
Some Insights into the Regulation of Cardiac Physiology and Pathology by the Hippo Pathway

 , , , , , , and
, , , , , , and 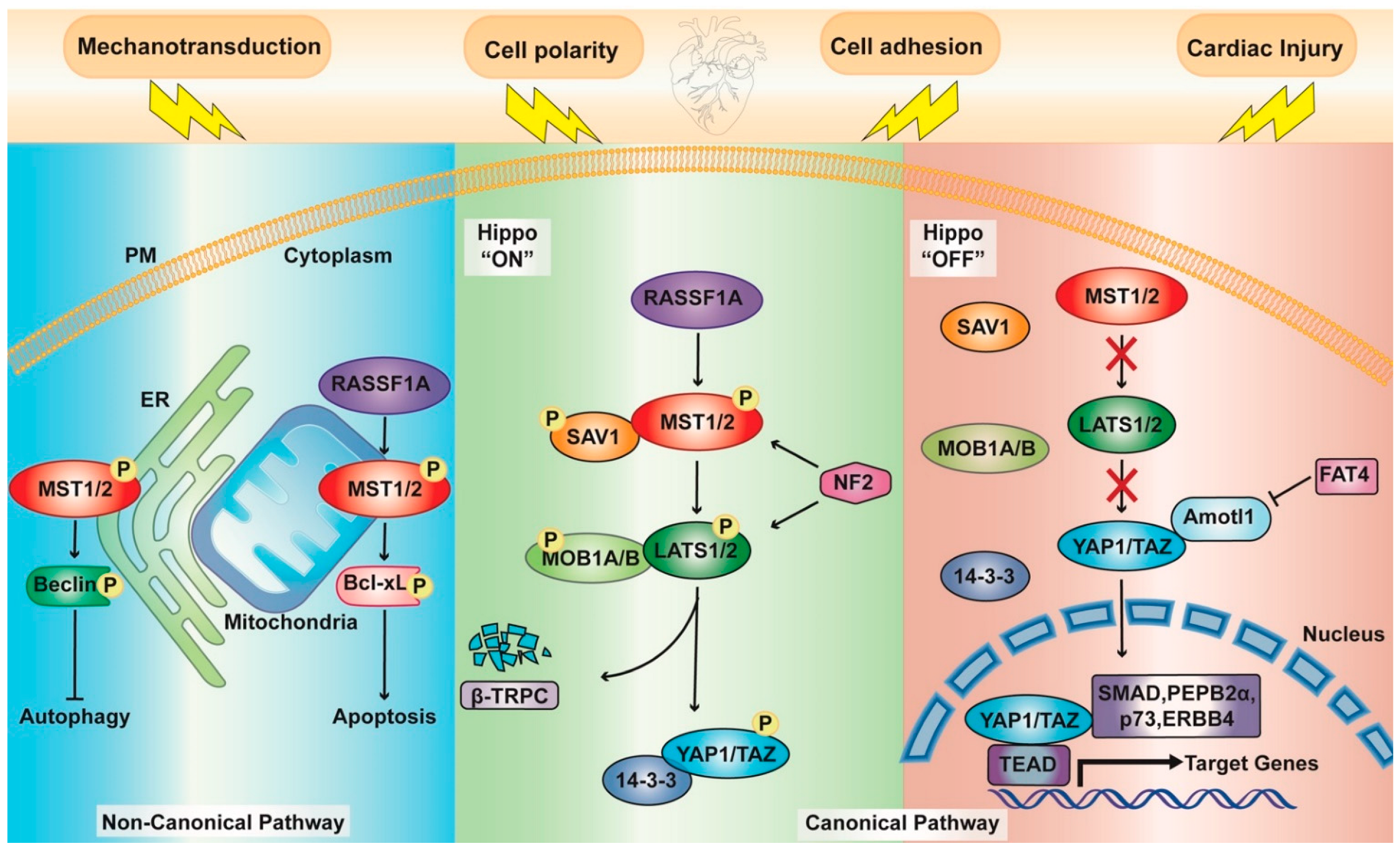{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Hippo Pathway Components
1.2. Upstream Effectors
1.3. A Noncanonical Point of View
2. The Hippo Pathway in Cardiac Physiology
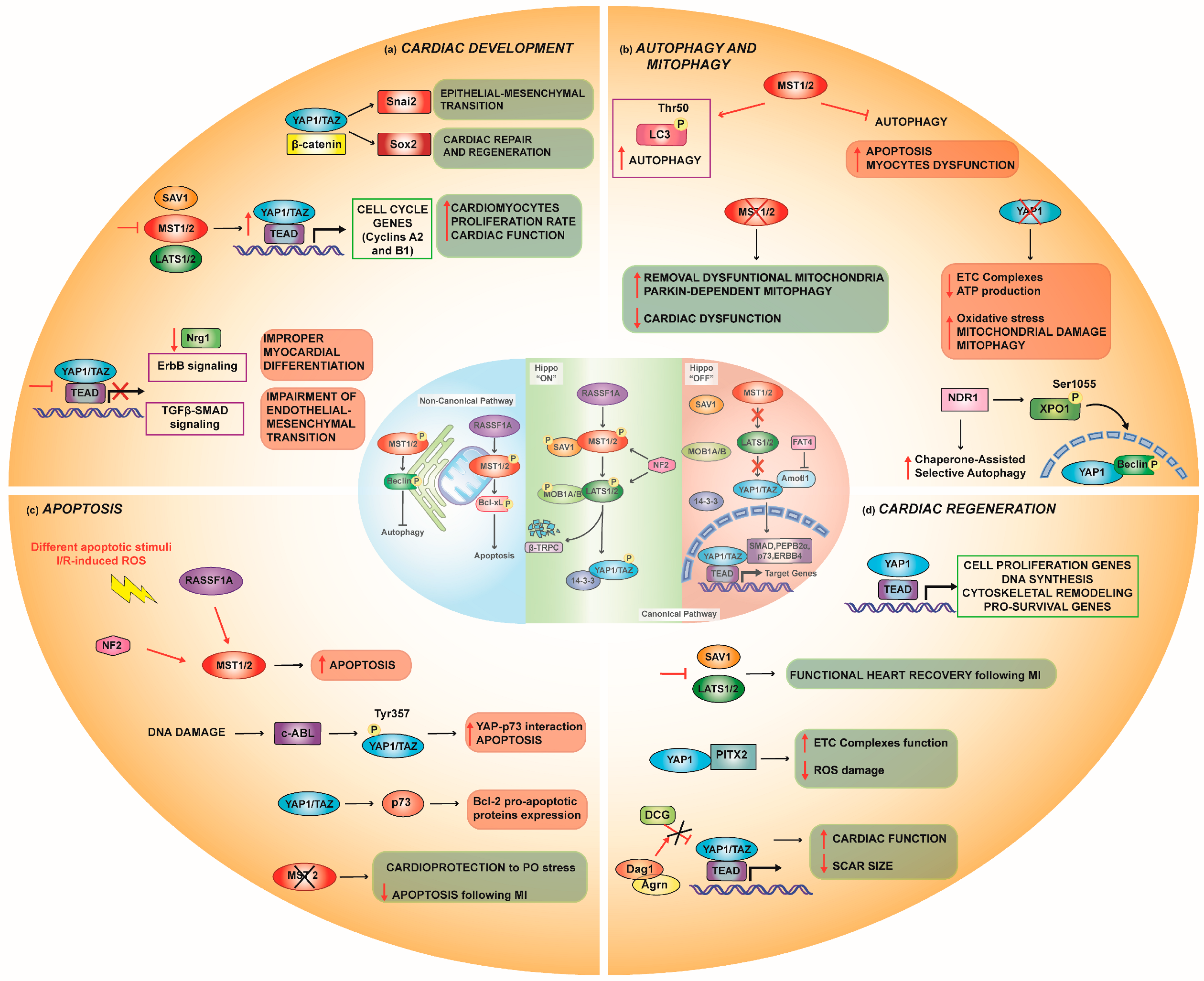
2.1. Cardiac Development
2.1.1. Interaction with Other Pathways in Cardiac Development
Wnt/β-Catenin Pathway
IGF Pathway
TGF-β-SMAD Pathway
RASSF1A Signaling
2.2. Autophagy
Mitophagy
2.3. Apoptosis
3. The Hippo Pathway in Cardiac Diseases
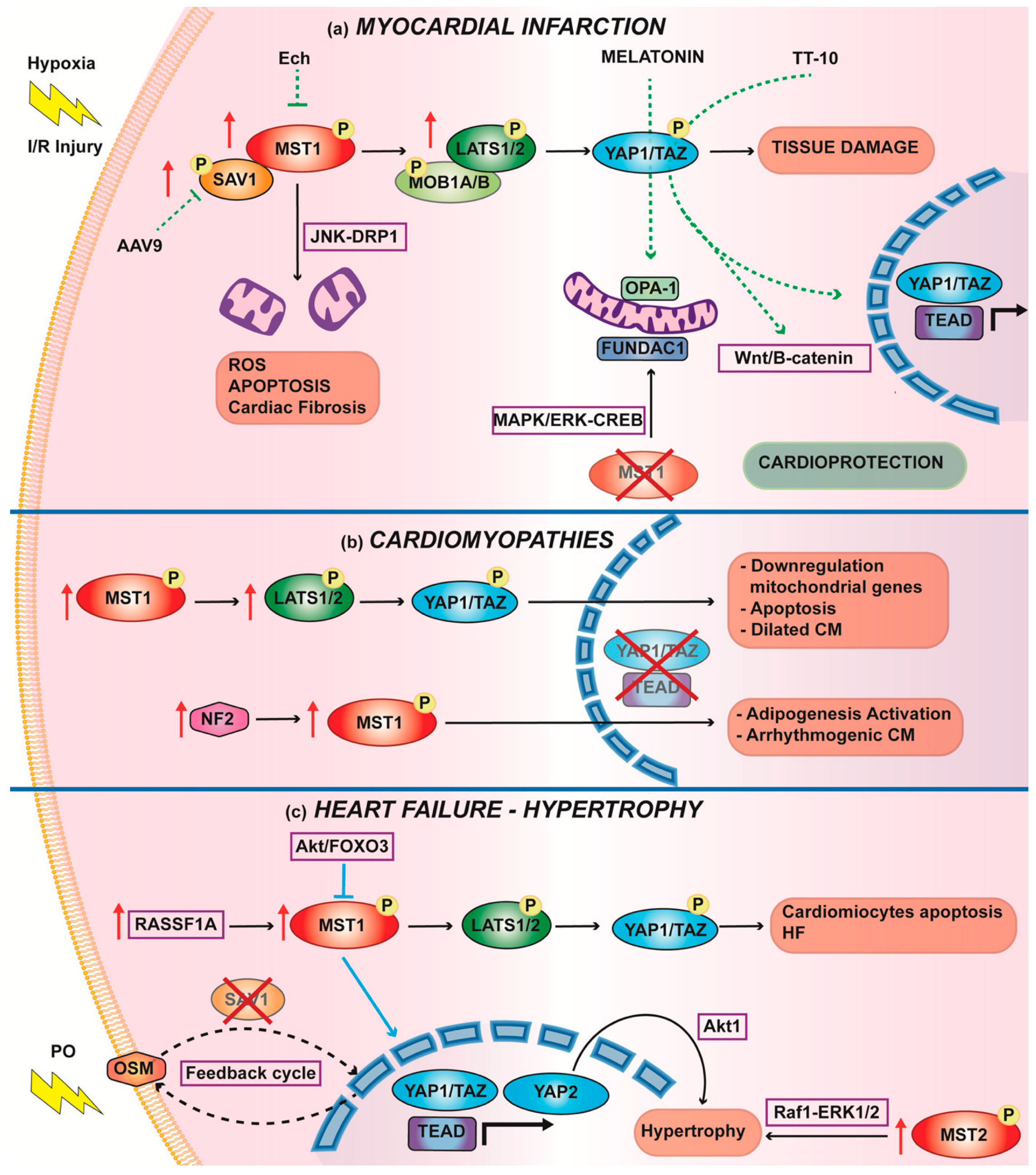
3.1. Myocardial Infarction
Therapies to Increase Nuclear YAP1 to Counteract IRI
3.2. Cardiomyopathies
3.3. Hypertrophy and Heart Failure
4. Cardiac Regeneration
5. Contribution of the Hippo Pathway in Inflammatory States
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xu, T.; Wang, W.; Zhang, S.; Stewart, R.A.; Yu, W. Identifying Tumor Suppressors in Genetic Mosaics: The Drosophila Lats Gene Encodes a Putative Protein Kinase. Development 1995, 121, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Zhao, B.; Guan, K.-L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.; Halder, G. The Two Faces of Hippo: Targeting the Hippo Pathway for Regenerative Medicine and Cancer Treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [Green Version]
- Harvey, K.F.; Pfleger, C.M.; Hariharan, I.K. The Drosophila Mst Ortholog, Hippo, Restricts Growth and Cell Proliferation and Promotes Apoptosis. Cell 2003, 114, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila Tumor Suppressor Gene Warts Encodes a Homolog of Human Myotonic Dystrophy Kinase and Is Required for the Control of Cell Shape and Proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morciano, G.; Vezzani, B.; Missiroli, S.; Boncompagni, C.; Pinton, P.; Giorgi, C. An Updated Understanding of the Role of YAP in Driving Oncogenic Responses. Cancers 2021, 13, 3100. [Google Scholar] [CrossRef]
- Halder, G.; Johnson, R.L. Hippo Signaling: Growth Control and Beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-Y.; Zha, Z.-Y.; Zhou, X.; Zhang, H.; Huang, W.; Zhao, D.; Li, T.; Chan, S.W.; Lim, C.J.; Hong, W.; et al. The Hippo Tumor Pathway Promotes TAZ Degradation by Phosphorylating a Phosphodegron and Recruiting the SCF{beta}-TrCP E3 Ligase. J. Biol. Chem. 2010, 285, 37159–37169. [Google Scholar] [CrossRef] [Green Version]
- Ardestani, A.; Lupse, B.; Maedler, K. Hippo Signaling: Key Emerging Pathway in Cellular and Whole-Body Metabolism. Trends Endocrinol. Metab. 2018, 29, 492–509. [Google Scholar] [CrossRef]
- Ferrigno, O.; Lallemand, F.; Verrecchia, F.; L’Hoste, S.; Camonis, J.; Atfi, A.; Mauviel, A. Yes-Associated Protein (YAP65) Interacts with Smad7 and Potentiates Its Inhibitory Activity against TGF-Beta/Smad Signaling. Oncogene 2002, 21, 4879–4884. [Google Scholar] [CrossRef] [Green Version]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Yaffe, M.B.; Zandstra, P.W.; Wrana, J.L. TAZ Controls Smad Nucleocytoplasmic Shuttling and Regulates Human Embryonic Stem-Cell Self-Renewal. Nat. Cell Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.; Zaromytidou, A.-I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs Drive Smad Transcriptional Activation and Turnover in BMP and TGF-Beta Pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strano, S.; Munarriz, E.; Rossi, M.; Castagnoli, L.; Shaul, Y.; Sacchi, A.; Oren, M.; Sudol, M.; Cesareni, G.; Blandino, G. Physical Interaction with Yes-Associated Protein Enhances P73 Transcriptional Activity. J. Biol. Chem. 2001, 276, 15164–15173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komuro, A.; Nagai, M.; Navin, N.E.; Sudol, M. WW Domain-Containing Protein YAP Associates with ErbB-4 and Acts as a Co-Transcriptional Activator for the Carboxyl-Terminal Fragment of ErbB-4 That Translocates to the Nucleus. J. Biol. Chem. 2003, 278, 33334–33341. [Google Scholar] [CrossRef] [Green Version]
- Yagi, R.; Chen, L.F.; Shigesada, K.; Murakami, Y.; Ito, Y. A WW Domain-Containing Yes-Associated Protein (YAP) Is a Novel Transcriptional Co-Activator. EMBO J. 1999, 18, 2551–2562. [Google Scholar] [CrossRef] [Green Version]
- Hamaratoglu, F.; Willecke, M.; Kango-Singh, M.; Nolo, R.; Hyun, E.; Tao, C.; Jafar-Nejad, H.; Halder, G. The Tumour-Suppressor Genes NF2/Merlin and Expanded Act through Hippo Signalling to Regulate Cell Proliferation and Apoptosis. Nat. Cell Biol. 2006, 8, 27–36. [Google Scholar] [CrossRef]
- Baumgartner, R.; Poernbacher, I.; Buser, N.; Hafen, E.; Stocker, H. The WW Domain Protein Kibra Acts Upstream of Hippo in Drosophila. Dev. Cell 2010, 18, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.; Yu, J.; Zheng, Y.; Chen, Q.; Zhang, N.; Pan, D. Spatial Organization of Hippo Signaling at the Plasma Membrane Mediated by the Tumor Suppressor Merlin/NF2. Cell 2013, 154, 1342–1355. [Google Scholar] [CrossRef] [Green Version]
- Genevet, A.; Tapon, N. The Hippo Pathway and Apico-Basal Cell Polarity. Biochem. J. 2011, 436, 213–224. [Google Scholar] [CrossRef]
- Schroeder, M.C.; Halder, G. Regulation of the Hippo Pathway by Cell Architecture and Mechanical Signals. Semin. Cell Dev. Biol. 2012, 23, 803–811. [Google Scholar] [CrossRef]
- Wada, K.-I.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo Pathway Regulation by Cell Morphology and Stress Fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Wang, K.-C.; Meng, Z. Mechanoregulation of YAP and TAZ in Cellular Homeostasis and Disease Progression. Front. Cell Dev. Biol. 2021, 9, 673599. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, Y.; Thang, B.Q.; Ramirez, K.; Shin, S.J.; Kohata, T.; Ohata, S.; Nguyen, T.A.V.; Ohtsuki, S.; Nagayama, K.; Yanagisawa, H. Matrix Mechanotransduction Mediated by Thrombospondin-1/Integrin/YAP in the Vascular Remodeling. Proc. Natl. Acad. Sci. USA 2020, 117, 9896–9905. [Google Scholar] [CrossRef]
- Arun, A.; Rayford, K.J.; Cooley, A.; Rachakonda, G.; Villalta, F.; Pratap, S.; Lima, M.F.; Sheibani, N.; Nde, P.N. Thrombospondin-1 Plays an Essential Role in Yes-Associated Protein Nuclear Translocation during the Early Phase of Trypanosoma Cruzi Infection in Heart Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 4912. [Google Scholar] [CrossRef]
- Arun, A.; Rayford, K.J.; Cooley, A.; Rana, T.; Rachakonda, G.; Villalta, F.; Pratap, S.; Lima, M.F.; Sheibani, N.; Nde, P.N. Thrombospondin-1 Expression and Modulation of Wnt and Hippo Signaling Pathways during the Early Phase of Trypanosoma Cruzi Infection of Heart Endothelial Cells. PLoS Negl. Trop. Dis. 2022, 16, e0010074. [Google Scholar] [CrossRef]
- Badouel, C.; Gardano, L.; Amin, N.; Garg, A.; Rosenfeld, R.; Le Bihan, T.; McNeill, H. The FERM-Domain Protein Expanded Regulates Hippo Pathway Activity via Direct Interactions with the Transcriptional Activator Yorkie. Dev. Cell 2009, 16, 411–420. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.-Y.; Lei, Q.; Guan, K.-L. Angiomotin Is a Novel Hippo Pathway Component That Inhibits YAP Oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef] [Green Version]
- An, L.; Nie, P.; Chen, M.; Tang, Y.; Zhang, H.; Guan, J.; Cao, Z.; Hou, C.; Wang, W.; Zhao, Y.; et al. MST4 Kinase Suppresses Gastric Tumorigenesis by Limiting YAP Activation via a Non-Canonical Pathway. J. Exp. Med. 2020, 217, e20191817. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Lu, J.; Li, W.; Wu, A.; Zhang, X.; Tong, W.; Ho, K.K.; Qin, L.; Song, H.; Mak, K.K. Reciprocal Inhibition of YAP/TAZ and NF-ΚB Regulates Osteoarthritic Cartilage Degradation. Nat. Commun. 2018, 9, 4564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.; Kim, M.H.; Hong, H.; Cho, H.; Park, S.; Kim, S.K.; Kim, J. MK5 Regulates YAP Stability and Is a Molecular Target in YAP-Driven Cancers. Cancer Res. 2019, 79, 6139–6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, J.-S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.-S.; Guan, K.-L. Cellular Energy Stress Induces AMPK-Mediated Regulation of YAP and the Hippo Pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Jiang, J. Hippo-Independent Regulation of Yki/Yap/Taz: A Non-Canonical View. Front. Cell Dev. Biol. 2021, 9, 658481. [Google Scholar] [CrossRef]
- Oudhoff, M.J.; Freeman, S.A.; Couzens, A.L.; Antignano, F.; Kuznetsova, E.; Min, P.H.; Northrop, J.P.; Lehnertz, B.; Barsyte-Lovejoy, D.; Vedadi, M.; et al. Control of the Hippo Pathway by Set7-Dependent Methylation of Yap. Dev. Cell 2013, 26, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Zhu, Y.; Zhang, W.; Liao, Q.; Chen, Y.; Zhao, X.; Guo, Q.; Shen, P.; Zhen, B.; Qian, X.; et al. Regulation of the Hippo-YAP Pathway by Glucose Sensor O-GlcNAcylation. Mol. Cell 2017, 68, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Stanger, B.Z. Organ Size Determination and the Limits of Regulation. Cell Cycle 2008, 7, 318–324. [Google Scholar] [CrossRef]
- Pan, D. The Hippo Signaling Pathway in Development and Cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.; Camargo, F.D. The Hippo Signaling Pathway and Stem Cell Biology. Trends Cell Biol. 2012, 22, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Von Gise, A.; Lin, Z.; Schlegelmilch, K.; Honor, L.B.; Pan, G.M.; Buck, J.N.; Ma, Q.; Ishiwata, T.; Zhou, B.; Camargo, F.D.; et al. YAP1, the Nuclear Target of Hippo Signaling, Stimulates Heart Growth through Cardiomyocyte Proliferation but Not Hypertrophy. Proc. Natl. Acad. Sci. USA 2012, 109, 2394–2399. [Google Scholar] [CrossRef] [Green Version]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo Pathway Inhibits Wnt Signaling to Restrain Cardiomyocyte Proliferation and Heart Size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.R.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo Pathway Effector Yap Promotes Cardiac Regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Qi, X.; McAnally, J.; Schwartz, R.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of Insulin-like Growth Factor Signaling by Yap Governs Cardiomyocyte Proliferation and Embryonic Heart Size. Sci. Signal. 2011, 4, ra70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Re, D.P.; Yang, Y.; Nakano, N.; Cho, J.; Zhai, P.; Yamamoto, T.; Zhang, N.; Yabuta, N.; Nojima, H.; Pan, D.; et al. Yes-Associated Protein Isoform 1 (Yap1) Promotes Cardiomyocyte Survival and Growth to Protect against Myocardial Ischemic Injury. J. Biol. Chem. 2013, 288, 3977–3988. [Google Scholar] [CrossRef] [Green Version]
- Artap, S.; Manderfield, L.J.; Smith, C.L.; Poleshko, A.; Aghajanian, H.; See, K.; Li, L.; Jain, R.; Epstein, J.A. Endocardial Hippo Signaling Regulates Myocardial Growth and Cardiogenesis. Dev. Biol. 2018, 440, 22–30. [Google Scholar] [CrossRef]
- Monroe, T.O.; Hill, M.C.; Morikawa, Y.; Leach, J.P.; Heallen, T.; Cao, S.; Krijger, P.H.L.; de Laat, W.; Wehrens, X.H.T.; Rodney, G.G.; et al. YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In Vivo. Dev. Cell 2019, 48, 765–779.e7. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo Pathway in the Heart: Pivotal Roles in Development, Disease, and Regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684. [Google Scholar] [CrossRef]
- Dammann, R.; Li, C.; Yoon, J.H.; Chin, P.L.; Bates, S.; Pfeifer, G.P. Epigenetic Inactivation of a RAS Association Domain Family Protein from the Lung Tumour Suppressor Locus 3p21.3. Nat. Genet. 2000, 25, 315–319. [Google Scholar] [CrossRef]
- Oceandy, D.; Pickard, A.; Prehar, S.; Zi, M.; Mohamed, T.M.A.; Stanley, P.J.; Baudoin-Stanley, F.; Nadif, R.; Tommasi, S.; Pfeifer, G.P.; et al. Tumor Suppressor Ras-Association Domain Family 1 Isoform A Is a Novel Regulator of Cardiac Hypertrophy. Circulation 2009, 120, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Lim, D.S.; Canman, C.E.; Kastan, M.B. Substrate Specificities and Identification of Putative Substrates of ATM Kinase Family Members. J. Biol. Chem. 1999, 274, 37538–37543. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Tommasi, S.; Liu, L.; Yee, J.-K.; Dammann, R.; Pfeifer, G.P. RASSF1A Is Part of a Complex Similar to the Drosophila Hippo/Salvador/Lats Tumor-Suppressor Network. Curr. Biol. 2007, 17, 700–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; O’neill, E. RASSF1A Elicits Apoptosis through an MST2 Pathway Directing Proapoptotic Transcription by the P73 Tumor Suppressor Protein. Mol. Cell 2007, 27, 962–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armesilla, A.L.; Williams, J.C.; Buch, M.H.; Pickard, A.; Emerson, M.; Cartwright, E.J.; Oceandy, D.; Vos, M.D.; Gillies, S.; Clark, G.J.; et al. Novel Functional Interaction between the Plasma Membrane Ca2+ Pump 4b and the Proapoptotic Tumor Suppressor Ras-Associated Factor 1 (RASSF1). J. Biol. Chem. 2004, 279, 31318–31328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in Human Health and Disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between Apoptosis, Necrosis and Autophagy. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, D.S.; Jariwala, J.S.; Anderson, E.; Mitra, K.; Meisenhelder, J.; Chang, J.T.; Ideker, T.; Hunter, T.; Nizet, V.; Dillin, A.; et al. Phosphorylation of LC3 by the Hippo Kinases STK3/STK4 Is Essential for Autophagy. Mol. Cell 2015, 57, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.-P.; et al. Mst1 Inhibits Autophagy by Promoting the Interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Millward, T.; Cron, P.; Hemmings, B.A. Molecular Cloning and Characterization of a Conserved Nuclear Serine(Threonine) Protein Kinase. Proc. Natl. Acad. Sci. USA 1995, 92, 5022–5026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hergovich, A. The Roles of NDR Protein Kinases in Hippo Signalling. Genes 2016, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hergovich, A. Regulation and Functions of Mammalian LATS/NDR Kinases: Looking beyond Canonical Hippo Signalling. Cell Biosci. 2013, 3, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, A.P.; Jacquemyn, M.; Lipecka, J.; Chhuon, C.; Aushev, V.N.; Meunier, B.; Singh, M.K.; Carpi, N.; Piel, M.; Codogno, P.; et al. STK38 Kinase Acts as XPO1 Gatekeeper Regulating the Nuclear Export of Autophagy Proteins and Other Cargoes. EMBO Rep. 2019, 20, e48150. [Google Scholar] [CrossRef]
- Ulbricht, A.; Eppler, F.J.; Tapia, V.E.; van der Ven, P.F.M.; Hampe, N.; Hersch, N.; Vakeel, P.; Stadel, D.; Haas, A.; Saftig, P.; et al. Cellular Mechanotransduction Relies on Tension-Induced and Chaperone-Assisted Autophagy. Curr. Biol. 2013, 23, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Tarocco, A.; Caroccia, N.; Morciano, G.; Wieckowski, M.R.; Ancora, G.; Garani, G.; Pinton, P. Melatonin as a Master Regulator of Cell Death and Inflammation: Molecular Mechanisms and Clinical Implications for Newborn Care. Cell Death Dis. 2019, 10, 317. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Lin, J.; Wang, S.; Cheng, Z.; Hu, J.; Wang, T.; Man, W.; Yin, T.; Guo, W.; Gao, E.; et al. Melatonin Protects against Diabetic Cardiomyopathy through Mst1/Sirt3 Signaling. J. Pineal Res. 2017, 63, e12418. [Google Scholar] [CrossRef]
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in Cardiovascular Diseases. J. Clin. Med. 2020, 9, 892. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Nah, J.; Oka, S.-I.; Mukai, R.; Monden, Y.; Maejima, Y.; Ikeda, Y.; Sciarretta, S.; Liu, T.; Li, H.; et al. An Alternative Mitophagy Pathway Mediated by Rab9 Protects the Heart against Ischemia. J. Clin. Investig. 2019, 129, 802–819. [Google Scholar] [CrossRef] [Green Version]
- Morciano, G.; Patergnani, S.; Pedriali, G.; Cimaglia, P.; Mikus, E.; Calvi, S.; Albertini, A.; Giorgi, C.; Campo, G.; Ferrari, R.; et al. Impairment of Mitophagy and Autophagy Accompanies Calcific Aortic Valve Stenosis Favoring Cell Death and the Severity of Disease. Cardiovasc. Res. 2021, cvab267. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Z.; Feng, X.; Cheng, Z.; Xiong, Z.; Wang, T.; Lin, J.; Zhang, M.; Hu, J.; Fan, Y.; et al. Melatonin Activates Parkin Translocation and Rescues the Impaired Mitophagy Activity of Diabetic Cardiomyopathy through Mst1 Inhibition. J. Cell Mol. Med. 2018, 22, 5132–5144. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Lin, K.; Zhang, Y.; Li, M.; Xu, J.; Chen, K.; Zhu, P.; Yu, R. Mst1 Deletion Reduces Septic Cardiomyopathy via Activating Parkin-Related Mitophagy. J. Cell Physiol. 2020, 235, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Cai, Y.; Li, Y.; Li, Y.; Hu, N.; Ma, S.; Hu, S.; Zhu, P.; Wang, W.; Zhou, H. Yap Promotes Hepatocellular Carcinoma Metastasis and Mobilization via Governing Cofilin/F-Actin/Lamellipodium Axis by Regulation of JNK/Bnip3/SERCA/CaMKII Pathways. Redox Biol. 2018, 14, 59–71. [Google Scholar] [CrossRef]
- Wu, W.; Ziemann, M.; Huynh, K.; She, G.; Pang, Z.-D.; Zhang, Y.; Duong, T.; Kiriazis, H.; Pu, T.-T.; Bai, R.-Y.; et al. Activation of Hippo Signaling Pathway Mediates Mitochondria Dysfunction and Dilated Cardiomyopathy in Mice. Theranostics 2021, 11, 8993–9008. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Programmed Cell Death in Animal Development and Disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [Green Version]
- Kiraz, Y.; Adan, A.; Yandim, M.K.; Baran, Y. Major Apoptotic Mechanisms and Genes Involved in Apoptosis. Tumor Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef] [Green Version]
- Staley, B.K.; Irvine, K.D. Hippo Signaling in Drosophila : Recent Advances and Insights: Hippo Signaling in Drosophila. Dev. Dyn. 2012, 241, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Jacob, J.; Hung, S.-H.; Wang, J. The Hippo Pathway in Cardiac Regeneration and Homeostasis: New Perspectives for Cell-Free Therapy in the Injured Heart. Biomolecules 2020, 10, 1024. [Google Scholar] [CrossRef]
- Zhao, B.; Tumaneng, K.; Guan, K.-L. The Hippo Pathway in Organ Size Control, Tissue Regeneration and Stem Cell Self-Renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. Bantam Encodes a Developmentally Regulated MicroRNA That Controls Cell Proliferation and Regulates the Proapoptotic Gene Hid in Drosophila. Cell 2003, 113, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Nolo, R.; Morrison, C.M.; Tao, C.; Zhang, X.; Halder, G. The Bantam MicroRNA Is a Target of the Hippo Tumor-Suppressor Pathway. Curr. Biol. 2006, 16, 1895–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, B.J.; Cohen, S.M. The Hippo Pathway Regulates the Bantam MicroRNA to Control Cell Proliferation and Apoptosis in Drosophila. Cell 2006, 126, 767–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulev, Y.; Fauny, J.D.; Gonzalez-Marti, B.; Flagiello, D.; Silber, J.; Zider, A. SCALLOPED Interacts with YORKIE, the Nuclear Effector of the Hippo Tumor-Suppressor Pathway in Drosophila. Curr. Biol. 2008, 18, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liu, Y.; Zheng, Y.; Dong, J.; Pan, D. The TEAD/TEF Family Protein Scalloped Mediates Transcriptional Output of the Hippo Growth-Regulatory Pathway. Dev. Cell 2008, 14, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Kornbluth, S.; White, K. Apoptosis in Drosophila: Neither Fish nor Fowl (nor Man, nor Worm). J. Cell Sci. 2005, 118, 1779–1787. [Google Scholar] [CrossRef] [Green Version]
- Steller, H. Regulation of Apoptosis in Drosophila. Cell Death Differ. 2008, 15, 1132–1138. [Google Scholar] [CrossRef] [Green Version]
- De Souza, P.M.; Lindsay, M.A. Mammalian Sterile20-like Kinase 1 and the Regulation of Apoptosis. Biochem. Soc. Trans. 2004, 32, 485–488. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms Underlying Acute Protection from Cardiac Ischemia-Reperfusion Injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Yang, G.; Zablocki, D.; Liu, J.; Hong, C.; Kim, S.-J.; Soler, S.; Odashima, M.; Thaisz, J.; Yehia, G.; et al. Activation of Mst1 Causes Dilated Cardiomyopathy by Stimulating Apoptosis without Compensatory Ventricular Myocyte Hypertrophy. J. Clin. Investig. 2003, 111, 1463–1474. [Google Scholar] [CrossRef] [Green Version]
- Odashima, M.; Usui, S.; Takagi, H.; Hong, C.; Liu, J.; Yokota, M.; Sadoshima, J. Inhibition of Endogenous Mst1 Prevents Apoptosis and Cardiac Dysfunction Without Affecting Cardiac Hypertrophy After Myocardial Infarction. Circ. Res. 2007, 100, 1344–1352. [Google Scholar] [CrossRef] [Green Version]
- Zi, M.; Maqsood, A.; Prehar, S.; Mohamed, T.M.A.; Abou-Leisa, R.; Robertson, A.; Cartwright, E.J.; Ray, S.G.; Oh, S.; Lim, D.-S.; et al. The Mammalian Ste20-like Kinase 2 (Mst2) Modulates Stress-Induced Cardiac Hypertrophy. J. Biol. Chem. 2014, 289, 24275–24288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Zhai, P.; Del Re, D.P.; Maejima, Y.; Sadoshima, J. Mst1-Mediated Phosphorylation of Bcl-XL Is Required for Myocardial Reperfusion Injury. JCI Insight 2016, 1, e86217. [Google Scholar] [CrossRef]
- Del Re, D.P.; Matsuda, T.; Zhai, P.; Maejima, Y.; Jain, M.R.; Liu, T.; Li, H.; Hsu, C.-P.; Sadoshima, J. Mst1 Promotes Cardiac Myocyte Apoptosis through Phosphorylation and Inhibition of Bcl-XL. Mol. Cell 2014, 54, 639–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Re, D.P.; Matsuda, T.; Zhai, P.; Gao, S.; Clark, G.J.; Van Der Weyden, L.; Sadoshima, J. Proapoptotic Rassf1A/Mst1 Signaling in Cardiac Fibroblasts Is Protective against Pressure Overload in Mice. J. Clin. Investig. 2010, 120, 3555–3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, Y.; Nakano, N.; Shao, D.; Gao, S.; Luo, W.; Hong, C.; Zhai, P.; Holle, E.; Yu, X.; Yabuta, N.; et al. Lats2 Is a Negative Regulator of Myocyte Size in the Heart. Circ. Res. 2008, 103, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The Transcriptional Coactivator Yes-Associated Protein Drives P73 Gene-Target Specificity in Response to DNA Damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 Phosphorylation by C-Abl Is a Critical Step in Selective Activation of Proapoptotic Genes in Response to DNA Damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef]
- Vigneron, A.M.; Ludwig, R.L.; Vousden, K.H. Cytoplasmic ASPP1 Inhibits Apoptosis through the Control of YAP. Genes Dev. 2010, 24, 2430–2439. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.-Y.; Yu, J.; Guan, K.-L. Cell Detachment Activates the Hippo Pathway via Cytoskeleton Reorganization to Induce Anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef] [Green Version]
- Campo, G.; Pavasini, R.; Morciano, G.; Lincoff, A.M.; Gibson, C.M.; Kitakaze, M.; Lonborg, J.; Ahluwalia, A.; Ishii, H.; Frenneaux, M.; et al. Clinical Benefit of Drugs Targeting Mitochondrial Function as an Adjunct to Reperfusion in ST-Segment Elevation Myocardial Infarction: A Meta-Analysis of Randomized Clinical Trials. Int. J. Cardiol. 2017, 244, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Li, K.; Florencio, L.W.; Tang, L.; Heallen, T.R.; Leach, J.P.; Wang, Y.; Grisanti, F.; Willerson, J.T.; Perin, E.C.; et al. Gene Therapy Knockdown of Hippo Signaling Induces Cardiomyocyte Renewal in Pigs after Myocardial Infarction. Sci. Transl. Med. 2021, 13, eabd6892. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Li, Y.; Song, X.; Liu, Y.; Li, Y.; Li, Y. Cardioprotective Effect of Echinatin Against Ischemia/Reperfusion Injury: Involvement of Hippo/Yes-Associated Protein Signaling. Front. Pharmacol. 2021, 11, 593225. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Makhoul, G.; Yu, B.; Schwertani, A.; Cecere, R. The Cytoprotective Impact of Yes-Associated Protein 1 after Ischemia-Reperfusion Injury in AC16 Human Cardiomyocytes. Exp. Biol. Med. 2019, 244, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, Q. Mst1 Regulates Post-Infarction Cardiac Injury through the JNK-Drp1-Mitochondrial Fission Pathway. Cell Mol. Biol. Lett. 2018, 23, 21. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Xu, M.; Zhang, T.; Zhang, Q.; Zou, C. Mst1 Promotes Cardiac Ischemia–Reperfusion Injury by Inhibiting the ERK-CREB Pathway and Repressing FUNDC1-Mediated Mitophagy. J. Physiol. Sci. 2019, 69, 113–127. [Google Scholar] [CrossRef]
- Lin, Z.; von Gise, A.; Zhou, P.; Gu, F.; Ma, Q.; Jiang, J.; Yau, A.L.; Buck, J.N.; Gouin, K.A.; van Gorp, P.R.R.; et al. Cardiac-Specific YAP Activation Improves Cardiac Function and Survival in an Experimental Murine MI Model. Circ. Res. 2014, 115, 354–363. [Google Scholar] [CrossRef]
- Ma, S.; Dong, Z. Melatonin Attenuates Cardiac Reperfusion Stress by Improving OPA1-Related Mitochondrial Fusion in a Yap-Hippo Pathway-Dependent Manner. J. Cardiovasc. Pharmacol. 2019, 73, 27–39. [Google Scholar] [CrossRef]
- Hara, H.; Takeda, N.; Kondo, M.; Kubota, M.; Saito, T.; Maruyama, J.; Fujiwara, T.; Maemura, S.; Ito, M.; Naito, A.T.; et al. Discovery of a Small Molecule to Increase Cardiomyocytes and Protect the Heart After Ischemic Injury. JACC Basic Transl. Sci. 2018, 3, 639–653. [Google Scholar] [CrossRef]
- Ito, M.; Hara, H.; Takeda, N.; Naito, A.T.; Nomura, S.; Kondo, M.; Hata, Y.; Uchiyama, M.; Morita, H.; Komuro, I. Characterization of a Small Molecule That Promotes Cell Cycle Activation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. J. Mol. Cell. Cardiol. 2019, 128, 90–95. [Google Scholar] [CrossRef] [Green Version]
- Mia, M.M.; Cibi, D.M.; Ghani, S.A.B.A.; Singh, A.; Tee, N.; Sivakumar, V.; Bogireddi, H.; Cook, S.A.; Mao, J.; Singh, M.K. Loss of Yap/Taz in Cardiac Fibroblasts Attenuates Adverse Remodeling and Improves Cardiac Function. Cardiovasc. Res. 2021, cvab205. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the Cardiomyopathies: A Position Statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 2020, 8, 624216. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lee, J.; Kim, B.S.; Wang, Q.; Buxton, S.K.; Balasubramanyam, N.; Kim, J.J.; Dong, J.; Zhang, A.; Li, S.; et al. Tead1 Is Required for Maintaining Adult Cardiomyocyte Function, and Its Loss Results in Lethal Dilated Cardiomyopathy. JCI Insight 2017, 2, 93343. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Jagannathan, R.; Sun, L.; Li, F.; Yang, P.; Lee, J.; Negi, V.; Perez-Garcia, E.M.; Shiva, S.; Yechoor, V.K.; et al. Tead1 Is Essential for Mitochondrial Function in Cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H89–H99. [Google Scholar] [CrossRef]
- Nguyen, M.-N.; Ziemann, M.; Kiriazis, H.; Su, Y.; Thomas, Z.; Lu, Q.; Donner, D.G.; Zhao, W.-B.; Rafehi, H.; Sadoshima, J.; et al. Galectin-3 Deficiency Ameliorates Fibrosis and Remodeling in Dilated Cardiomyopathy Mice with Enhanced Mst1 Signaling. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H45–H60. [Google Scholar] [CrossRef]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular Mechanisms of Arrhythmogenic Cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The Hippo Pathway Is Activated and Is a Causal Mechanism for Adipogenesis in Arrhythmogenic Cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef]
- DeAlmeida, A.C.; van Oort, R.J.; Wehrens, X.H.T. Transverse Aortic Constriction in Mice. J. Vis. Exp. 2010, 38, 1729. [Google Scholar] [CrossRef]
- Byun, J.; Del Re, D.P.; Zhai, P.; Ikeda, S.; Shirakabe, A.; Mizushima, W.; Miyamoto, S.; Brown, J.H.; Sadoshima, J. Yes-Associated Protein (YAP) Mediates Adaptive Cardiac Hypertrophy in Response to Pressure Overload. J. Biol. Chem. 2019, 294, 3603–3617. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Mao, B.; Luo, W.; Wei, B.; Jiang, W.; Liu, D.; Song, L.; Ji, G.; Yang, Z.; Lai, Y.-Q.; et al. The Alteration of Hippo/YAP Signaling in the Development of Hypertrophic Cardiomyopathy. Basic Res. Cardiol. 2014, 109, 435. [Google Scholar] [CrossRef]
- Song, B.; Dang, H.; Dong, R. The Role and the Signal Pathways of Yes-Associated Protein 2 in Hypertrophic Cardiomyopathy. Gen. Physiol. Biophys. 2021, 40, 419–426. [Google Scholar] [CrossRef]
- Ikeda, S.; Mizushima, W.; Sciarretta, S.; Abdellatif, M.; Zhai, P.; Mukai, R.; Fefelova, N.; Oka, S.-I.; Nakamura, M.; Del Re, D.P.; et al. Hippo Deficiency Leads to Cardiac Dysfunction Accompanied by Cardiomyocyte Dedifferentiation During Pressure Overload. Circ. Res. 2019, 124, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Perestrelo, A.R.; Silva, A.C.; Oliver-De La Cruz, J.; Martino, F.; Horváth, V.; Caluori, G.; Polanský, O.; Vinarský, V.; Azzato, G.; de Marco, G.; et al. Multiscale Analysis of Extracellular Matrix Remodeling in the Failing Heart. Circ. Res. 2021, 128, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Rolle, I.G.; Crivellari, I.; Zanello, A.; Mazzega, E.; Dalla, E.; Bulfoni, M.; Avolio, E.; Battistella, A.; Lazzarino, M.; Cellot, A.; et al. Heart Failure Impairs the Mechanotransduction Properties of Human Cardiac Pericytes. J. Mol. Cell Cardiol. 2021, 151, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, S.; Schenke-Layland, K. Cardiac Fibrosis-A Short Review of Causes and Therapeutic Strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-β, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef]
- Działo, E.; Tkacz, K.; Błyszczuk, P. Crosstalk between the TGF-β and WNT Signalling Pathways during Cardiac Fibrogenesis. Acta Biochim. Pol. 2018, 65, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Sharifi-Sanjani, M.; Berman, M.; Goncharov, D.; Alhamaydeh, M.; Avolio, T.G.; Baust, J.; Chang, B.; Kobir, A.; Ross, M.; St Croix, C.; et al. Yes-Associated Protein (Yap) Is Up-Regulated in Heart Failure and Promotes Cardiac Fibroblast Proliferation. Int. J. Mol. Sci. 2021, 22, 6164. [Google Scholar] [CrossRef]
- Xiao, Y.; Hill, M.C.; Li, L.; Deshmukh, V.; Martin, T.J.; Wang, J.; Martin, J.F. Hippo Pathway Deletion in Adult Resting Cardiac Fibroblasts Initiates a Cell State Transition with Spontaneous and Self-Sustaining Fibrosis. Genes Dev. 2019, 33, 1491–1505. [Google Scholar] [CrossRef] [Green Version]
- Gamba, L.; Harrison, M.; Lien, C.-L. Cardiac Regeneration in Model Organisms. Curr. Treat. Options Cardiovasc. Med. 2014, 16, 288. [Google Scholar] [CrossRef] [Green Version]
- Witman, N.; Murtuza, B.; Davis, B.; Arner, A.; Morrison, J.I. Recapitulation of Developmental Cardiogenesis Governs the Morphological and Functional Regeneration of Adult Newt Hearts Following Injury. Dev. Biol. 2011, 354, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart Regeneration in Zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient Regenerative Potential of the Neonatal Mouse Heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of Neonatal and Adult Mammalian Heart Regeneration by the MiR-15 Family. Proc. Natl. Acad. Sci. USA 2013, 110, 187–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uygur, A.; Lee, R.T. Mechanisms of Cardiac Regeneration. Dev. Cell 2016, 36, 362–374. [Google Scholar] [CrossRef] [Green Version]
- Cui, B.; Zheng, Y.; Sun, L.; Shi, T.; Shi, Z.; Wang, L.; Huang, G.; Sun, N. Heart Regeneration in Adult Mammals after Myocardial Damage. Acta Cardiol. Sin. 2018, 34, 115–123. [Google Scholar] [CrossRef]
- Flinn, M.A.; Link, B.A.; O’Meara, C.C. Upstream Regulation of the Hippo-Yap Pathway in Cardiomyocyte Regeneration. Semin. Cell Dev. Biol. 2020, 100, 11–19. [Google Scholar] [CrossRef]
- Morikawa, Y.; Zhang, M.; Heallen, T.; Leach, J.; Tao, G.; Xiao, Y.; Bai, Y.; Li, W.; Willerson, J.T.; Martin, J.F. Actin Cytoskeletal Remodeling with Protrusion Formation Is Essential for Heart Regeneration in Hippo-Deficient Mice. Sci. Signal. 2015, 8, ra41. [Google Scholar] [CrossRef] [Green Version]
- Heallen, T.; Morikawa, Y.; Leach, J.; Tao, G.; Willerson, J.T.; Johnson, R.L.; Martin, J.F. Hippo Signaling Impedes Adult Heart Regeneration. Development 2013, 140, 4683–4690. [Google Scholar] [CrossRef] [Green Version]
- Leach, J.P.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.C.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo Pathway Deficiency Reverses Systolic Heart Failure after Infarction. Nature 2017, 550, 260–264. [Google Scholar] [CrossRef] [Green Version]
- Tao, G.; Kahr, P.C.; Morikawa, Y.; Zhang, M.; Rahmani, M.; Heallen, T.R.; Li, L.; Sun, Z.; Olson, E.N.; Amendt, B.A.; et al. Pitx2 Promotes Heart Repair by Activating the Antioxidant Response after Cardiac Injury. Nature 2016, 534, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Bassat, E.; Mutlak, Y.E.; Genzelinakh, A.; Shadrin, I.Y.; Umansky, K.B.; Yifa, O.; Kain, D.; Rajchman, D.; Leach, J.; Riabov Bassat, D.; et al. The Extracellular Matrix Protein Agrin Promotes Heart Regeneration in Mice. Nature 2017, 547, 179–184. [Google Scholar] [CrossRef]
- Morikawa, Y.; Heallen, T.; Leach, J.; Xiao, Y.; Martin, J.F. Dystrophin-Glycoprotein Complex Sequesters Yap to Inhibit Cardiomyocyte Proliferation. Nature 2017, 547, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, T.; Zhang, J.; Cheng, A.S.L.; Yu, J.; Kang, W.; To, K.F. Emerging Roles of Hippo Signaling in Inflammation and YAP-Driven Tumor Immunity. Cancer Lett. 2018, 426, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, Y.; Yin, F.; Yu, J.; Silverman, N.; Pan, D. Toll Receptor-Mediated Hippo Signaling Controls Innate Immunity in Drosophila. Cell 2016, 164, 406–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaCanna, R.; Liccardo, D.; Zhang, P.; Tragesser, L.; Wang, Y.; Cao, T.; Chapman, H.A.; Morrisey, E.E.; Shen, H.; Koch, W.J.; et al. Yap/Taz Regulate Alveolar Regeneration and Resolution of Lung Inflammation. J. Clin. Investig. 2019, 129, 2107–2122. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Han, X.; Chen, J.; Xie, X.; Xu, J.; Zhao, Y.; Shen, J.; Hu, L.; Xu, P.; Song, H.; et al. Yes-Associated Protein (YAP) and Transcriptional Coactivator with PDZ-Binding Motif (TAZ) Mediate Cell Density-Dependent Proinflammatory Responses. J. Biol. Chem. 2018, 293, 18071–18085. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Ong, N.; An, H.; Zheng, Y. The Emerging Roles of NDR1/2 in Infection and Inflammation. Front. Immunol. 2020, 11, 534. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Medoff, B.D.; Chen, L.; Li, L.; Zhang, X.; Praskova, M.; Liu, M.; Landry, A.; Blumberg, R.S.; Boussiotis, V.A.; et al. The Nore1B/Mst1 Complex Restrains Antigen Receptor-Induced Proliferation of Naïve T Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 20321–20326. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Ramesh, S.; Cibi, D.M.; Yun, L.S.; Li, J.; Li, L.; Manderfield, L.J.; Olson, E.N.; Epstein, J.A.; Singh, M.K. Hippo Signaling Mediators Yap and Taz Are Required in the Epicardium for Coronary Vasculature Development. Cell Rep. 2016, 15, 1384–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramjee, V.; Li, D.; Manderfield, L.J.; Liu, F.; Engleka, K.A.; Aghajanian, H.; Rodell, C.B.; Lu, W.; Ho, V.; Wang, T.; et al. Epicardial YAP/TAZ Orchestrate an Immunosuppressive Response Following Myocardial Infarction. J. Clin. Investig. 2017, 127, 899–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Wang, Z.; Wu, Z.; Li, X.; Wang, L.; Liu, C. Calibration of Laser Beam Direction for Inner Diameter Measuring Device. Sensors 2017, 17, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, X.; Tao, J.; Barbi, J.; Chen, Q.; Park, B.V.; Li, Z.; Zhang, N.; Lebid, A.; Ramaswamy, A.; Wei, P.; et al. YAP Is Essential for Treg-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2018, 8, 1026–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Li, W.; Wang, S.; Zhang, P.; Wang, Q.; Xiao, J.; Zhang, C.; Zheng, X.; Xu, X.; Xue, S.; et al. YAP Aggravates Inflammatory Bowel Disease by Regulating M1/M2 Macrophage Polarization and Gut Microbial Homeostasis. Cell Rep. 2019, 27, 1176–1189.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, J.; Yu, S.; Zhao, H.; Sun, X.; Li, X.; Wang, P.; Xiong, X.; Hong, L.; Xie, C.; Gao, J.; et al. The Transcriptional Coactivator TAZ Regulates Reciprocal Differentiation of TH17 Cells and Treg Cells. Nat. Immunol. 2017, 18, 800–812. [Google Scholar] [CrossRef]
- Mia, M.M.; Cibi, D.M.; Ghani, S.A.B.A.; Song, W.; Tee, N.; Ghosh, S.; Mao, J.; Olson, E.N.; Singh, M.K. YAP/TAZ Deficiency Reprograms Macrophage Phenotype and Improves Infarct Healing and Cardiac Function after Myocardial Infarction. PLoS Biol. 2020, 18, e3000941. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramaccini, D.; Pedriali, G.; Perrone, M.; Bouhamida, E.; Modesti, L.; Wieckowski, M.R.; Giorgi, C.; Pinton, P.; Morciano, G. Some Insights into the Regulation of Cardiac Physiology and Pathology by the Hippo Pathway. Biomedicines 2022, 10, 726. https://doi.org/10.3390/biomedicines10030726
Ramaccini D, Pedriali G, Perrone M, Bouhamida E, Modesti L, Wieckowski MR, Giorgi C, Pinton P, Morciano G. Some Insights into the Regulation of Cardiac Physiology and Pathology by the Hippo Pathway. Biomedicines. 2022; 10(3):726. https://doi.org/10.3390/biomedicines10030726
Chicago/Turabian StyleRamaccini, Daniela, Gaia Pedriali, Mariasole Perrone, Esmaa Bouhamida, Lorenzo Modesti, Mariusz R. Wieckowski, Carlotta Giorgi, Paolo Pinton, and Giampaolo Morciano. 2022. "Some Insights into the Regulation of Cardiac Physiology and Pathology by the Hippo Pathway" Biomedicines 10, no. 3: 726. https://doi.org/10.3390/biomedicines10030726