
Oxidative Stress-Induced Growth Inhibitor (OSGIN1), a Target of X-Box-Binding Protein 1, Protects Palmitic Acid-Induced Vascular Lipotoxicity through Maintaining Autophagy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Bovine Serum Albumin (BSA)-Conjugated Palmitate Preparation
2.3. MTS Assay
2.4. Oil Red O Staining
2.5. Wound Healing Migration Assay
2.6. Tube Formation Assay
2.7. Small Interfering RNA (siRNA) Transfection
2.8. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.9. Western Blotting Analysis
2.10. Immunohistochemistry (IHC) Staining
2.11. Intracellular Reactive Oxygen Species (ROS) Assay
2.12. Statistical Analysis
3. Results
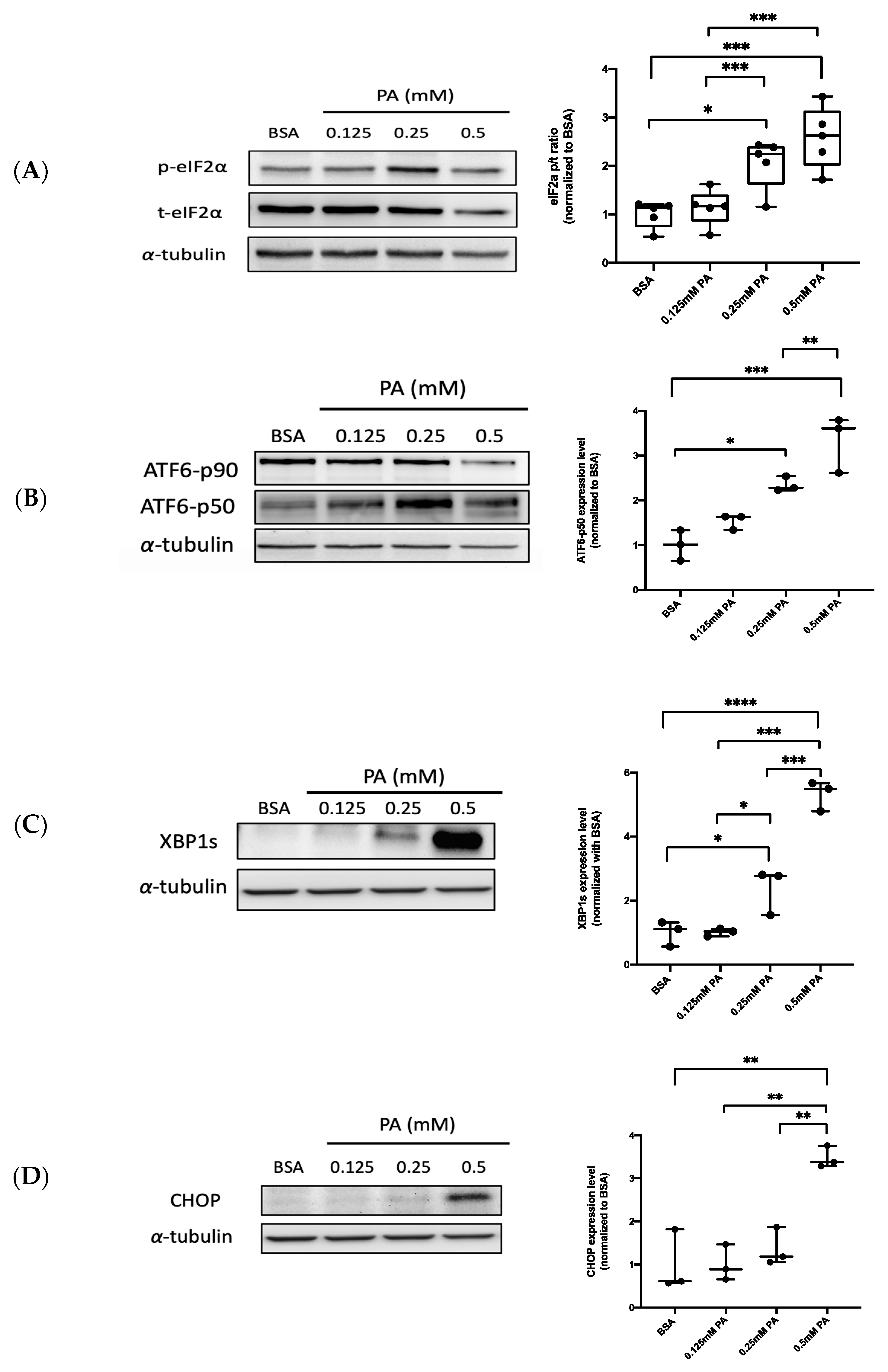
3.1. PA Activates ER Stress-Related UPR Signaling in Endothelial Cells
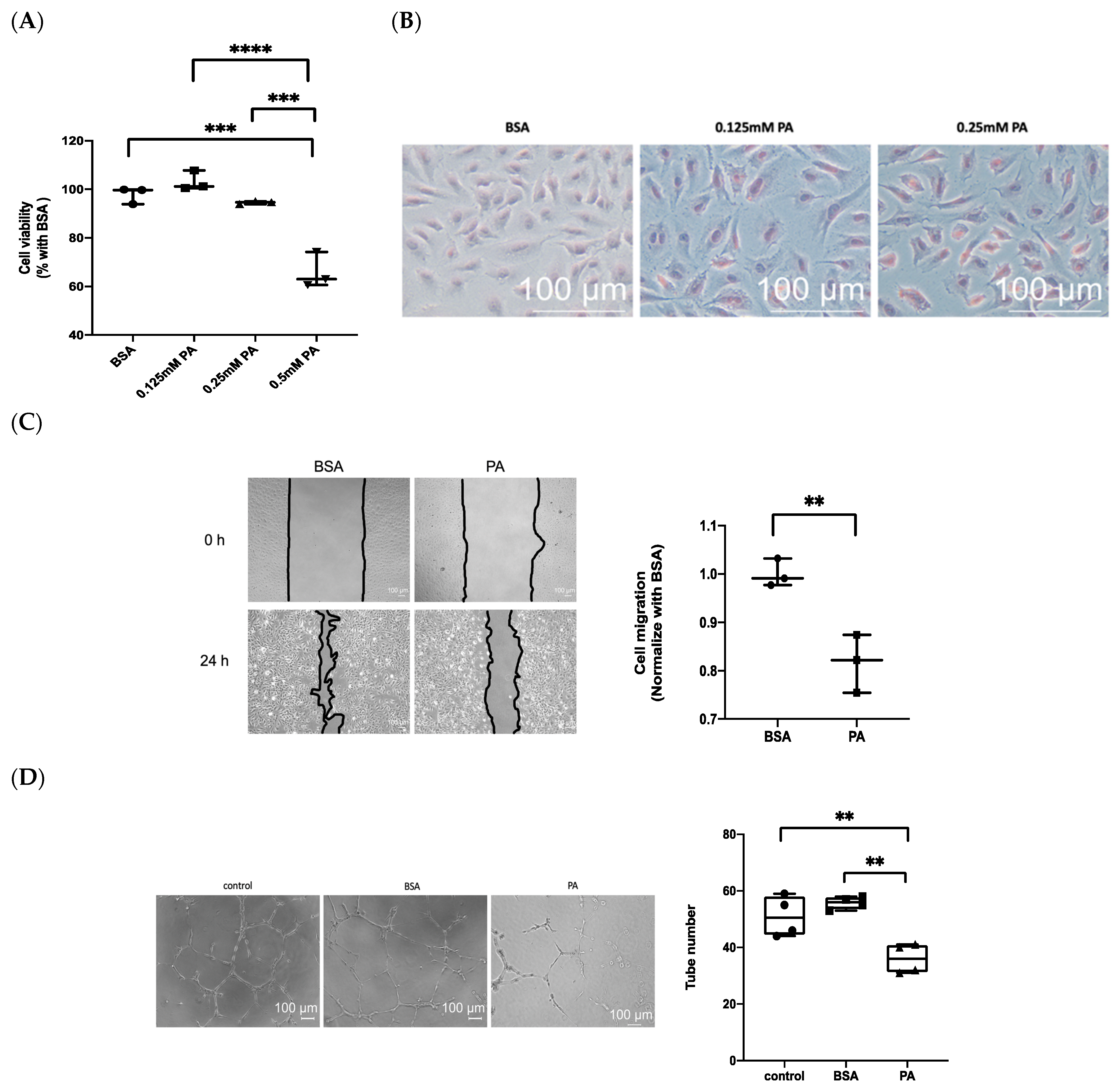
3.2. Treatment with PA Results in Endothelial Dysfunction
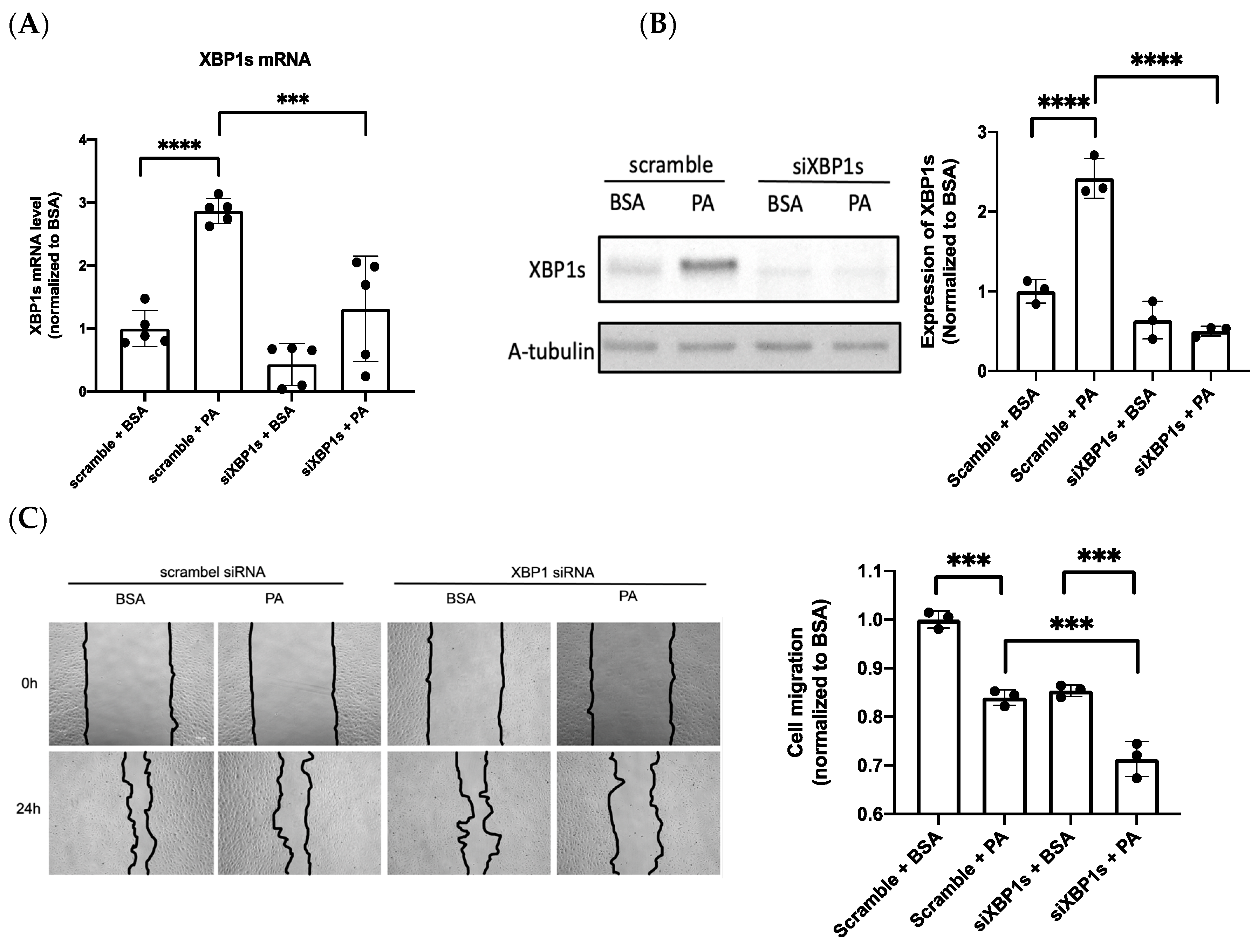
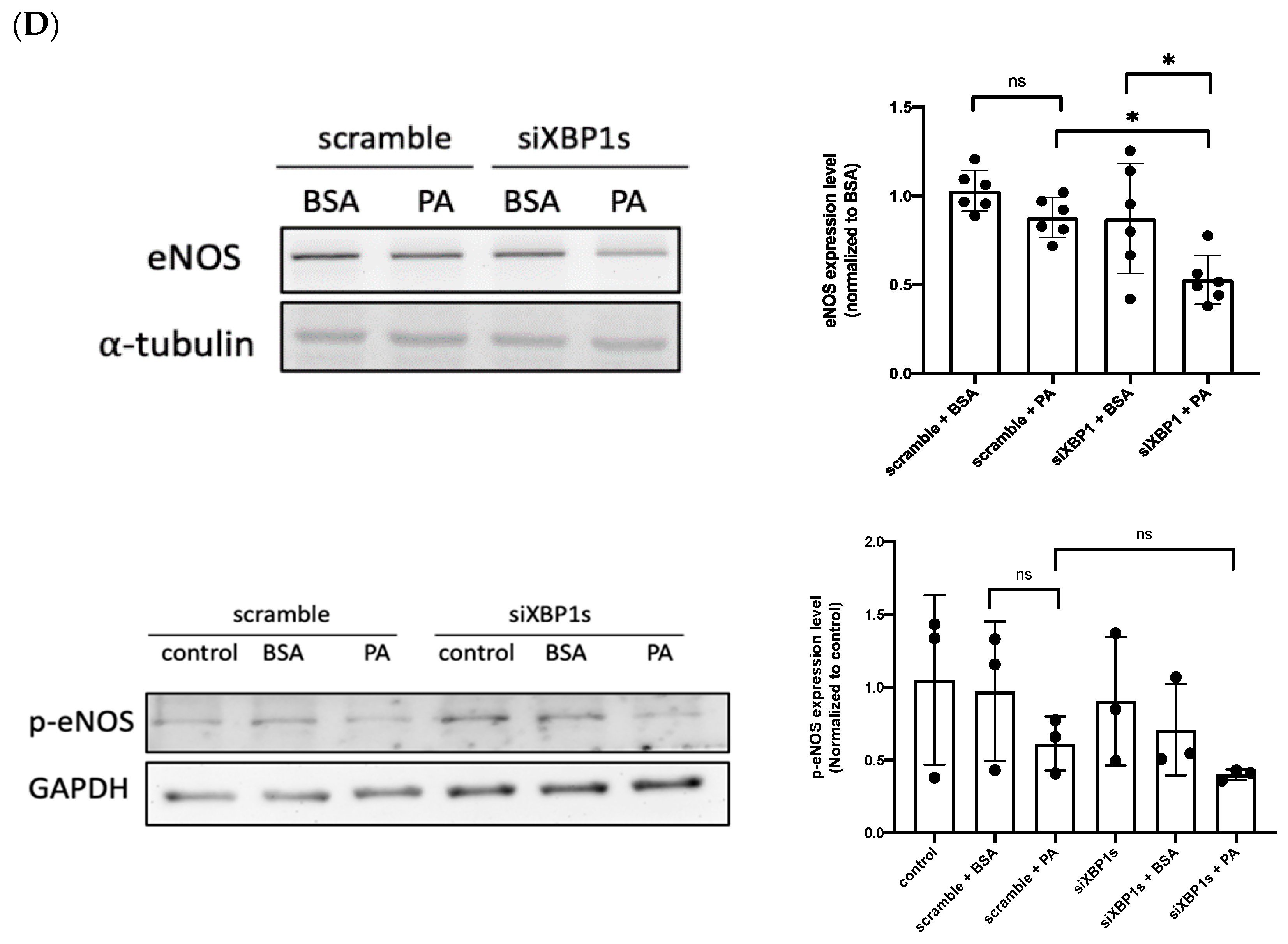
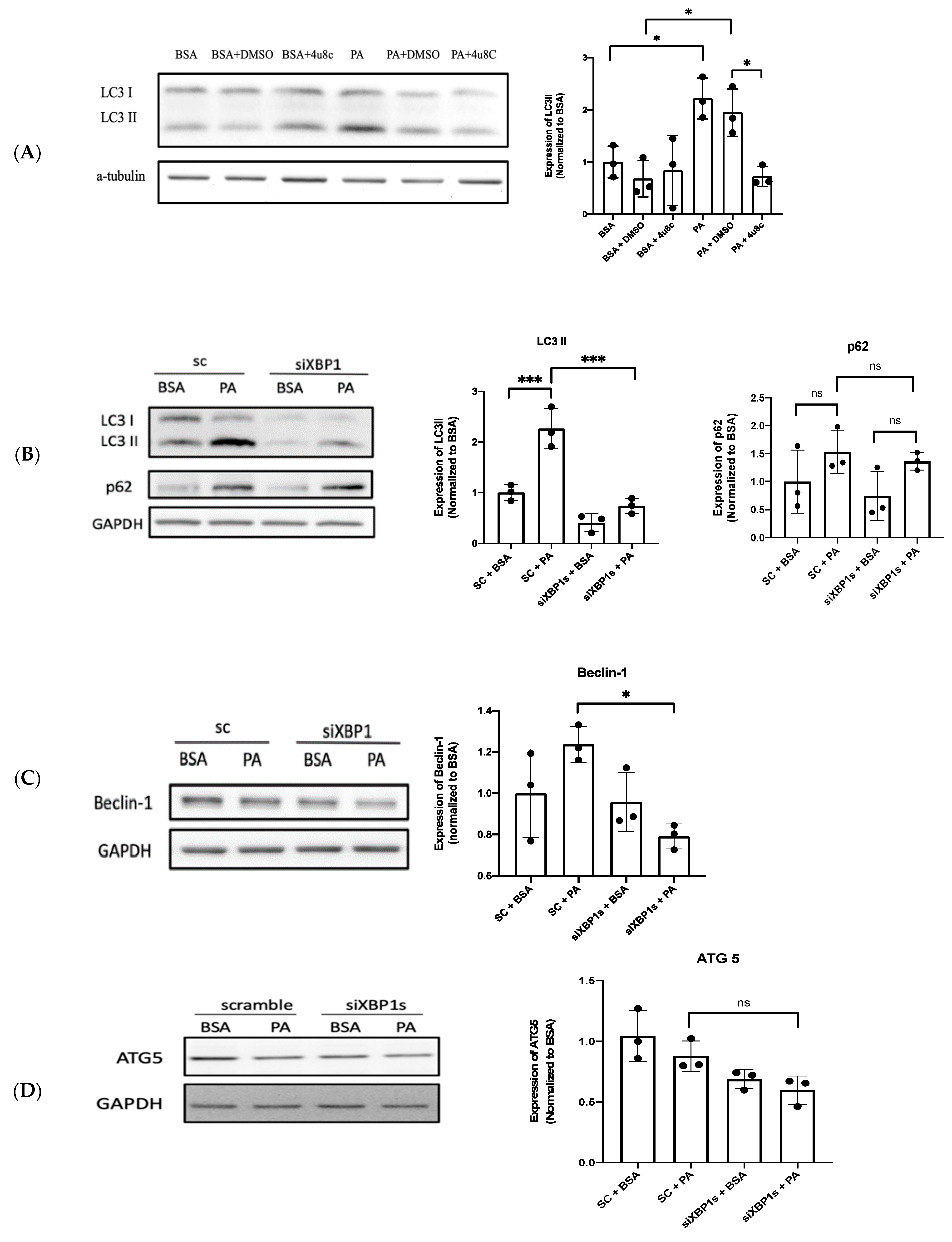
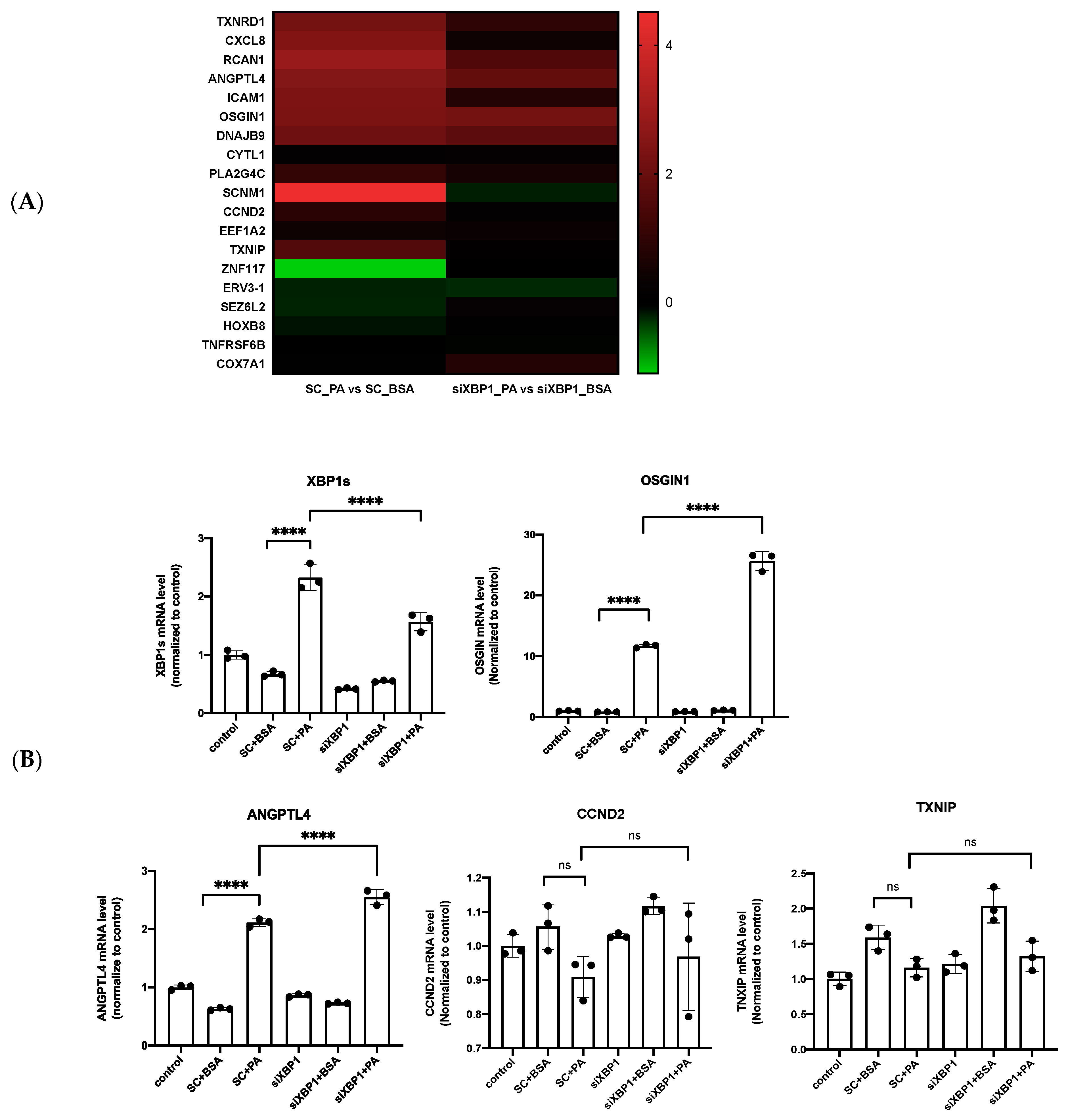
3.3. IRE1α–XBP1s Axis Activation Protects Endothelial Dysfunction Following PA Exposure
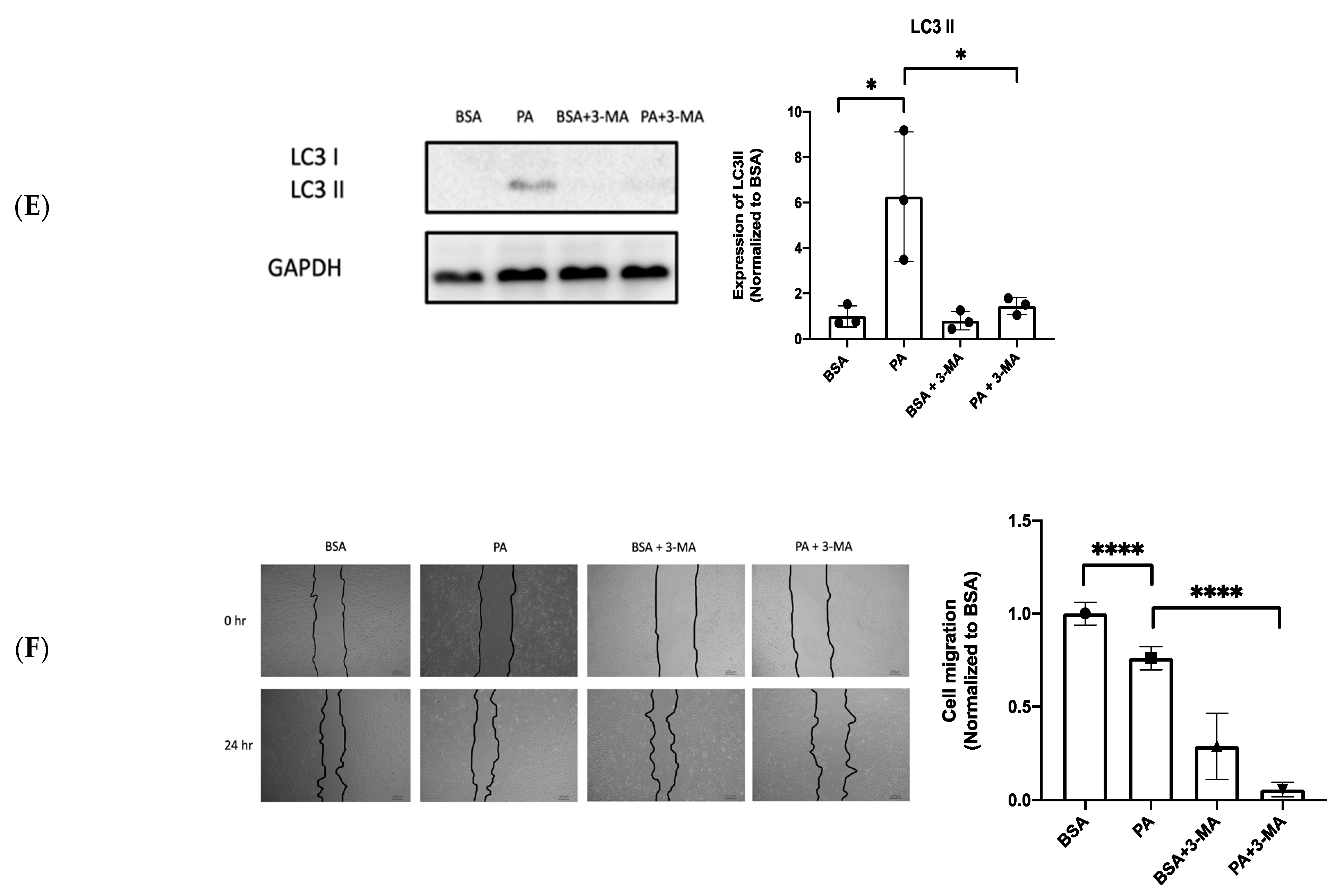
3.4. XBP1s Protects Endothelial Cells from Lipotoxicity via Autophagy
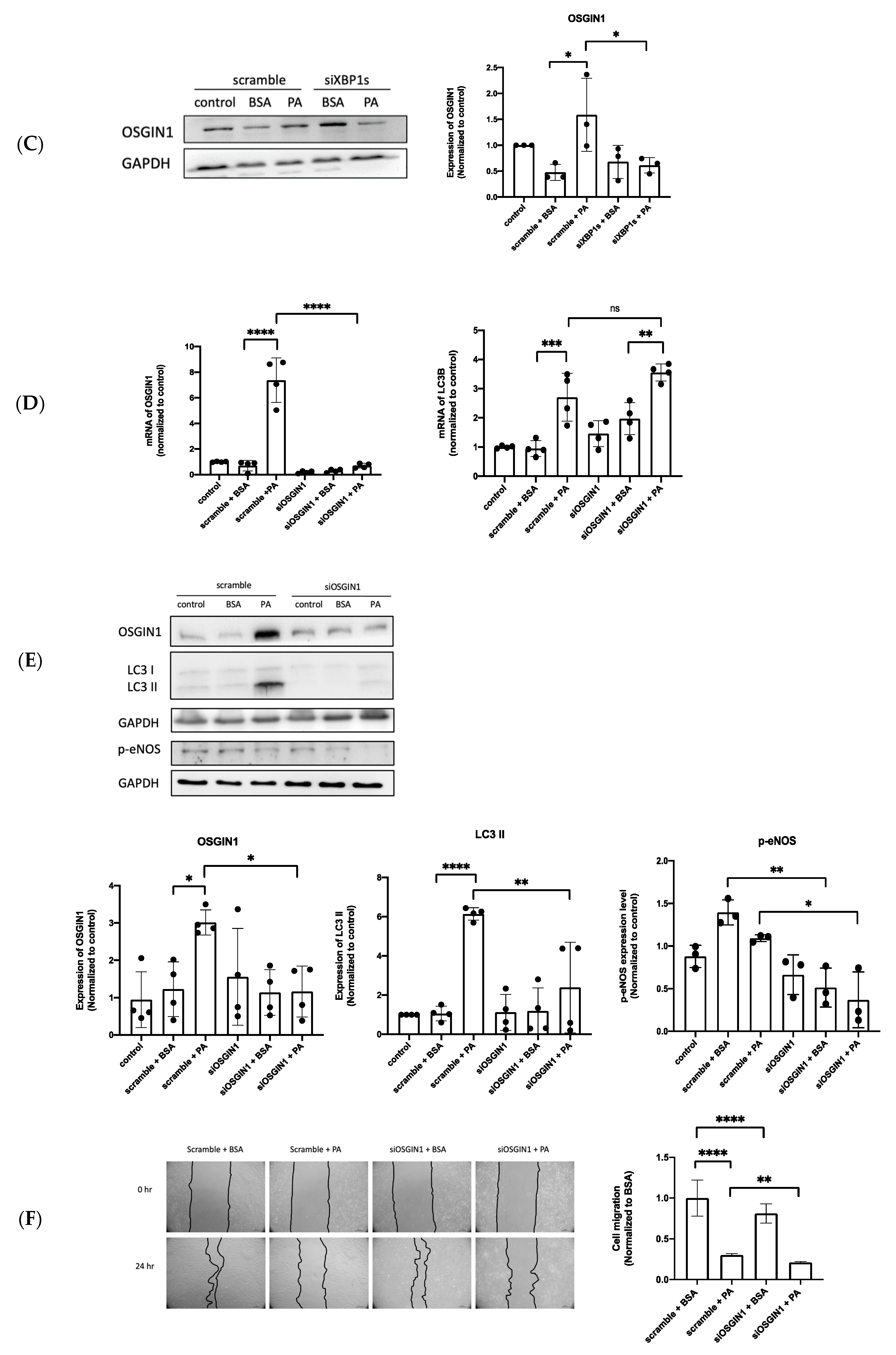
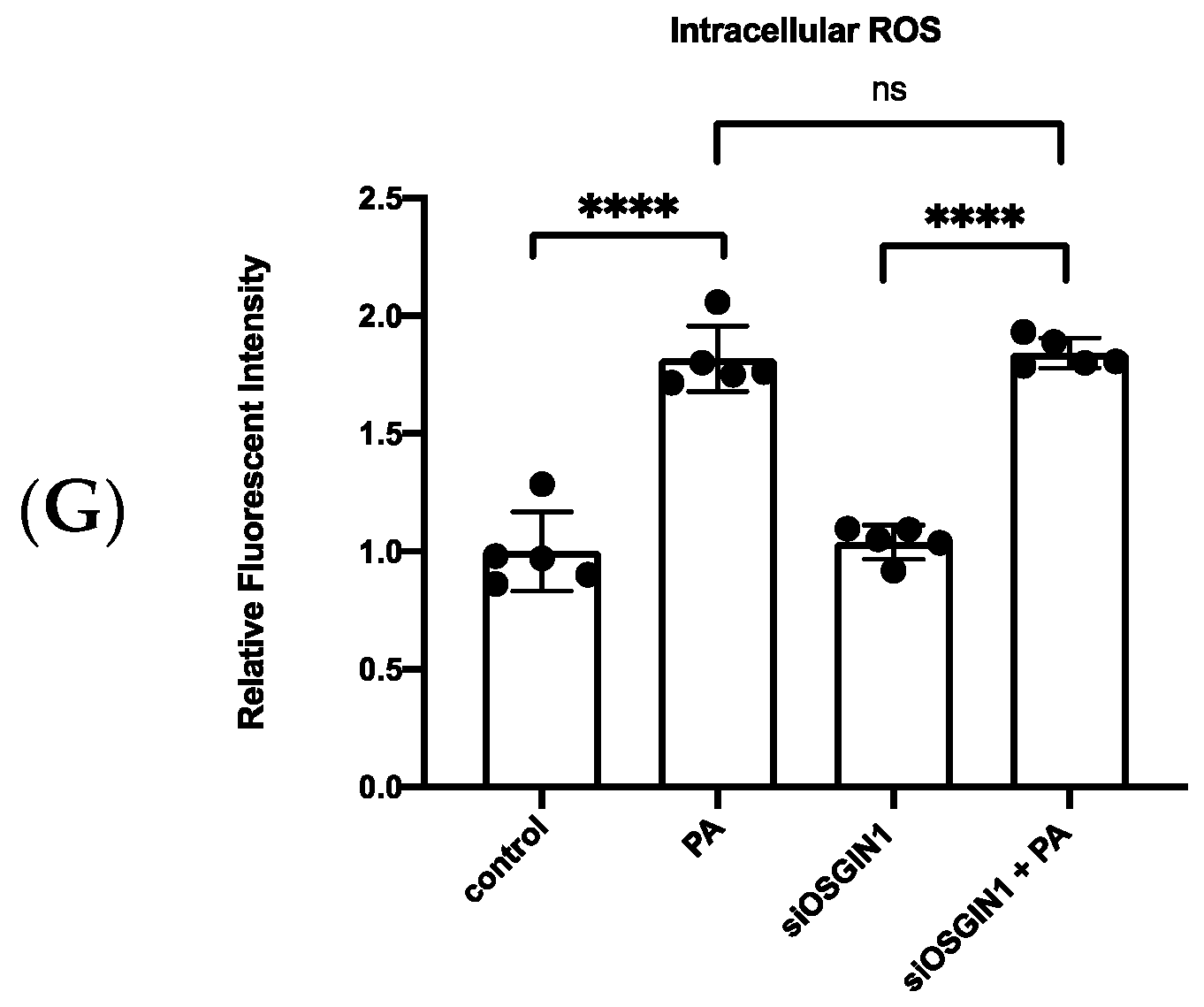
3.5. XBP1s Regulated OSGIN1 against Endothelial Dysfunction via Autophagy
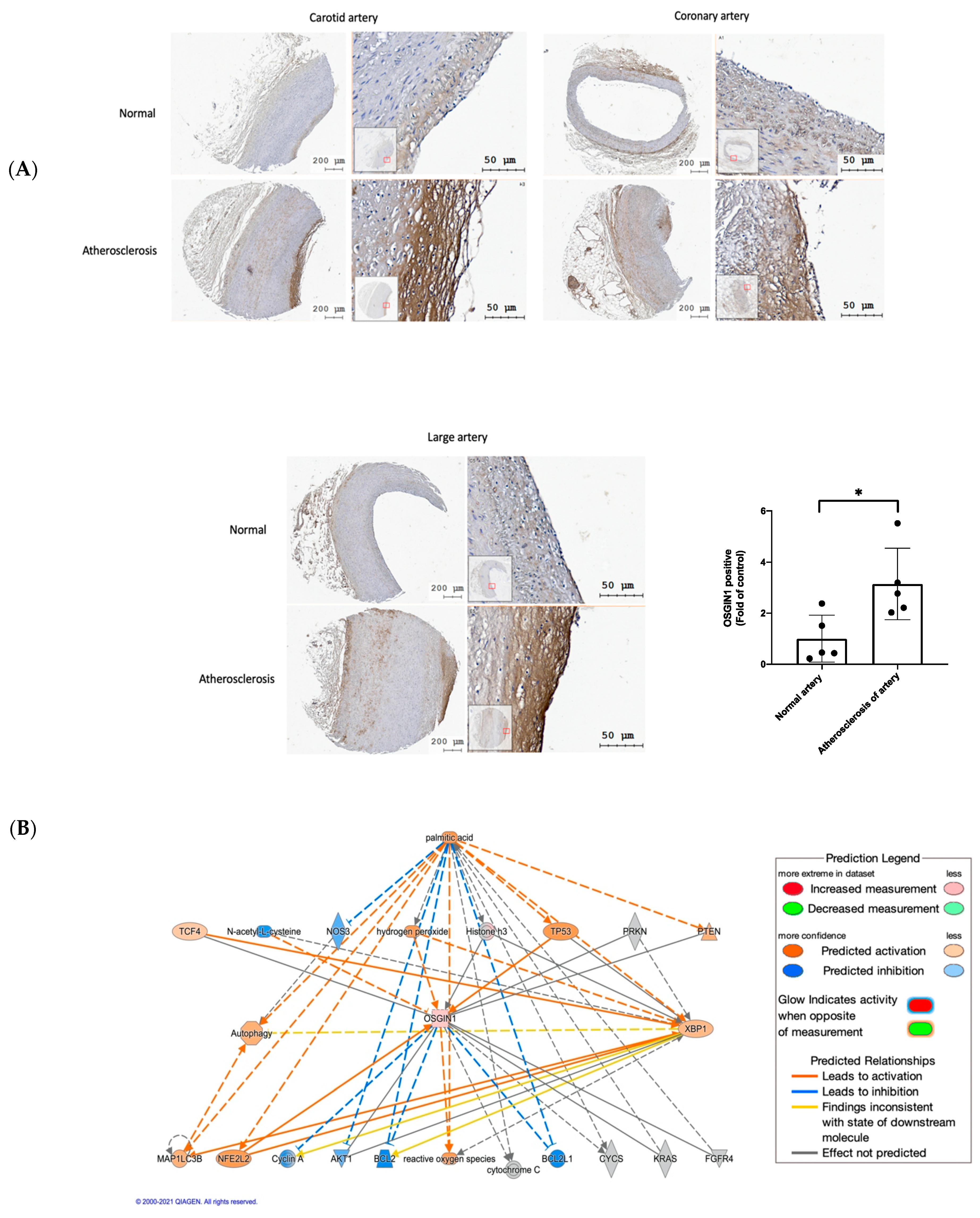
3.6. OSGIN1 Is Involved in the Atherosclerosis Process
4. Discussion
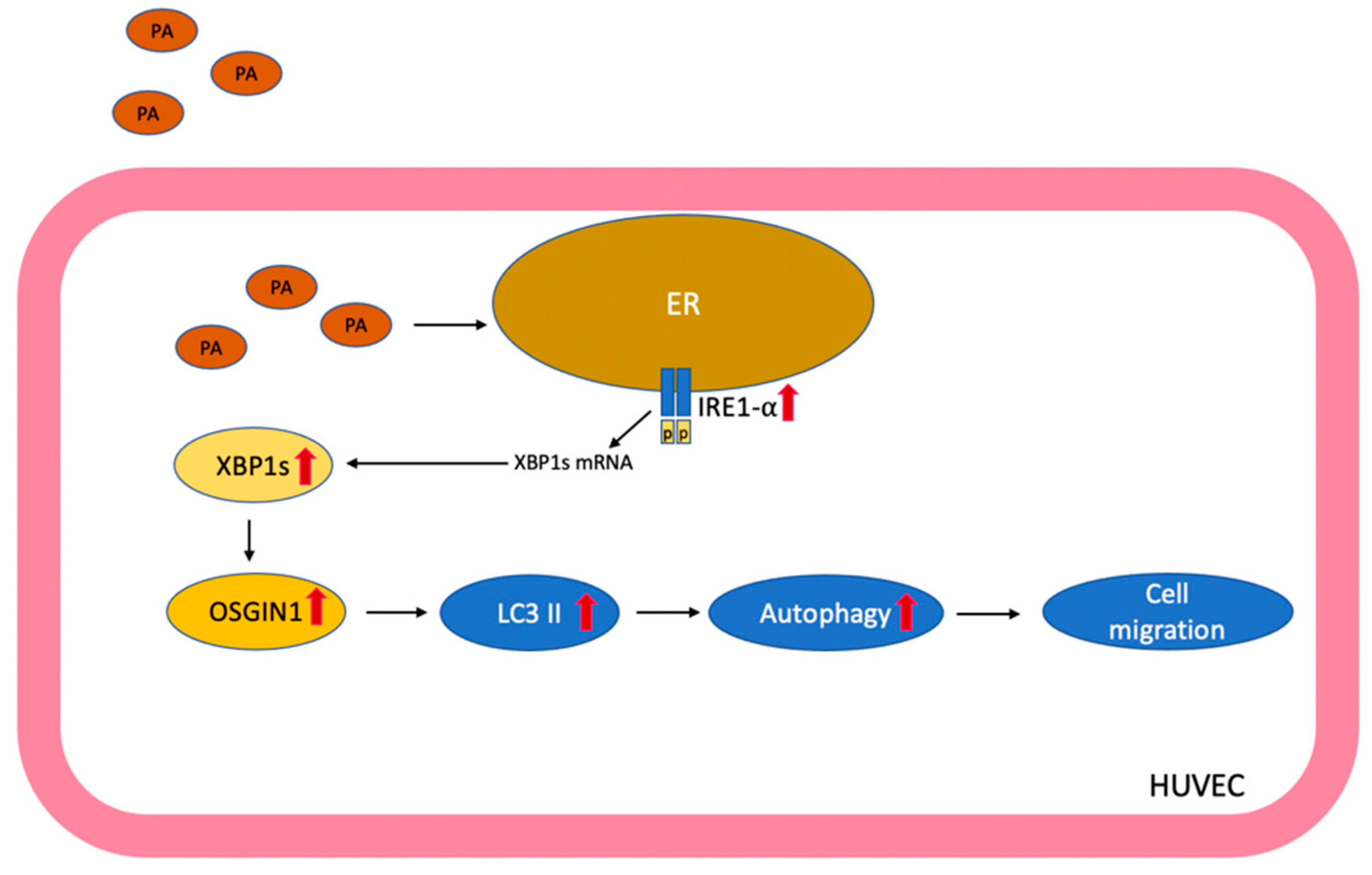
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | Serine/threonine-specific protein kinases |
| ANGPTL4 | Angiopoietin-like 4 |
| AMPK | AMP-activated protein kinase |
| ATF6 | Activating transcription factor 6 |
| ATG5 | Autophagy-related protein 5 |
| BiP | Binding immunoglobulin protein |
| CCND2 | Cyclin D2 |
| CHOP | CAAT/enhancer-binding protein (C/EBP) homologous protein |
| eIF2α | Eukaryotic translation initiation factor 2α |
| eNOS | Endothelial nitric oxide synthase |
| ER | Endoplasmic reticulum |
| ERK1/2 | Extracellular signal-regulated kinases 1/2 |
| ER stress | Endoplasmic reticulum stress |
| GRP78 | Glucose regulatory protein 78 |
| HCC | Hepatocellular carcinoma |
| IRE1α | Inositol-requiring enzyme 1 α |
| JNK | c-Jun amino-terminal kinases |
| LC3B | Microtubule-associated protein 1 light chain 3B |
| LC3 II | LC3-phosphatidylethanolamine conjugate |
| MEK | Mitogen-activated protein kinase |
| mTOR | Mammalian target of rapamycin |
| NO | Nitric oxide |
| NOX2 | NADPH oxidase 2 |
| NOX4 | NADPH oxidase 4 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| OKL38 | Ovary kidney and liver protein 38 |
| OSGIN1 | Oxidative stress-induced growth inhibitor 1 |
| PARP | Poly(ADP-Ribose) Polymerase |
| PERK | Protein kinase R (PKR)-like endoplasmic reticulum kinase |
| PI3K | Phosphoinositide 3-kinases |
| PKCα | Protein kinase C-alpha |
| RNase | Ribonuclease |
| ROS | Reactive oxygen species |
| TNFα | Tumor necrosis factor alpha |
| TXNIP | Thioredoxin-interacting protein |
References
- Garbarino, J.; Sturley, S.L. Saturated with fat: New perspectives on lipotoxicity. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J. Lipotoxicity. Kidney Int. 2006, 70, 1560–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Liu, H.; Xiang, H.; Zhou, J.; Zeng, Z.; Chen, R.; Zhao, S.; Xiao, J.; Shu, Z.; Chen, S.; et al. Palmitic acid-induced autophagy increases reactive oxygen species via the Ca2+/PKCα/NOX4 pathway and impairs endothelial function in human umbilical vein endothelial cells. Exp. Ther. Med. 2019, 17, 2425–2432. [Google Scholar] [CrossRef]
- Zhao, Q.; Yang, H.; Liu, F.; Luo, J.; Li, X.; Yang, Y. Naringenin Exerts Cardiovascular Protective Effect in a Palmitate-Induced Human Umbilical Vein Endothelial Cell Injury Model via Autophagy Flux Improvement. Mol. Nutr. Food Res. 2019, 63, e1900601. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szendroedi, J.; Frossard, M.; Klein, N.; Bieglmayer, C.; Wagner, O.; Pacini, G.; Decker, J.; Nowotny, P.; Müller, M.; Roden, M. Lipid-Induced Insulin Resistance Is Not Mediated by Impaired Transcapillary Transport of Insulin and Glucose in Humans. Diabetes 2012, 61, 3176–3180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Wang, N.; Pagliassotti, M.J. Saturated fatty acid-mediated endoplasmic reticulum stress and apoptosis are augmented by trans-10, cis-12-conjugated linoleic acid in liver cells. Mol. Cell. Biochem. 2007, 303, 105–113. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R.; Ishimoto, Y.; Nangaku, M. Proteostasis in endoplasmic reticulum—new mechanisms in kidney disease. Nat. Rev. Nephrol. 2014, 10, 369–378. [Google Scholar] [CrossRef]
- Waldherr, S.M.; Strovas, T.J.; Vadset, T.; Liachko, N.; Kraemer, B.C. Constitutive XBP-1s-mediated activation of the endoplasmic reticulum unfolded protein response protects against pathological tau. Nat. Commun. 2019, 10, 4443. [Google Scholar] [CrossRef]
- Imanikia, S.; Özbey, N.P.; Krueger, C.; Casanueva, M.O.; Taylor, R.C. Neuronal XBP-1 Activates Intestinal Lysosomes to Improve Proteostasis in C. elegans. Curr. Biol. 2019, 29, 2322–2338.e7. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.; Li, Y.; Yang, J.; Wang, G.; Margariti, A.; Jiang, Z.; Yu, H.; Zampetaki, A.; Hu, Y.; Xu, Q.; et al. Unspliced X-box-binding Protein 1 (XBP1) Protects Endothelial Cells from Oxidative Stress through Interaction with Histone Deacetylase 3. J. Biol. Chem. 2014, 289, 30625–30634. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, J.J.; Zhang, S.X. Preconditioning with Endoplasmic Reticulum Stress Mitigates Retinal Endothelial Inflammation via Activation of X-box Binding Protein 1. J. Biol. Chem. 2011, 286, 4912–4921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Q.; Yang, L.; Gong, W.; Chaugai, S.; Wang, F.; Chen, C.; Wang, P.; Zou, M.; Wang, D.W. MicroRNA-214 Is Upregulated in Heart Failure Patients and Suppresses XBP1-Mediated Endothelial Cells Angiogenesis. J. Cell. Physiol. 2015, 230, 1964–1973. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, T.; Nakashima, M.; Ozawa, K. Incorporation of the Endoplasmic Reticulum Stress-Induced Spliced Form of XBP1 mRNA in the Exosomes. Front. Physiol. 2018, 9, 1357. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Zampetaki, A.; Margariti, A.; Pepe, A.E.; Alam, S.; Martin, D.; Xiao, Q.; Wang, W.; Jin, Z.-G.; Cockerill, G.; et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2009, 106, 8326–8331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, C.K.; Ng, C.Y.; Leong, C.; Ng, C.P.; Foo, K.T.; Tan, P.H.; Huynh, H. Genomic Structure of Human OKL38 Gene and Its Differential Expression in Kidney Carcinogenesis. J. Biol. Chem. 2004, 279, 743–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Yao, H.; Gan, F.; Tokarski, A.; Wang, Y. Interaction of OKL38 and p53 in Regulating Mitochondrial Structure and Function. PLoS ONE 2012, 7, e43362. [Google Scholar] [CrossRef]
- Li, R.; Chen, W.; Yanes, R.; Lee, S.; Berliner, J.A. OKL38 is an oxidative stress response gene stimulated by oxidized phospholipids. J. Lipid Res. 2007, 48, 709–715. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Lee, S.; Gugiu, B.G.; Koroniak, L.; Jung, M.E.; Berliner, J.; Cheng, J.; Li, R. Fatty acid epoxyisoprostane E2 stimulates an oxidative stress response in endothelial cells. Biochem. Biophys. Res. Commun. 2014, 444, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Romanoski, C.E.; Che, N.; Yin, F.; Mai, N.; Pouldar, D.; Civelek, M.; Pan, C.; Lee, S.; Vakili, L.; Yang, W.-P.; et al. Network for Activation of Human Endothelial Cells by Oxidized Phospholipids: A critical role of heme oxygenase 1. Circ. Res. 2011, 109, e27–e41. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Zhu, M.; Liu, X.; Chen, X.; Yuan, Y.; Li, L.; Liu, J.; Lu, Y.; Cheng, J.; Chen, Y. Oleic acid ameliorates palmitic acid induced hepatocellular lipotoxicity by inhibition of ER stress and pyroptosis. Nutr. Metab. 2020, 17, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escoula, Q.; Bellenger, S.; Narce, M.; Bellenger, J. Docosahexaenoic and Eicosapentaenoic Acids Prevent Altered-Muc2 Secretion Induced by Palmitic Acid by Alleviating Endoplasmic Reticulum Stress in LS174T Goblet Cells. Nutrients 2019, 11, 2179. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Lei, L.; Wen, D.; Yang, L. Melatonin attenuates palmitic acid-induced mouse granulosa cells apoptosis via endoplasmic reticulum stress. J. Ovarian Res. 2019, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Miao, L.; Zhang, H.; Wu, G.; Zhang, Z.; Lv, J. Chlorogenic acid against palmitic acid in endoplasmic reticulum stress-mediated apoptosis resulting in protective effect of primary rat hepatocytes. Lipids Health Dis. 2018, 17, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Baumeister, P.; Roy, B.; Phan, T.; Foti, D.; Luo, S.; Lee, A.S. ATF6 as a Transcription Activator of the Endoplasmic Reticulum Stress Element: Thapsigargin Stress-Induced Changes and Synergistic Interactions with NF-Y and YY1. Mol. Cell. Biol. 2000, 20, 5096–5106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.J.; Alam, M.R.; Waldeck-Weiermair, M.; Karsten, F.; Groschner, L.; Riederer, M.; Hallström, S.; Rockenfeller, P.; Konya, V.; Heinemann, A.; et al. Inhibition of Autophagy Rescues Palmitic Acid-induced Necroptosis of Endothelial Cells. J. Biol. Chem. 2012, 287, 21110–21120. [Google Scholar] [CrossRef] [Green Version]
- Lenna, S.; Han, R.; Trojanowska, M. Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life 2014, 66, 530–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icli, B.; Wu, W.; Ozdemir, D.; Li, H.; Cheng, H.S.; Haemmig, S.; Liu, X.; Giatsidis, G.; Avci, S.N.; Lee, N.; et al. MicroRNA-615-5p Regulates Angiogenesis and Tissue Repair by Targeting AKT/eNOS (Protein Kinase B/Endothelial Nitric Oxide Synthase) Signaling in Endothelial Cells. Arter. Thromb. Vasc. Biol. 2019, 39, 1458–1474. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Huang, Y.; Zheng, W.; Yan, J.; Cheng, M.; Zhao, R.; Chen, L.; Hu, C.; Jia, W. Resveratrol reduces intracellular reactive oxygen species levels by inducing autophagy through the AMPK-mTOR pathway. Front. Med. 2018, 12, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-H.Y.A.H.-J.C.W.-S.; Yoo, W.-H.; Chae, H.-J. ER Stress and Autophagy. Curr. Mol. Med. 2015, 15, 735–745. [Google Scholar] [CrossRef]
- Schläfli, A.M.; Adams, O.; Galván, J.A.; Gugger, M.; Savic, S.; Bubendorf, L.; Schmid, R.A.; Becker, K.-F.; Tschan, M.P.; Langer, R.; et al. Prognostic value of the autophagy markers LC3 and p62/SQSTM1 in early-stage non-small cell lung cancer. Oncotarget 2016, 7, 39544–39555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Wilkinson, F.L.; Jones, A.M.; Wilkinson, J.A.; Romero, M.; Duarte, J.; Alexander, M.Y. A novel role for small molecule glycomimetics in the protection against lipid-induced endothelial dysfunction: Involvement of Akt/eNOS and Nrf2/ARE signaling. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3311–3322. [Google Scholar] [CrossRef] [PubMed]
- Wouters, K.; Shiri-Sverdlov, R.; van Gorp, P.; Van Bilsen, M.; Hofker, M.H. Understanding hyperlipidemia and atherosclerosis: Lessons from genetically modified apoe and ldlr mice. Clin. Chem. Lab. Med. 2005, 43, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced Apoptosis Can Occur through a Ceramide-independent Pathway. J. Biol. Chem. 2001, 276, 14890–14895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, W.; Ma, J.; Wang, X.; Yang, W.; Zhang, J.; Ji, Q. Free Fatty Acid Induces Endoplasmic Reticulum Stress and Apoptosis of beta-cells by Ca2+/Calpain-2 Pathways. PLoS ONE 2013, 8, e59921. [Google Scholar] [CrossRef]
- Lu, X.; Drocco, J.; Wieschaus, E.F. Cell cycle regulation via inter-nuclear communication during the early embryonic development of Drosophila melanogaster. Cell Cycle 2010, 9, 2908–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F. Autophagy in vascular endothelial cells. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1021–1028. [Google Scholar] [CrossRef]
- Vion, A.-C.; Kheloufi, M.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc. Natl. Acad. Sci. USA 2017, 114, E8675–E8684. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Liu, Y.; Li, B.; Zheng, Z.; Ke, X.; Hao, Y.; Li, X.; Li, X.; Liu, F.; Zhang, Z. Autophagy attenuates endothelial-to-mesenchymal transition by promoting Snail degradation in human cardiac microvascular endothelial cells. Biosci. Rep. 2017, 37, BSR20171049. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Yoo, J.H.; Kim, H.S.; Cho, Y.K.; La Lee, Y.; Lee, W.J.; Park, J.-Y.; Jung, C.H. C1q/TNF-related protein-9 attenuates palmitic acid-induced endothelial cell senescence via increasing autophagy. Mol. Cell. Endocrinol. 2021, 521, 111114. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Xiao, Q.; Chen, M.; Margariti, A.; Martin, D.; Ivetic, A.; Xu, H.; Mason, J.; Wang, W.; Cockerill, G.; et al. Vascular Endothelial Cell Growth—Activated XBP1 Splicing in Endothelial Cells Is Crucial for Angiogenesis. Circulation 2013, 127, 1712–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Qimuge, A.; Wang, H.; Xing, C.; Gu, Y.; Liu, S.; Xu, H.; Hu, M.; Song, L. IRE1α/XBP1s branch of UPR links HIF1α activation to mediate ANGII-dependent endothelial dysfunction under particulate matter (PM) 2.5 exposure. Sci. Rep. 2017, 7, 13507. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Qian, Q.; Li, M.; Shao, F.; Ding, W.-X.; Lira, V.A.; Chen, S.X.; Sebag, S.C.; Hotamisligil, G.S.; Cao, H.; et al. The unfolded protein response regulates hepatic autophagy by sXBP1-mediated activation of TFEB. Autophagy 2021, 17, 1841–1855. [Google Scholar] [CrossRef]
- Sharma, M.; Bhattacharyya, S.; Sharma, K.B.; Chauhan, S.; Asthana, S.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J. Gen. Virol. 2017, 98, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, P.; Ding, S.-Y.; Sun, T.; Liu, L.; Han, S.; DeLeo, A.B.; Sadagopan, A.; Guo, W.; Wang, X. Induction of autophagy-dependent apoptosis in cancer cells through activation of ER stress: An uncovered anti-cancer mechanism by anti-alcoholism drug disulfiram. Am. J. Cancer Res. 2019, 9, 1266–1281. [Google Scholar] [PubMed]
- Zhao, Y.; Li, Y.; Luo, P.; Gao, Y.; Yang, J.; Lao, K.-H.; Wang, G.; Cockerill, G.; Hu, Y.; Xu, Q.; et al. XBP1 splicing triggers miR-150 transfer from smooth muscle cells to endothelial cells via extracellular vesicles. Sci. Rep. 2016, 6, 28627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Guo, Y.; Zeng, W.; Huang, L.; Pang, Q.; Nie, L.; Mu, J.; Yuan, F.; Feng, B. ER stress triggers MCP-1 expression through SET7/9-induced histone methylation in the kidneys of db/db mice. Am. J. Physiol. Physiol. 2014, 306, F916–F925. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Li, Y.; Chen, L.; Chan, T.H.M.; Song, Y.; Fu, L.; Zeng, T.; Dai, Y.; Zhu, Y.; Chen, J.; et al. Allele-Specific Imbalance of Oxidative Stress-Induced Growth Inhibitor 1 Associates with Progression of Hepatocellular Carcinoma. Gastroenterol. 2014, 146, 1084–1096.e5. [Google Scholar] [CrossRef]
- Tsai, C.-H.; Lii, C.-K.; Wang, T.-S.; Liu, K.-L.; Chen, H.-W.; Huang, C.-S.; Li, C.-C. Docosahexaenoic acid promotes the formation of autophagosomes in MCF-7 breast cancer cells through oxidative stress-induced growth inhibitor 1 mediated activation of AMPK/mTOR pathway. Food Chem. Toxicol. 2021, 154, 112318. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Zhu, H.; Liu, H.; Wang, M.; Chu, H.; Zhang, Z. METTL3 regulates PM2.5-induced cell injury by targeting OSGIN1 in human airway epithelial cells. J. Hazard. Mater. 2021, 415, 125573. [Google Scholar] [CrossRef]
- Gao, M.; Li, C.; Xu, M.; Liu, Y.; Liu, S. LncRNA UCA1 attenuates autophagy-dependent cell death through blocking autophagic flux under arsenic stress. Toxicol. Lett. 2018, 284, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.-J.; Zhao, L.-M.; Pang, Z.-D.; She, G.; Song, Z.; Cheng, X.; Du, X.-J.; Deng, X.-L. Oxidative stress induced by palmitic acid modulates KCa2.3 channels in vascular endothelium. Exp. Cell Res. 2019, 383, 111552. [Google Scholar] [CrossRef]
- Li, X.; Jin, S.-J.; Su, J.; Li, X.-X.; Xu, M. Acid Sphingomyelinase Down-regulation Alleviates Vascular Endothelial Insulin Resistance in Diabetic Rats. Basic Clin. Pharmacol. Toxicol. 2018, 123, 645–659. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Yang, Q.; Ma, W.; Li, J.; Qu, L.-Z.; Wang, M. Protocatechuic Acid Ameliorated Palmitic-Acid-Induced Oxidative Damage in Endothelial Cells through Activating Endogenous Antioxidant Enzymes via an Adenosine-Monophosphate-Activated-Protein-Kinase-Dependent Pathway. J. Agric. Food Chem. 2018, 66, 10400–10409. [Google Scholar] [CrossRef]
- Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Münzel, T.; Li, H. New Therapeutic Implications of Endothelial Nitric Oxide Synthase (eNOS) Function/Dysfunction in Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.; Yang, Y.; Wei, S.; Huang, X.; Peng, Z.; Ke, X.; Zeng, Z.; Song, Y. Hydrogen Sulfide Protects Against High Glucose-Induced Human Umbilical Vein Endothelial Cell Injury Through Activating PI3K/Akt/eNOS Pathway. Drug Des. Dev. Ther. 2020, 14, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Duan, M.-X.; Zhou, H.; Wu, Q.-Q.; Liu, C.; Xiao, Y.; Deng, W.; Tang, Q.-Z. Andrographolide Protects against HG-Induced Inflammation, Apoptosis, Migration, and Impairment of Angiogenesis via PI3K/AKT-eNOS Signalling in HUVECs. Mediat. Inflamm. 2019, 2019, 6168340. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Mueller, R.; Li, Y.; Ruan, T.; Kunz, D.; Goodrich, R.; Mills, T.; Deeter, L.; Sargsyan, A.; Babu, P.V.A.; et al. Impairment of autophagy in endothelial cells prevents shear-stress-induced increases in nitric oxide bioavailability. Can. J. Physiol. Pharmacol. 2014, 92, 605–612. [Google Scholar] [CrossRef]
- Guo, F.; Li, X.; Peng, J.; Tang, Y.; Yang, Q.; Liu, L.; Wang, Z.; Jiang, Z.; Xiao, M.; Ni, C.; et al. Autophagy Regulates Vascular Endothelial Cell eNOS and ET-1 Expression Induced by Laminar Shear Stress in an Ex Vivo Perfused System. Ann. Biomed. Eng. 2014, 42, 1978–1988. [Google Scholar] [CrossRef]
- Liu, D.; Sun, W.-P.; Chen, J.-W.; Jiang, Y.; Xue, R.; Wang, L.-H.; Murao, K.; Zhang, G.-X. Autophagy contributes to angiotensin II induced dysfunction of HUVECs. Clin. Exp. Hypertens. 2021, 43, 462–473. [Google Scholar] [CrossRef]
- Shao, J.; Miao, C.; Geng, Z.; Gu, M.; Wu, Y.; Li, Q. Effect of eNOS on Ischemic Postconditioning-Induced Autophagy against Ischemia/Reperfusion Injury in Mice. BioMed Res. Int. 2019, 2019, 5201014. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis as an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoi, C.-S.; Xiao, C.-Q.; Hung, K.-Y.; Lin, T.-Y.; Chiang, C.-K. Oxidative Stress-Induced Growth Inhibitor (OSGIN1), a Target of X-Box-Binding Protein 1, Protects Palmitic Acid-Induced Vascular Lipotoxicity through Maintaining Autophagy. Biomedicines 2022, 10, 992. https://doi.org/10.3390/biomedicines10050992
Khoi C-S, Xiao C-Q, Hung K-Y, Lin T-Y, Chiang C-K. Oxidative Stress-Induced Growth Inhibitor (OSGIN1), a Target of X-Box-Binding Protein 1, Protects Palmitic Acid-Induced Vascular Lipotoxicity through Maintaining Autophagy. Biomedicines. 2022; 10(5):992. https://doi.org/10.3390/biomedicines10050992
Chicago/Turabian StyleKhoi, Chong-Sun, Cai-Qin Xiao, Kuan-Yu Hung, Tzu-Yu Lin, and Chih-Kang Chiang. 2022. "Oxidative Stress-Induced Growth Inhibitor (OSGIN1), a Target of X-Box-Binding Protein 1, Protects Palmitic Acid-Induced Vascular Lipotoxicity through Maintaining Autophagy" Biomedicines 10, no. 5: 992. https://doi.org/10.3390/biomedicines10050992