. 2019, 126, S83–S84. [Google Scholar] [CrossRef]
- Okuyama, T.; Eto, Y.; Sakai, N.; Minami, K.; Yamamoto, T.; Sonoda, H.; Yamaoka, M.; Tachibana, K.; Hirato, T.; Sato, Y. Iduronate-2-Sulfatase with Anti-human Transferrin Receptor Antibody for Neuropathic Mucopolysaccharidosis II: A Phase 1/2 Trial. Mol. Ther. 2019, 27, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Hultqvist, G.; Syvänen, S.; Fang, X.T.; Lannfelt, L.; Sehlin, D. Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor. Theranostics 2017, 7, 308–318. Available online: https://www.thno.org/v07p0308.htm (accessed on 27 June 2022). [CrossRef] [PubMed]
- Thom, G.; Burrell, M.; Haqqani, A.S.; Yogi, A.; Lessard, E.; Brunette, E.; Delaney, C.; Baumann, E.; Callaghan, D.; Rodrigo, N.; et al. Enhanced Delivery of Galanin Conjugates to the Brain through Bioengineering of the Anti-Transferrin Receptor Antibody OX26. Mol. Pharm. 2018, 15, 1420–1431. [Google Scholar] [CrossRef] [PubMed]
- Bien-Ly, N.; Yu, Y.J.; Bumbaca, D.; Elstrott, J.; Boswell, C.A.; Zhang, Y.; Luk, W.; Lu, Y.; Dennis, M.S.; Weimer, R.M.; et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 2014, 11, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Chang, R.; Maghribi, A.A.; Vanderpoel, V.; Vasilevko, V.; Cribbs, D.H.; Boado, R.; Pardridge, W.M.; Sumbria, R.K. Brain Penetrating Bifunctional Erythropoietin-Transferrin Receptor Antibody Fusion Protein for Alzheimer’s Disease. Mol. Pharm. 2018, 5, 4963–4973. [Google Scholar] [CrossRef] [PubMed]
- Niewoehner, J.; Bohrmann, B.; Collin, L.; Urich, E.; Sade, H.; Maier, P.; Rueger, P.; Stracke, J.; Lau, W.; Tissot, A.; et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 2014, 81, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, M.A.; Banks, W.A. Transcellular routes of blood–brain barrier disruption. Exp. Biol. Med. 2022, 247, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenesis Res. 2010, 2, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, F.; Pellerino, A.; Soffietti, R.; Rudà, R. Blood–Brain Barrier in Brain Tumors: Biology and Clinical Relevance. Int. J. Mol. Sci. 2021, 22, 12654. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Goerne, R.; Bogdahn, U.; Hau, P. Procarbazine-a traditional drug in the treatment of malignant gliomas. Curr. Med. Chem. 2008, 15, 1376–1387. [Google Scholar] [CrossRef] [PubMed]
- Happold, C.; Roth, P.; Wick, W.; Steinbach, J.P.; Linnebank, M.; Weller, M.; Eisele, G. ACNU-based chemotherapy for recurrent glioma in the temozolomide era. J. Neurooncol. 2009, 92, 45–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Zhou, F.; Kruh, G.D.; Gallo, J.M. Influence of blood-brain barrier efflux pumps on the distribution of vincristine in brain and brain tumors. Neuro-Oncology 2010, 12, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Estrada, C.C.; Maldonado, A.; Mallipattu, S.K. Therapeutic Inhibition of VEGF Signaling and Associated Nephrotoxicities. J. Am. Soc. Nephrol. 2019, 30, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Sareddy, G.R.; Viswanadhapalli, S.; Surapaneni, P.; Suzuki, T.; Brenner, A.; Vadlamudi, R.K. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene 2017, 36, 2423–2434. [Google Scholar] [CrossRef] [Green Version]
- Goss, G.D.; Vokes, E.E.; Gordon, M.S.; Gandhi, L.; Papadopoulos, K.P.; Rasco, D.W.; Fischer, J.S.; Chu, K.L.; Ames, W.W.; Mittapalli, R.K.; et al. Efficacy and safety results of depatuxizumab mafodotin (ABT-414) in patients with advanced solid tumors likely to overexpress epidermal growth factor receptor. Cancer 2018, 124, 2174–2183. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Kozlosky, C.J.; Siu, S.; Chang, W.S.; Liu, H.; Foltz, I.N.; Trueblood, E.S.; Meininger, D.; Arora, T.; Twomey, B.; et al. AMG 595, an Anti-EGFRvIII Antibody-Drug Conjugate, Induces Potent Antitumor Activity against EGFRvIII-Expressing Glioblastoma. Mol. Cancer Ther. 2015, 14, 1614–1624. [Google Scholar] [CrossRef] [Green Version]
- Parakh, S.; Nicolazzo, J.; Scott, A.M.; Gan, H.K. Antibody Drug Conjugates in Glioblastoma—Is There a Future for Them? Mol. Cancer Ther. 2015, 14, 1614–1624. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front. Aging Neurosci. 2020, 11, 373. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Jacob, A.P.; Gurgel, J.L.; Tometsko, M.E.; Rock, B.M.; Patel, S.K.; Milburn, R.R.; Siu, S.; Ragan, S.P.; Rock, D.A.; et al. SLC46A3 is required to transport catabolites of noncleavable antibody maytansine conjugates from the lysosome to the cytoplasm. Cancer Res. 2015, 75, 5329–5340. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanamiya, R.; Sano, M.; Asano, T.; Yanaka, M.; Nakamura, T.; Saito, M.; Tanaka, T.; Hosono, H.; Tateyama, N.; Kaneko, M.K.; et al. Epitope Mapping of an Anti-Human Epidermal Growth Factor Receptor Monoclonal Antibody (EMab-51) Using the RIEDL Insertion for Epitope Mapping Method. Monoclon. Antibodies Immunodiagn. Immunother. 2021, 40, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Kaneko, M.K.; Asano, T.; Kato, Y. Epitope Mapping of an Anti-human EGFR Monoclonal Antibody (EMab-134) using the REMAP Method. Monoclon. Antibodies Immunodiagn. Immunother. 2021, 40, 191–195. [Google Scholar] [CrossRef]
- Cai, W.Q.; Zeng, L.S.; Wang, L.F.; Wang, Y.Y.; Cheng, J.T.; Zhang, Y.; Han, Z.W.; Zhou, Y.; Huang, S.L.; Wang, X.W.; et al. The Latest Battles Between EGFR Monoclonal Antibodies and Resistant Tumor Cells. Front. Oncol. 2020, 10, 1249. [Google Scholar] [CrossRef]
- Mihara, E.; Watanabe, S.; Bashiruddin, N.K.; Nakamura, N.; Matoba, K.; Sano, Y.; Maini, R.; Yin, Y.; Sakai, K.; Arimori, T.; et al. Lasso-grafting of macrocyclic peptide pharmacophores yields multi-functional proteins. Nat. Commun. 2021, 9, 1543. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Passioura, T.; Suga, H. Technologies for the Synthesis of mRNA-Encoding Libraries and Discovery of Bioactive Natural Product-Inspired Non-Traditional Macrocyclic Peptides. Molecules 2013, 18, 3502–3528. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Merchan, E.C.; Valentino, L.A. Emicizumab: Review of the literature and critical appraisal. Haemophilia 2019, 25, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, D.; Fujii, I. Phage Selection of Peptide “Microantibodies”. Curr. Protoc. Chem. Biol. 2013, 5, 171–194. [Google Scholar] [CrossRef]
- Caculitan, N.G.; Chuh, D.C.J.; Ma, Y.; Zhang, D.; Kozak, K.R.; Liu, Y.; Pillow, T.H.; Sadowsky, J.; Cheung, T.K.; Phung, Q.; et al. Cathepsin B Is Dispensable for Cellular Processing of Cathepsin B-Cleavable Antibody–Drug Conjugates. Cancer Res. 2017, 77, 7027–7037. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Administrated Drug | Formulation/ Co-Administrated Drug | Disease | Vector | Cargo | Linker | Group | Status | References |
|---|---|---|---|---|---|---|---|---|---|
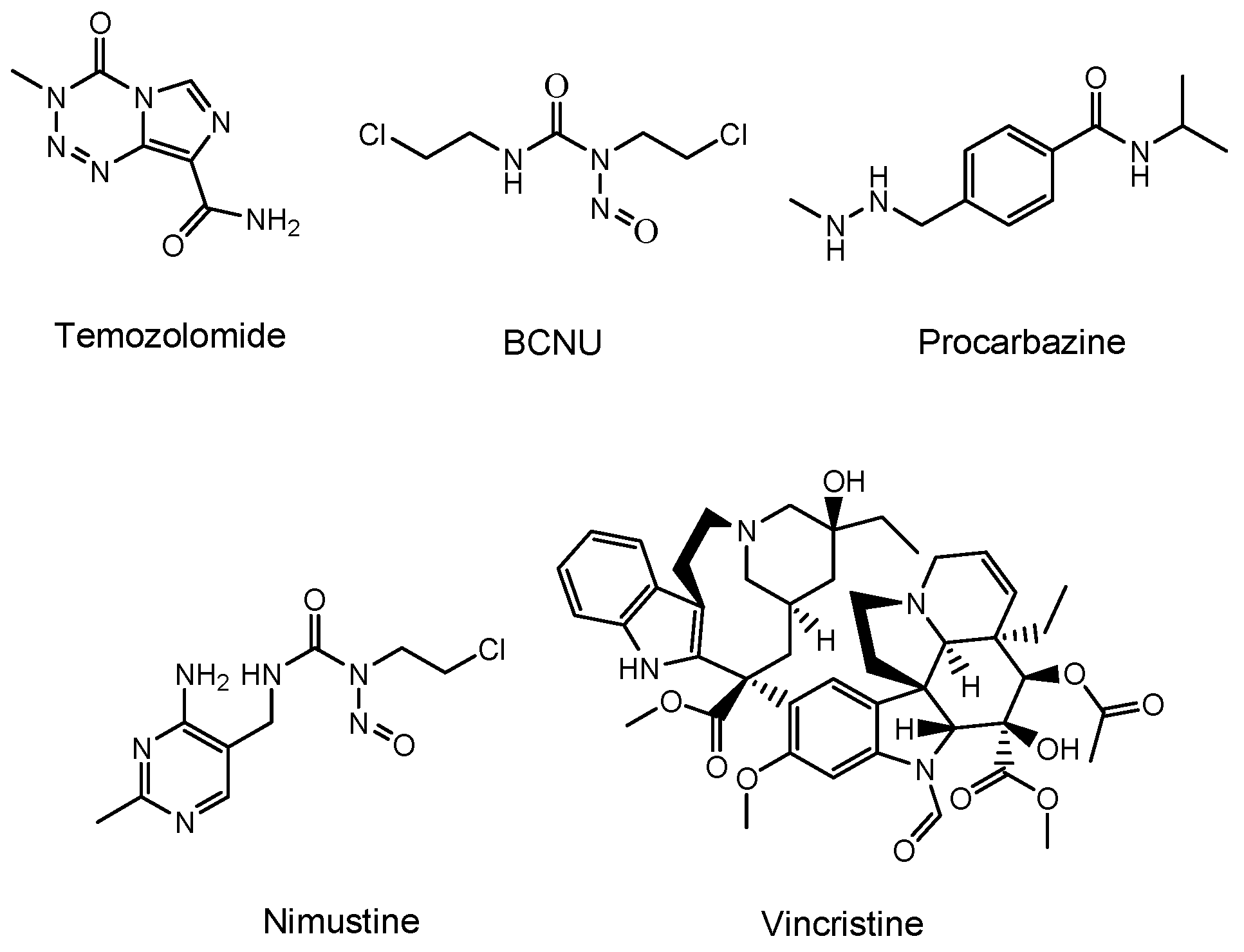
| (i) | Temozolomide | Low-molecular compound | Glioma | - | Temozolomide | - | - | Launched | [11] |
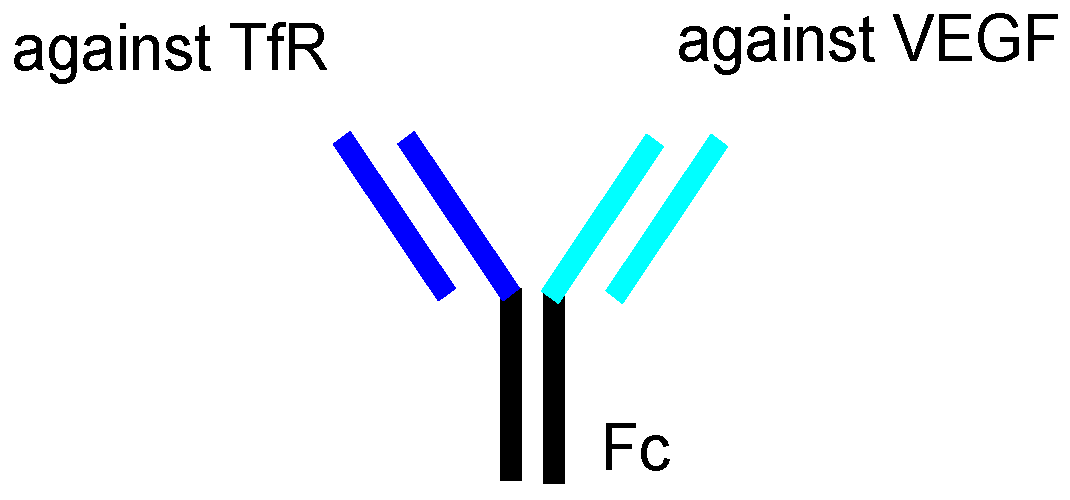
| (ii) | Bevacizumab | Anti-VEGFmAb | Glioma | - | Bevacizumab | - | - | Launched | [12] |
| (iii) | BCNU wafer | Low-molecular compound/polifeprosan 20 polymer | Glioma | - | BCNU | - | - | Launched | [13] |
| (iv) | Procarbazine | Low-molecular compound | Brain cancers | - | Procarbazine | - | - | Launched | [41] |
| (v) | Nimustine (ACNU) | Low-molecular compound | Brain cancers | - | Nimustine | - | Daiichi Sankyo | Launched | [42] |
| (vi) | Vincristine | Low-molecular compound | Brain cancers | - | Vincristine | - | - | Launched | [43] |
| (vii) | Tocilizumab (Actemra®) | Anti-IL-6 mAb | Rheumatoid arthritis | - | Tocilizumab | - | Chugai | Launched | [17] |
| (viii) | Nivolumab (Opdivo®) | Anti-PD-1 mAb | Metastatic lung squamous cell carcinoma | - | Nivolumab | - | Ono | Launched | [18] |
| (ix) | Mogamulizumab (Poteligeo®) | Anti-CCR4 mAb | Relapsed or refractory mycosis fungoides and Sézary disease | - | Mogamulizumab | - | Kyowa Kirin | Launched | [19] |
| (x) | Burosumab (Crysvita®) | Anti-FGF23 mAb | X-linked hypophosphatemic rickets | - | Burosumab | - | Kyowa Kirin | Launched | [20] |
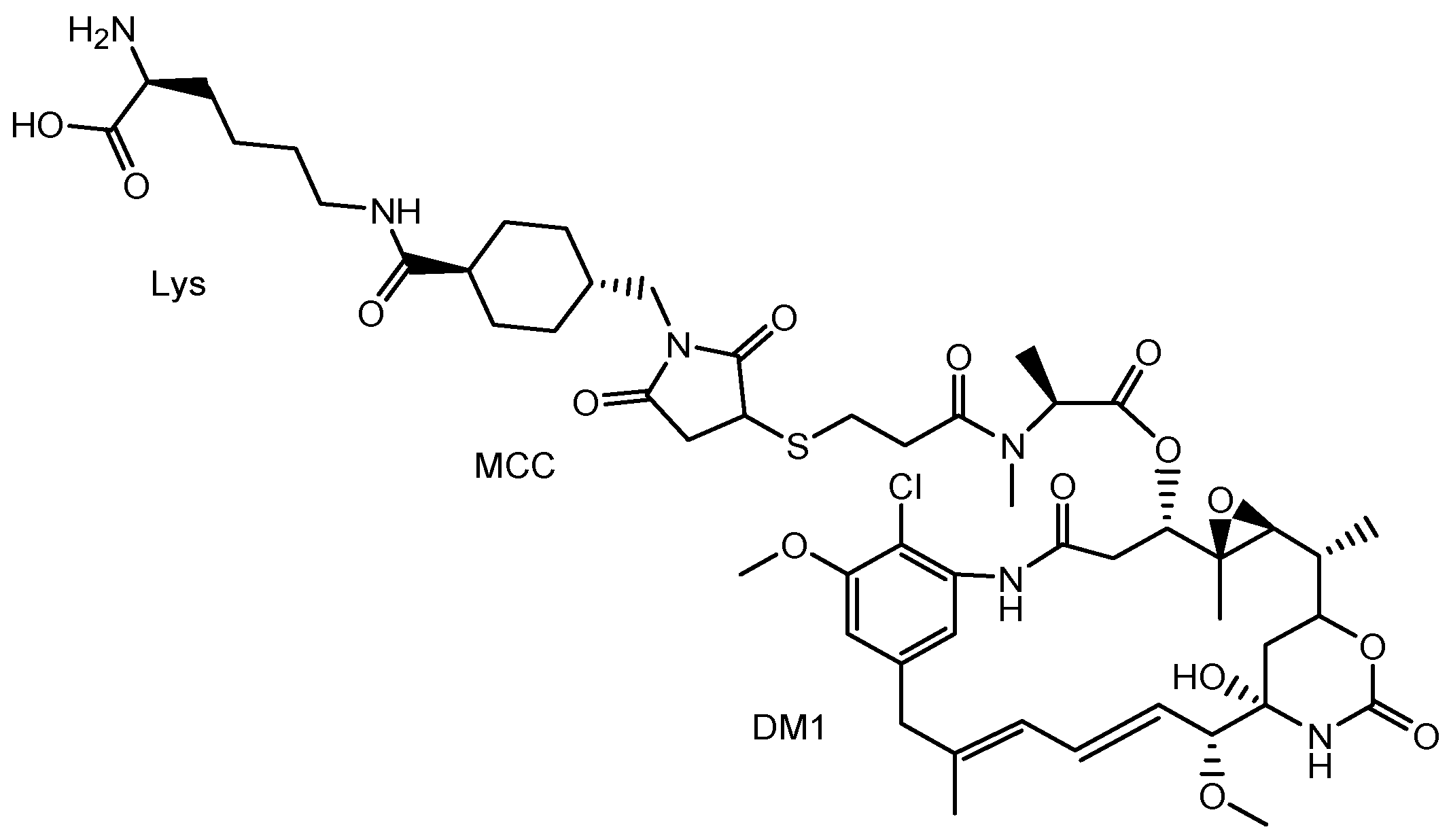
| (xi) | Trastuzumab deruxtecan (Enhertu®) | Anti-HER2 ADC | HER2 positive breast cancer | Anti-HER2 mAb | Deruxtecan | Linker | Daiichi Sankyo | Launched | [21,23] |
| (xii) | Datopotamab deruxtecan (Dato-DXd) | Anti-ROP2 ADC | Solid cancers | Anti-ROP2 mAb | Deruxtecan | Linker | Daiichi Sankyo | Clinical trial | [24] |
| (xiii) | Patritumab deruxtecan (HER3-DXd) | Anti-HER3 ADC | Solid cancers | Anti-HER3 mAb | Deruxtecan | Linker | Daiichi Sankyo | Clinical trial | [25] |
| (xiv) | DS-7300 | Anti-B7-H3 ADC | Solid cancers | Anti-B7-H mAb | Deruxtecan | Linker | Daiichi Sankyo | Clinical trial | [26] |
| (xv) | DS-6000 | Anti-CDH6 ADC | Solid cancers | Anti-CDH6 | Deruxtecan | Linker | Daiichi Sankyo | Clinical trial | [27] |
| (xvi) | DS-3939 | Anti-TA-MUC1 ADC | Solid cancers | Anti-TA-MUC1 | Deruxtecan | Linker | Daiichi Sankyo | Pre-clinical | [27] |
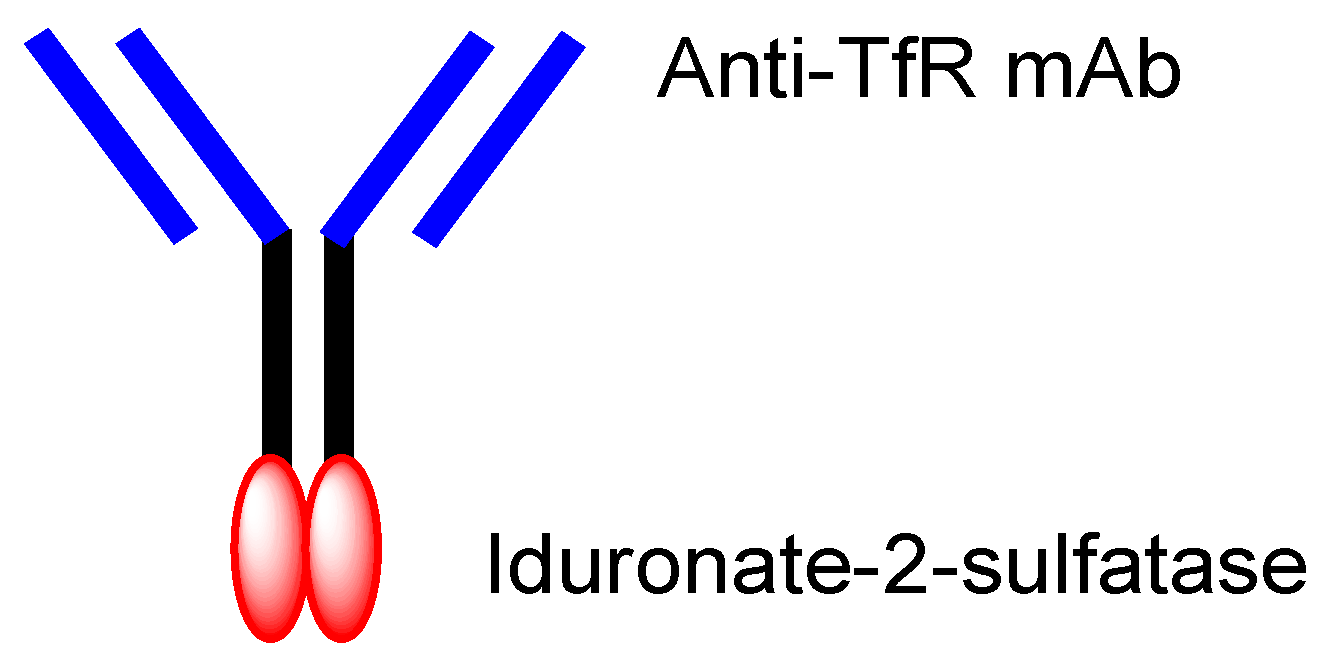
| (xvii) | Idursulfase beta | Anti-TfR ADC with-iduronate-2-sulfatase | Hunter syndrome | Anti-TfR mAb | Iduronate-2-sulfatase | Fusion protein | JCR Pharmaceuticals | Launched | [30] |
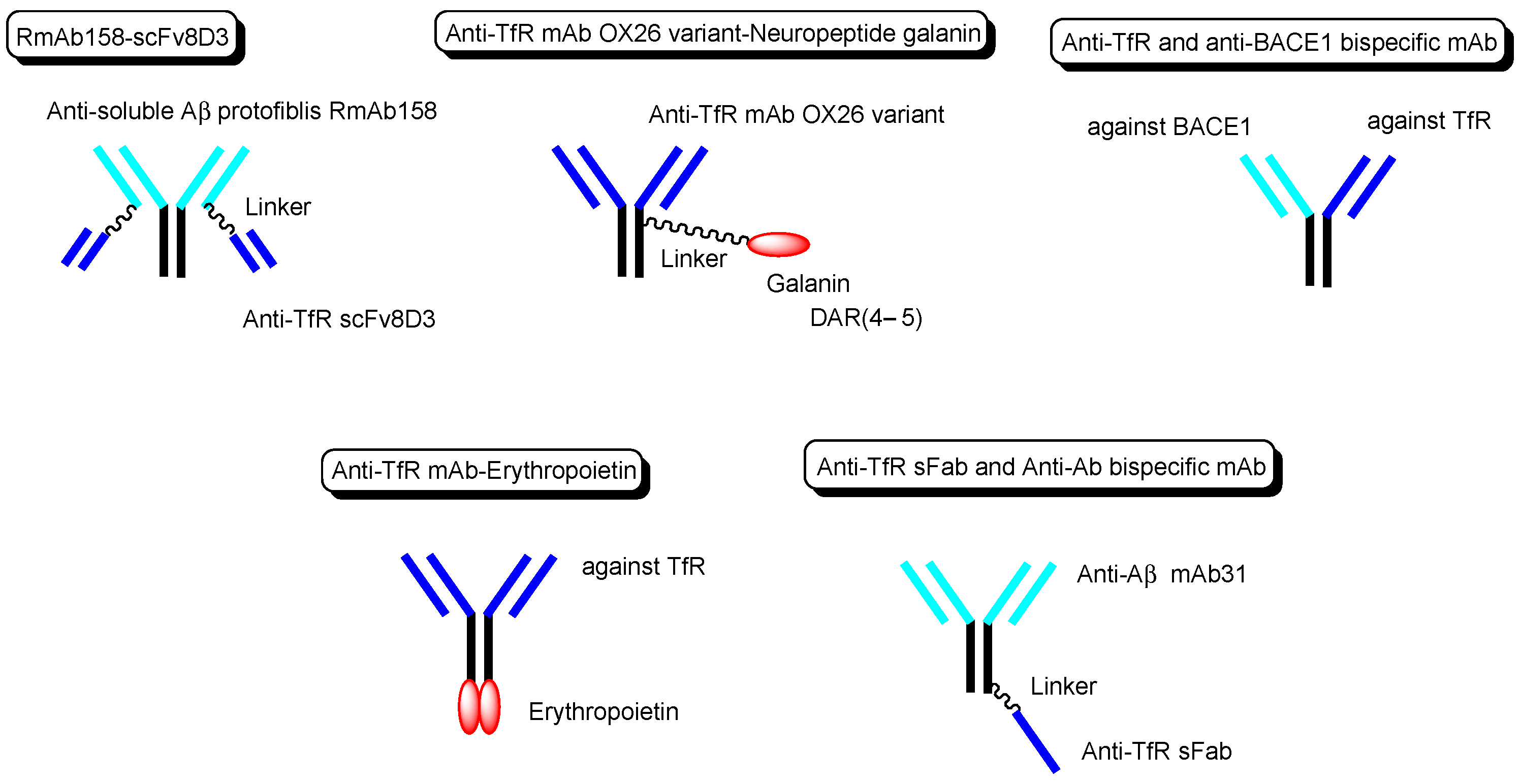
| (xviii) | Bispecific RmAb158-scFv8D3 | Bispecific RmAb158-scFv8D3 | Alzheimer’s disease | Anti-TfR scFv8D3 | Anti-soluble AβRmAb158 | Linker | - | Basic research | [31] |
| (xix) | Anti-TfR mAb OX26 variant with galanin | Anti-TfR ADC with galanin | Induction of homeostatic rebound sleep | Anti-TfR mAb | Neuropeptide galanin | Linker | - | Basic research | [32] |
| (xx) | Anti-TfR and anti-BACE1 bispecific mAb | Anti-TfR and anti-BACE1 bispecific mAb | Alzheimer’s disease | Anti-TfR mAb | Anti-BACE1 mAb | Fusion protein | - | Basic research | [33] |
| (xxi) | Anti-TfR mAb with erythropoietin | Anti-TfR mAb fused to erythropoietin | Alzheimer’s disease | Anti-TfR mAb | erythropoietin | Fusion protein | - | Basic research | [34] |
| (xxii) | scFab of anti-TfR mAb and anti-Aβ mAb31 | scFab of anti-TfR mAb and anti-Aβ mAb31 | Alzheimer’s disease | scFab of anti-TfR mAb | anti-Aβ mAb31 | Linker | - | Basic research | [35] |
| (xxiii) | ABT-414 (depatuxizumab mafodotin) | Anti-EGFR ADC with MMAF | Glioblastoma | Depatuxizumab | MMAF | Linker | - | Basic research | [46] |
| (xxiv) | AMG-595 | Anti-EGFR ADC with DM1 | Glioblastoma | Anti-EGFR mAb | DM1 (mertansine) | Linker | - | Basic research | [47] |
| (xxv) | Aducanumab | Anti-Aβ mAb | Alzheimer’s disease | - | Anti-Aβ mAb | - | Eisai | Launched | [9,49] |
| (xxvi) | Emicizumab (Hemlibra®) | Anti-blood coagulation factor IX and X bispecific mAb | Hemophilia A | - | Anti-blood coagulation factor IX and X bispecific mAb | Chugai | Launched | [57] | |
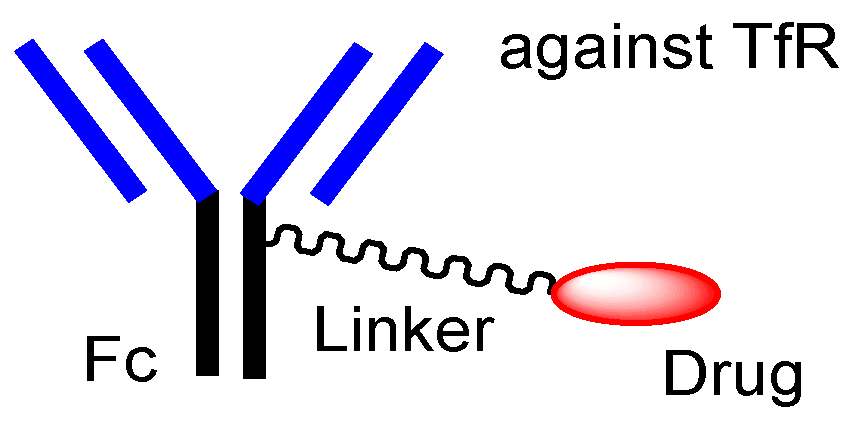
| (xxvii) | Anti-TfR ADCs with linked low-molecular cargos | Anti-TfR ADCs with linked low-molecular cargos | Glioma | Anti-TfR mAb | Brain cancer drugs | pH-sensitive cleavable linker | Tashima lab | Under analysis in Tashima lab | - |
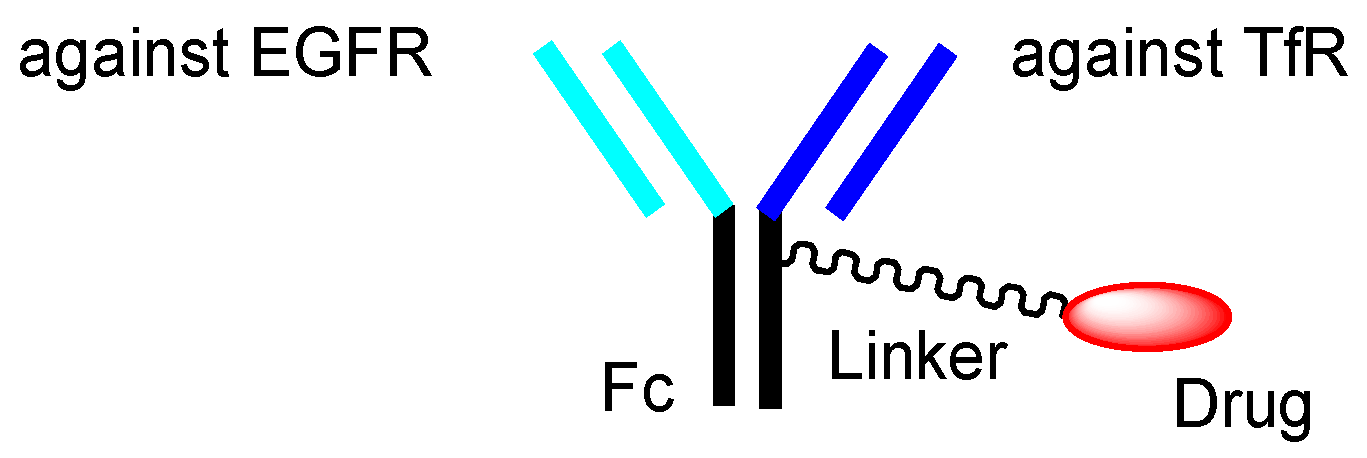
| (xxviii) | Anti-TfR and anti-EGFR bispecific ADCs with low-molecular payloads | Anti-TfR and anti-EGFR bispecific ADCs with low-molecular payloads | Glioma | Anti-TfR mAb Anti-EGFR mAb | Brain cancer drugs | Linker | Tashima lab | Under analysis in Tashima lab | - |
| (xxix) | Trispecific mAbs against TfR, EGFR, and tumor-specific molecules | Trispecific mAbs against TfR, EGFR, and tumor-specific molecules | Glioma | Anti-TfR and anti-EGFR mAbs | Anti-tumor-specific molecules unit | - | Tashima lab | Under analysis in Tashima lab | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tashima, T. Brain Cancer Chemotherapy through a Delivery System across the Blood-Brain Barrier into the Brain Based on Receptor-Mediated Transcytosis Using Monoclonal Antibody Conjugates. Biomedicines 2022, 10, 1597. https://doi.org/10.3390/biomedicines10071597
Tashima T. Brain Cancer Chemotherapy through a Delivery System across the Blood-Brain Barrier into the Brain Based on Receptor-Mediated Transcytosis Using Monoclonal Antibody Conjugates. Biomedicines. 2022; 10(7):1597. https://doi.org/10.3390/biomedicines10071597
Chicago/Turabian StyleTashima, Toshihiko. 2022. "Brain Cancer Chemotherapy through a Delivery System across the Blood-Brain Barrier into the Brain Based on Receptor-Mediated Transcytosis Using Monoclonal Antibody Conjugates" Biomedicines 10, no. 7: 1597. https://doi.org/10.3390/biomedicines10071597