
Machine Learning-Based Retention Time Prediction of Trimethylsilyl Derivatives of Metabolites

 , and
, and 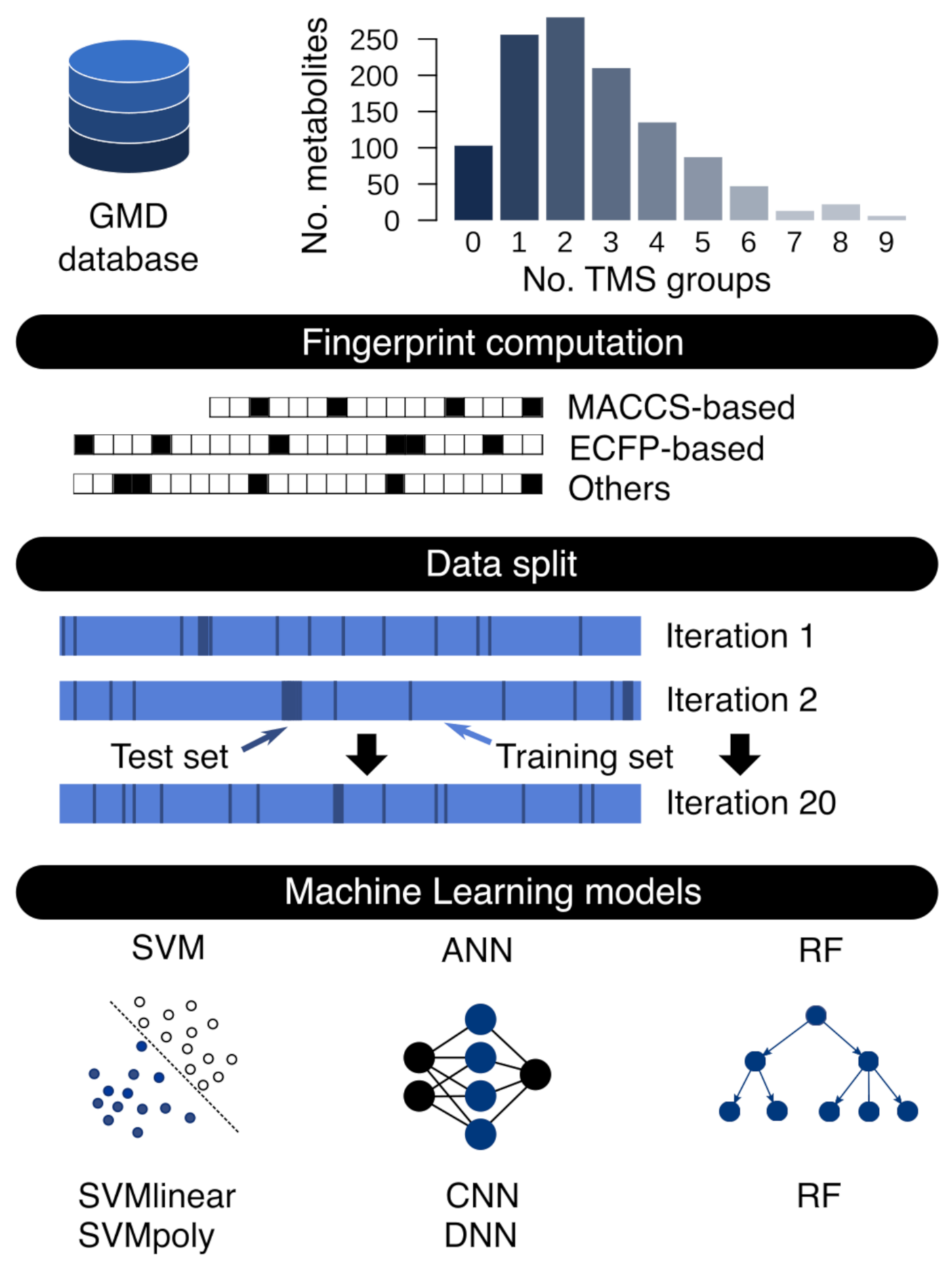{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
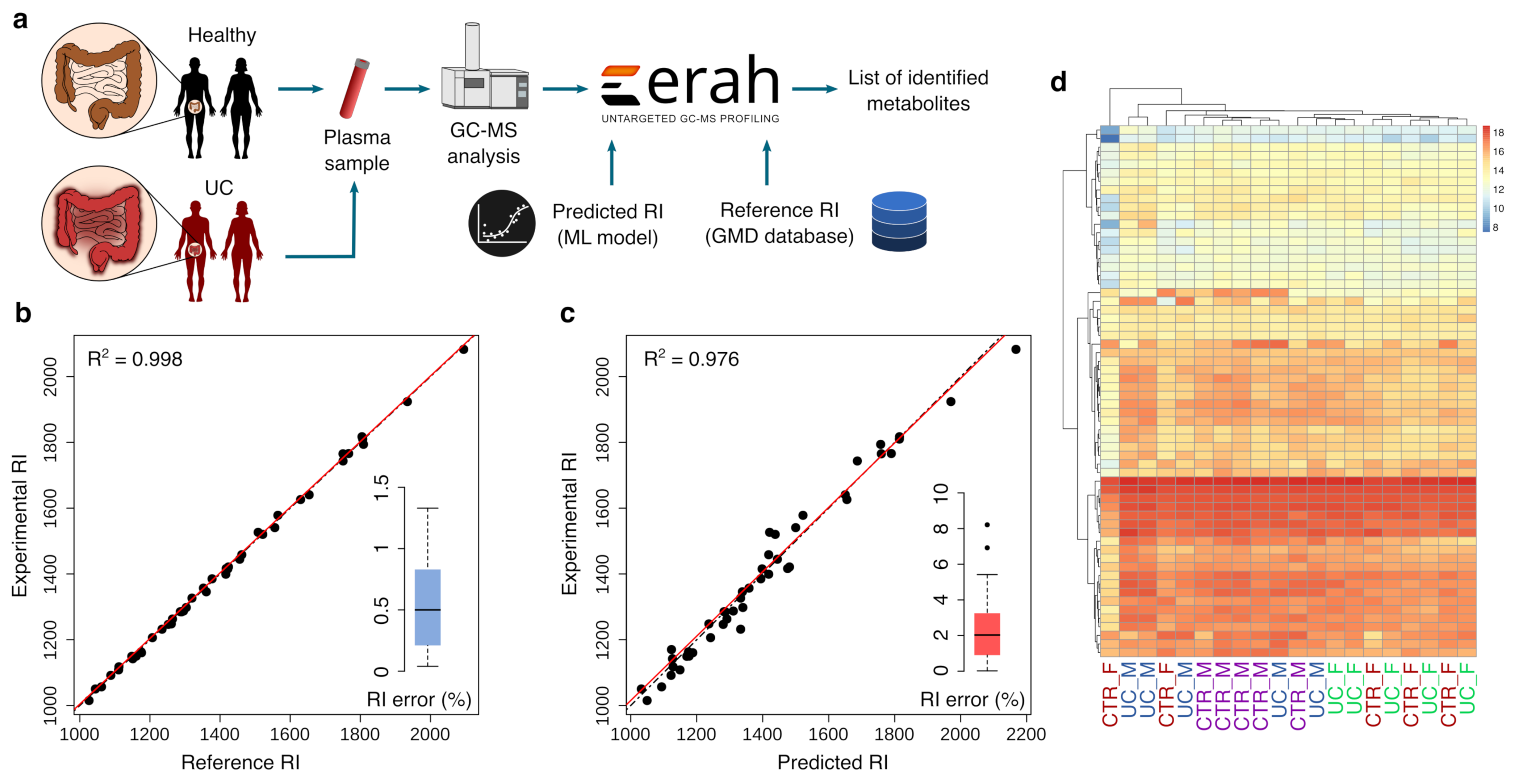
Abstract
:1. Introduction
2. Methods
2.1. Fingerprint Calculation and Dataset Generation
2.2. Machine Learning Model Training and Test
2.3. GC-MS Plasma Analysis
3. Results
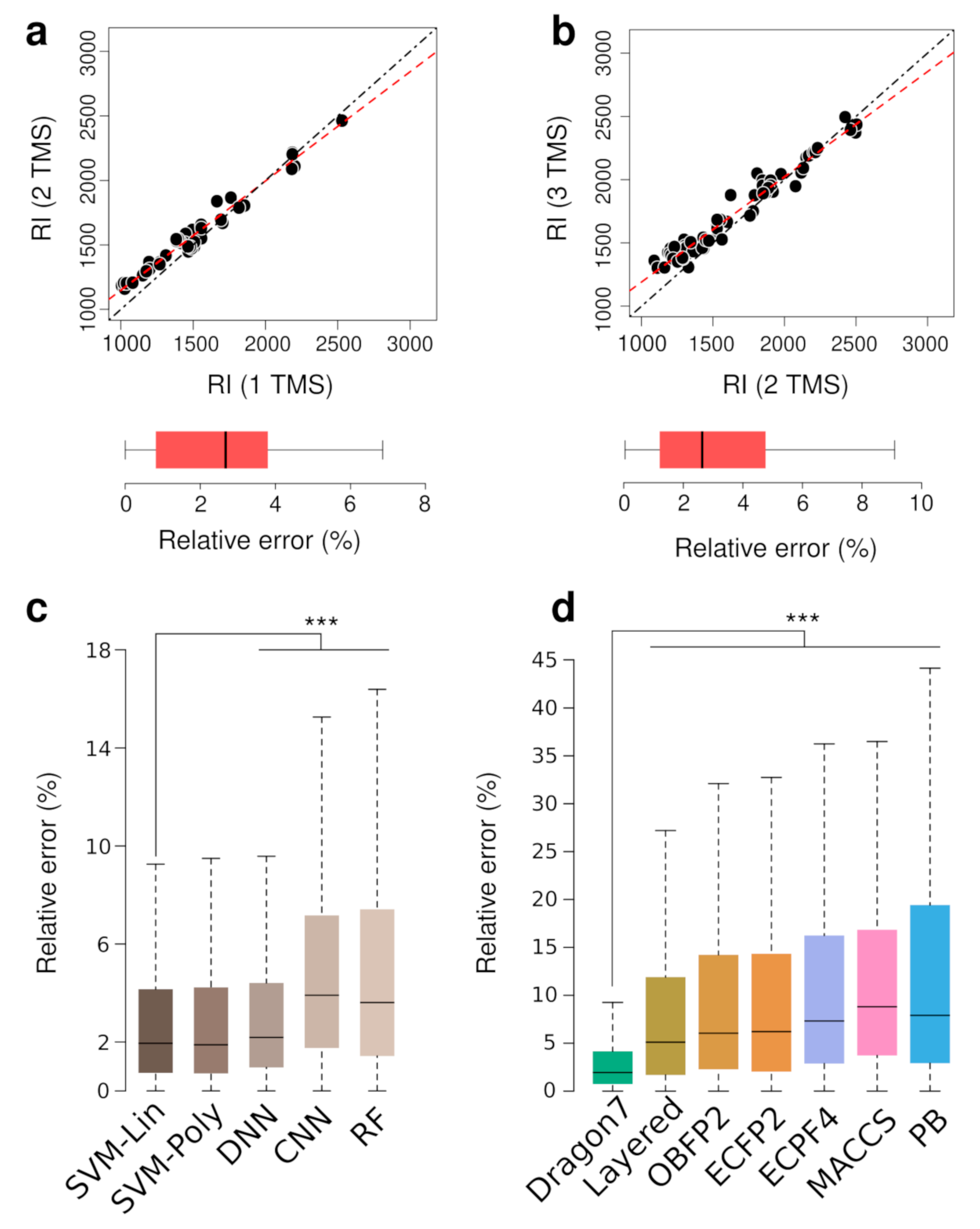
3.1. TMS Derivatives of Metabolites RI Prediction via Machine Learning Models
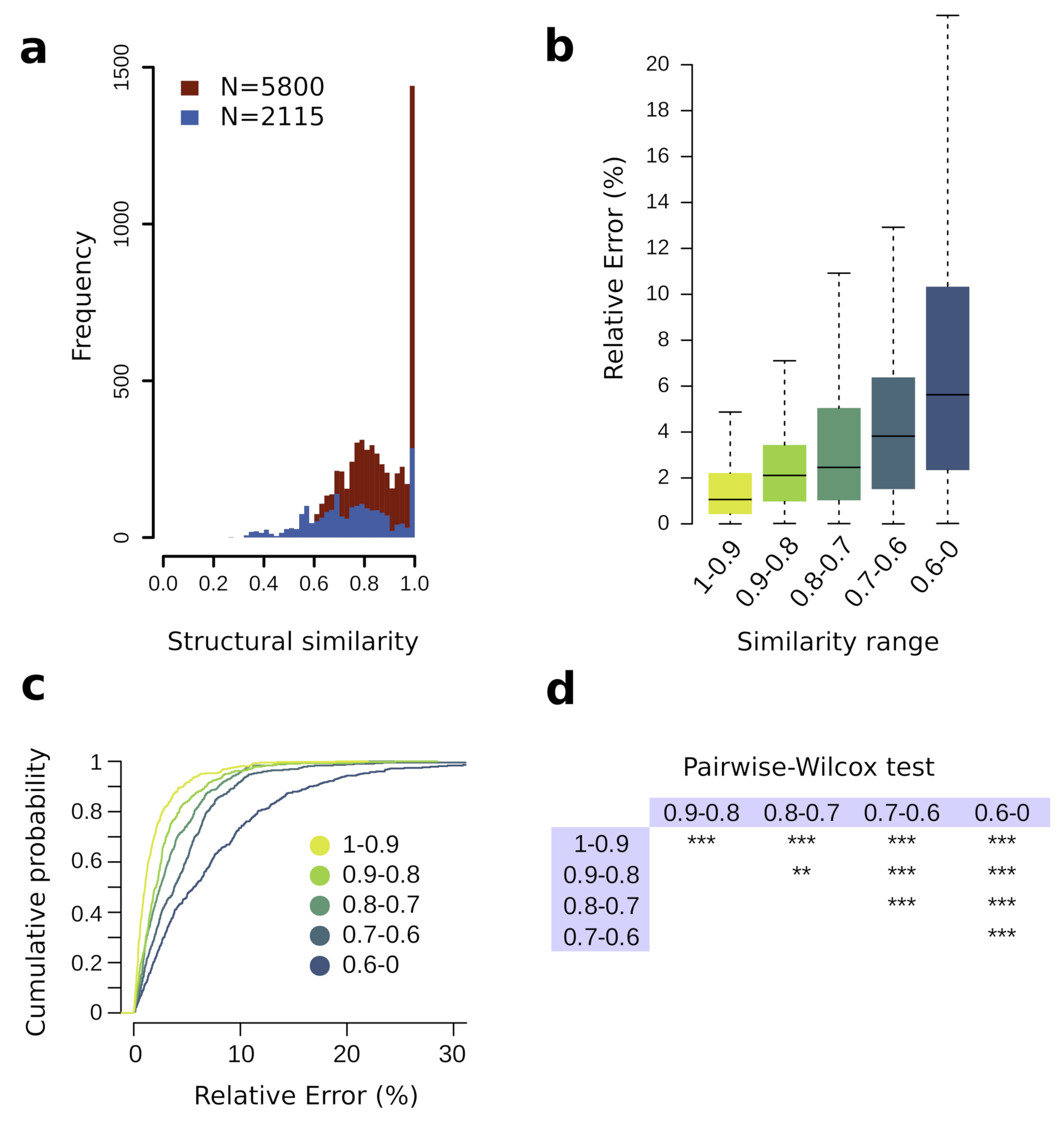
3.2. Training Set Structural Similarity Influence on Prediction Performance
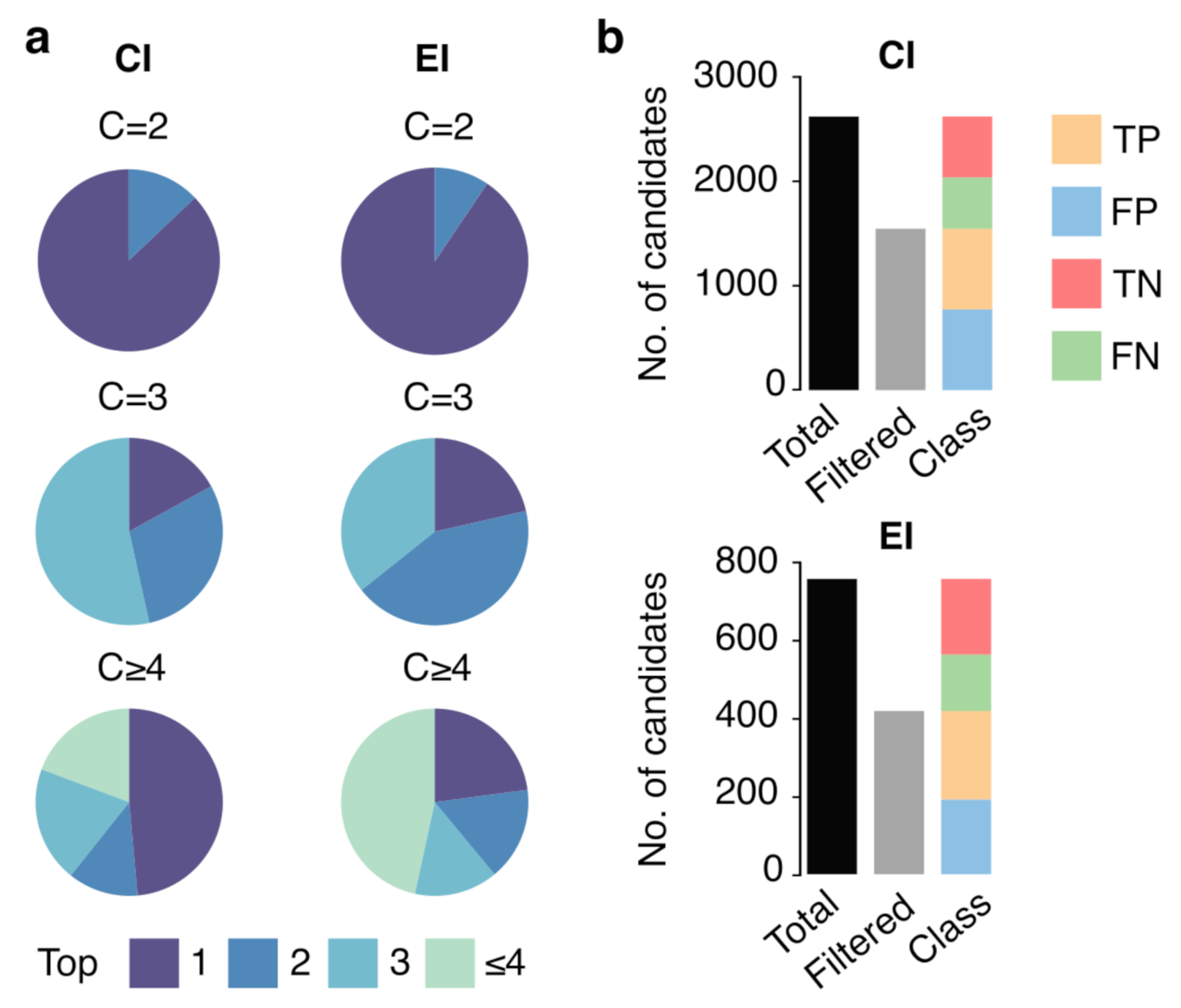
3.3. Ranking and Filtering Capability of the ML Model
3.4. Application of the Prediction Model for Metabolite Identification in Plasma from Patients with Ulcerative Colitis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinschen, M.M.; Ivanisevic, J.; Giera, M.; Siuzdak, G. Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell Biol. 2019, 20, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Capellades, J.; Junza, A.; Samino, S.; Brunner, J.S.; Schabbauer, G.; Vinaixa, M.; Yanes, O. Exploring the Use of Gas Chromatography Coupled to Chemical Ionization Mass Spectrometry (GC-CI-MS) for Stable Isotope Labeling in Metabolomics. Anal. Chem. 2021, 93, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Almenara, X.; Montenegro-Burke, J.R.; Ivanisevic, J.; Thomas, A.; Sidibé, J.; Teav, T.; Guijas, C.; Aisporna, A.E.; Rinehart, D.; Hoang, L.; et al. XCMS-MRM and METLIN-MRM: A cloud library and public resource for targeted analysis of small molecules. Nat. Methods 2018, 15, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC–MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass Spectral and Retention Index Libraries for Metabolomics Based on Quadrupole and Time-of-Flight Gas Chromatography/Mass Spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Almenara, X.; Brezmes, J.; Venturini, G.; Vivó-Truyols, G.; Perera, A.; Vinaixa, M. Baitmet, a computational approach for GC–MS library-driven metabolite profiling. Metabolomics 2017, 13, 93. [Google Scholar] [CrossRef] [Green Version]
- Vrzal, T.; Malečková, M.; Olšovská, J. DeepReI: Deep learning-based gas chromatographic retention index predictor. Anal. Chim. Acta 2021, 1147, 64–71. [Google Scholar] [CrossRef]
- Matyushin, D.D.; Buryak, A.K. Gas Chromatographic Retention Index Prediction Using Multimodal Machine Learning. IEEE Access 2020, 8, 223140–223155. [Google Scholar] [CrossRef]
- Dossin, E.; Martin, E.; Diana, P.; Castellon, A.; Monge, A.; Pospisil, P.; Bentley, M.; Guy, P.A. Prediction Models of Retention Indices for Increased Confidence in Structural Elucidation during Complex Matrix Analysis: Application to Gas Chromatography Coupled with High-Resolution Mass Spectrometry. Anal. Chem. 2016, 88, 7539–7547. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Lei, Z.; Sumner, L.W. MetExpert: An expert system to enhance gas chromatography‐mass spectrometry-based metabolite identifications. Anal. Chim. Acta 2018, 1037, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Veenaas, C.; Linusson, A.; Haglund, P. Retention-time prediction in comprehensive two-dimensional gas chromatography to aid identification of unknown contaminants. Anal. Bioanal. Chem. 2018, 410, 7931–7941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Li, Y.; Han, L.; Li, J.; Liu, J.; Zhao, Z.; Nie, W.; Liu, Y.; Wang, R. PDB-wide collection of binding data: Current status of the PDBbind database. Bioinformatics 2015, 31, 405–412. [Google Scholar] [CrossRef]
- Domingo-Almenara, X.; Guijas, C.; Billings, E.; Montenegro-Burke, J.R.; Uritboonthai, W.; Aisporna, A.E.; Chen, E.; Benton, H.P.; Siuzdak, G. The METLIN small molecule dataset for machine learning-based retention time prediction. Nat. Commun. 2019, 10, 5811. [Google Scholar] [CrossRef] [PubMed]
- Zarate, E.; Boyle, V.; Rupprecht, U.; Green, S.; Villas-Boas, S.G.; Baker, P.; Pinu, F.R. Fully Automated Trimethylsilyl (TMS) Derivatisation Protocol for Metabolite Profiling by GC-MS. Metabolites 2016, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Fiehn, O.; Garvey, W.T.; Newman, J.W.; Lok, K.H.; Hoppel, C.L.; Adams, S.H. Plasma metabolomic profiles reflective of glucose homeostasis in non-diabetic and type 2 diabetic obese African-American women. PLoS ONE 2010, 5, e15234. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S.; Stevens, D.; Kind, T.; Denkert, C.; Fiehn, O. Applying in-silico retention index and mass spectra matching for identification of unknown metabolites in accurate mass GC-TOF mass spectrometry. Anal. Chem. 2011, 83, 5895–5902. [Google Scholar] [CrossRef] [Green Version]
- Fiehn, O. Metabolomics by Gas Chromatography-Mass Spectrometry: Combined Targeted and Untargeted Profiling. Curr. Protoc. Mol. Biol. 2016, 114, 30.4.1–30.4.32. [Google Scholar] [CrossRef]
- Héberger, K. Quantitative structure-(chromatographic) retention relationships. J. Chromatogr. A 2007, 1158, 273–305. [Google Scholar] [CrossRef] [Green Version]
- Zhokhov, A.K.; Loskutov, A.Y.; Rybal’chenko, I.V. Methodological Approaches to the Calculation and Prediction of Retention Indices in Capillary Gas Chromatography. J. Anal. Chem. 2018, 73, 207–220. [Google Scholar] [CrossRef]
- Matyushin, D.D.; Sholokhova, A.Y.; Buryak, A.K. A deep convolutional neural network for the estimation of gas chromatographic retention indices. J. Chromatogr. A 2019, 1607, 460395. [Google Scholar] [CrossRef] [PubMed]
- Samaraweera, M.A.; Hall, L.M.; Hill, D.W.; Grant, D.F. Evaluation of an Artificial Neural Network Retention Index Model for Chemical Structure Identification in Nontargeted Metabolomics. Anal. Chem. 2018, 90, 12752–12760. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef]
- Lantz, B. Machine Learning with R, 2nd ed.; Packt Publishing: Birmingham, UK, 2015. [Google Scholar]
- Chambers, M.C.; MacLean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Taraji, M.; Haddad, P.R.; Amos, R.I.J.; Talebi, M.; Szucs, R.; Dolan, J.W.; Pohl, C.A. Use of dual-filtering to create training sets leading to improved accuracy in quantitative structure-retention relationships modelling for hydrophilic interaction liquid chromatographic systems. J. Chromatogr. A 2017, 1507, 53–62. [Google Scholar] [CrossRef]
- Wen, Y.; Amos, R.I.J.; Talebi, M.; Szucs, R.; Dolan, J.W.; Pohl, C.A.; Haddad, P.R. Retention Index Prediction Using Quantitative Structure-Retention Relationships for Improving Structure Identification in Nontargeted Metabolomics. Anal. Chem. 2018, 90, 9434–9440. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Goudarzi, N.; Shahsavani, D.; Emadi-Gandaghi, F.; Chamjangali, M.A. Application of random forests method to predict the retention indices of some polycyclic aromatic hydrocarbons. J. Chromatogr. A 2014, 1333, 25–31. [Google Scholar] [CrossRef]
- Goudarzi, N.; Shahsavani, D.; Emadi-Gandaghi, F.; Chamjangali, M.A. Quantitative structure-property relationships of retention indices of some sulfur organic compounds using random forest technique as a variable selection and modeling method. J. Sep. Sci. 2016, 39, 3835–3842. [Google Scholar] [CrossRef]
- Matyushin, D.D.; Sholokhova, A.Y.; Karnaeva, A.E.; Buryak, A.K. Various aspects of retention index usage for GC-MS library search: A statistical investigation using a diverse data set. Chemom. Intell. Lab. Syst. 2020, 202, 104042. [Google Scholar] [CrossRef]
- Landrum, G. RDKit (Open-Source Cheminformatics Software). 2021. Available online: https://www.rdkit.org/ (accessed on 1 March 2022).
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalke, A. The chemfp project. J. Cheminformatics 2019, 11, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheridan, R.P.; Feuston, B.P.; Maiorov, V.N.; Kearsley, S.K. Similarity to molecules in the training set is a good discriminator for prediction accuracy in QSAR. J. Chem. Inf. Comput. Sci. 2004, 44, 1912–1928. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Almenara, X.; Brezmes, J.; Vinaixa, M.; Samino, S.; Ramirez, N.; Ramon-Krauel, M.; Lerin, C.; Díaz, M.; Ibáñez, L.; Correig, X.; et al. eRah: A Computational Tool Integrating Spectral Deconvolution and Alignment with Quantification and Identification of Metabolites in GC/MS-Based Metabolomics. Anal. Chem. 2016, 88, 9821–9829. [Google Scholar] [CrossRef]
- Domingo-Almenara, X.; Montenegro-Burke, J.R.; Guijas, C.; Majumder, E.L.; Benton, H.P.; Siuzdak, G. Autonomous METLIN-Guided In-source Fragment Annotation for Untargeted Metabolomics. Anal. Chem. 2019, 91, 3246–3253. [Google Scholar] [CrossRef]
- Scheubert, K.; Hufsky, F.; Petras, D.; Wang, M.; Nothias, L.F.; Dührkop, K.; Bandeira, N.; Dorrestein, P.C.; Böcker, S. Significance estimation for large scale metabolomics annotations by spectral matching. Nat. Commun. 2017, 8, 1494. [Google Scholar] [CrossRef] [Green Version]
- Vinaixa, M.; Samino, S.; Saez, I.; Duran, J.; Guinovart, J.J.; Yanes, O. A Guideline to Univariate Statistical Analysis for LC/MS-Based Untargeted Metabolomics-Derived Data. Metabolites 2012, 2, 775–795. [Google Scholar] [CrossRef]
- Opialla, T.; Kempa, S.; Pietzke, M. Towards a More Reliable Identification of Isomeric Metabolites Using Pattern Guided Retention Validation. Metabolites 2020, 10, 457. [Google Scholar] [CrossRef]
- Strehmel, N.; Hummel, J.; Erban, A.; Strassburg, K.; Kopka, J. Retention index thresholds for compound matching in GC-MS metabolite profiling. J. Chromatogr. Anal. Technol. Biomed. Life Sci. 2008, 871, 182–190. [Google Scholar] [CrossRef]
- Yang, Q.; Ji, H.; Lu, H.; Zhang, Z. Prediction of Liquid Chromatographic Retention Time with Graph Neural Networks to Assist in Small Molecule Identification. Anal. Chem. 2021, 93, 2200–2206. [Google Scholar] [CrossRef] [PubMed]
- Riniker, S.; Landrum, G.A. Open-source platform to benchmark fingerprints for ligand-based virtual screening. J. Cheminformatics 2013, 5, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- K.srl.Dragon (Software for Molecular Descriptor Calculation) Version 7.0. Available online: https://chm.kode-solutions.net/ (accessed on 1 March 2022).
- Dalke, A. ChemFP (Software for Fingerprint Generation and High-Performance Similarity Search). Available online: https://chemfp.com/ (accessed on 1 March 2022).





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Cripan, S.M.; Cereto-Massagué, A.; Herrero, P.; Barcaru, A.; Canela, N.; Domingo-Almenara, X. Machine Learning-Based Retention Time Prediction of Trimethylsilyl Derivatives of Metabolites. Biomedicines 2022, 10, 879. https://doi.org/10.3390/biomedicines10040879
de Cripan SM, Cereto-Massagué A, Herrero P, Barcaru A, Canela N, Domingo-Almenara X. Machine Learning-Based Retention Time Prediction of Trimethylsilyl Derivatives of Metabolites. Biomedicines. 2022; 10(4):879. https://doi.org/10.3390/biomedicines10040879
Chicago/Turabian Stylede Cripan, Sara M., Adrià Cereto-Massagué, Pol Herrero, Andrei Barcaru, Núria Canela, and Xavier Domingo-Almenara. 2022. "Machine Learning-Based Retention Time Prediction of Trimethylsilyl Derivatives of Metabolites" Biomedicines 10, no. 4: 879. https://doi.org/10.3390/biomedicines10040879