
Emerging Therapeutic Strategies for Parkinson’s Disease and Future Prospects: A 2021 Update



Abstract
:1. Introduction
2. Current Treatments
3. Pathophysiology
3.1. Genetic Basis
3.2. Mitochondrial Dysfunction and Oxidative Stress
3.3. Autophagy-Lysosome System Dysfunction
4. Novel Therapeutic Strategies in PD Management
4.1. Targeting α-Syn Aggregation
4.1.1. α-Syn Misfolding Inhibitors
4.1.2. Antisense Oligonucleotides (ASOs)
4.1.3. Beta2-Adrenoreceptor (β2AR) Agonists
4.1.4. Lymphocyte-Activation Gene 3 (LAG3) Receptor
4.2. Enhancing Autophagy
4.2.1. Mammalian Target of Rapamycin (mTOR) Signaling
4.2.2. Inhibition of Cellular Homolog of ABL1 (c-Abl)
4.2.3. RhoA-ROCK Signaling
4.2.4. Lysosomal Acid Sphingomyelinase Enzyme (ASMase)
4.3. Promoting Neuroprotection
4.3.1. L-type Voltage-dependent Ca2+ Channel (L-VDCC)
4.3.2. Glucagon-like Peptide-1 (GLP-1) Agonists
4.3.3. Peroxisome Proliferator-Activated Receptors (PPARs) Agonists
4.3.4. PGC-1α
4.3.5. Iron Chelators
4.4. Targeting Mutated Genes
4.4.1. LRKK2 Inhibitors
4.4.2. GCase Agonists
4.5. Targeting Neuroinflammation
4.5.1. Phosphodiesterase 10A (PDE10A)
4.5.2. TLRs
4.6. Others
4.6.1. Adenosine A2A Receptor (A2AR) Antagonists
4.6.2. 5-HT1A Receptor Agonists
4.7. Immunization
5. Stem Cell Research
Stem Cell-Derived Dopamine Replacement Therapies in PD Clinical Trials
6. Growing Orientation: Biomarker Discovery
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stoker, T.B.; Torsney, K.M.; Barker, R.A. Emerging Treatment Approaches for Parkinson’s Disease. Front. Neurosci. 2018, 12, 693. [Google Scholar] [CrossRef] [Green Version]
- Aarsland, D.; Batzu, L.; Halliday, G.M.; Geurtsen, G.J.; Ballard, C.; Chaudhuri, K.R.; Weintraub, D. Parkinson disease-associated cognitive impairment. Nat. Rev. Dis. Primers 2021, 7, 47. [Google Scholar] [CrossRef]
- Hoyert, D.L.; Xu, J. Deaths: Preliminary data for 2011. Natl. Vital Stat. Rep. 2012, 61, 1–51. [Google Scholar] [PubMed]
- Bridi, J.; Hirth, F. Mechanisms of α-Synuclein Induced Synaptopathy in Parkinson’s Disease. Front. Neurosci. 2018, 12, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Tong, J.; Rathitharan, G.; Meyer, J.H.; Furukawa, Y.; Ang, L.-C.; Boileau, I.; Guttman, M.; Hornykiewicz, O.; Kish, S.J. Brain monoamine oxidase B and A in human parkinsonian dopamine deficiency disorders. Brain 2017, 140, 2460–2474. [Google Scholar] [CrossRef]
- Tabar, V.; Studer, L. Pluripotent stem cells in regenerative medicine: Challenges and recent progress. Nat. Rev. Genet. 2014, 15, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Elkamhawy, A.; Woo, J.; Gouda, N.A.; Kim, J.; Nada, H.; Roh, E.J.; Park, K.D.; Cho, J.; Lee, K. Melatonin Analogues Potently Inhibit MAO-B and Protect PC12 Cells against Oxidative Stress. Antioxidants 2021, 10, 1604. [Google Scholar] [CrossRef]
- Elsherbeny, M.H.; Kim, J.; Gouda, N.A.; Gotina, L.; Cho, J.; Pae, A.N.; Lee, K.; Park, K.D.; Elkamhawy, A.; Roh, E.J. Highly Potent, Selective, and Competitive Indole-Based MAO-B Inhibitors Protect PC12 Cells against 6-Hydroxydopamine- and Rotenone-Induced Oxidative Stress. Antioxidants 2021, 10, 1641. [Google Scholar] [CrossRef]
- Blandini, F.; Porter, R.H.P.; Greenamyre, J.T. Glutamate and Parkinson’s disease. Mol. Neurobiol. 1996, 12, 73–94. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.J.; Sargent, L.; Nawaz, H.; Weintraub, D.; Price, E.T.; Willis, A.W. Antimuscarinic Anticholinergic Medications in Parkinson Disease: To Prescribe or Deprescribe? Mov. Disord. Clin. Pract. 2021, 8, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Alcalay, R.N. Precision medicine in Parkinson’s disease: Emerging treatments for genetic Parkinson’s disease. J. Neurol. 2020, 267, 860–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Fernández, R.; Rodriguez-Rojas, R.; del Álamo, M.; Hernández-Fernández, F.; Pineda-Pardo, J.; Dileone, M.; Alonso-Frech, F.; Foffani, G.; Obeso, I.; Gasca-Salas, C.; et al. Focused ultrasound subthalamotomy in patients with asymmetric Parkinson’s disease: A pilot study. Lancet Neurol. 2018, 17, 54–63. [Google Scholar] [CrossRef]
- Pfeiffer, R.F. Non-motor symptoms in Parkinson’s disease. Park. Relat. Disord. 2015, 22, S119–S122. [Google Scholar] [CrossRef]
- Lees, A.J.; Tolosa, E.; Olanow, C.W. Four pioneers of L-dopa treatment: Arvid Carlsson, Oleh Hornykiewicz, George Cotzias, and Melvin Yahr. Mov. Disord. 2014, 30, 19–36. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Cherian, A.; Divya, K.P. Genetics of Parkinson’s disease. Acta Neurol. Belg. 2020, 120, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Lunati, A.; Lesage, S.; Brice, A. The genetic landscape of Parkinson’s disease. Rev. Neurol. 2018, 174, 628–643. [Google Scholar] [CrossRef] [PubMed]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; De Bernard, M. Triggering of Inflammasome by Aggregated α–Synuclein, an Inflammatory Response in Synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [Green Version]
- Brundin, P.; Li, J.-Y.; Holton, J.L.; Lindvall, O.; Revesz, T. Research in motion: The enigma of Parkinson’s disease pathology spread. Nat. Rev. Neurosci. 2008, 9, 741–745. [Google Scholar] [CrossRef]
- Sundal, C.; Fujioka, S.; Uitti, R.J.; Wszolek, Z.K. Autosomal dominant Parkinson’s disease. Park. Relat. Disord. 2012, 18, S7–S10. [Google Scholar] [CrossRef]
- Piccoli, G.; Volta, M. LRRK2 along the Golgi and lysosome connection: A jamming situation. Biochem. Soc. Trans. 2021, 49, 2063–2072. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef] [Green Version]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.-M.; Ramet, L.; Fahmy, A.; Ducrot, C.; Laplante, A.; Bourque, M.-J.; Zhu, L. Parkinson’s disease related proteins PINK1 and Parkin are major regulators of the immune system. Am. Assoc. Immnol. 2019, 202, 177–197. [Google Scholar]
- Lee, Y.; Stevens, D.A.; Kang, S.-U.; Jiang, H.; Lee, Y.-I.; Ko, H.S.; Scarffe, L.A.; Umanah, G.E.; Kang, H.; Ham, S.; et al. PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival. Cell Rep. 2017, 18, 918–932. [Google Scholar] [CrossRef] [Green Version]
- Ryan, E.; Seehra, G.; Sharma, P.; Sidransky, E. GBA1-associated parkinsonism: New insights and therapeutic opportunities. Curr. Opin. Neurol. 2019, 32, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Amaral, C.E.D.M.; Lopes, P.F.; Ferreira, J.C.C.; Alves, E.A.C.; Montenegro, M.V.B.; Da Costa, E.T.; Yamada, E.S.; Cavalcante, F.O.Q.; Santana-Da-Silva, L.C. GBA mutations p.N370S and p.L444P are associated with Parkinson’s disease in patients from Northern Brazil. Arq. Neuro Psiquiatr. 2019, 77, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Kaidery, N.A.; Thomas, B. Current perspective of mitochondrial biology in Parkinson’s disease. Neurochem. Int. 2018, 117, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Modesti, L.; Danese, A.; Vitto, V.A.M.; Ramaccini, D.; Aguiari, G.; Gafà, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef]
- Rakovic, A.; Ziegler, J.; Mårtensson, C.U.; Prasuhn, J.; Shurkewitsch, K.; König, P.; Paulson, H.L.; Klein, C. PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ. 2018, 26, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamoto, E.M.; Vivar, C.; Camandola, S. Physiology and Pathology of Calcium Signaling in the Brain. Front. Pharmacol. 2012, 3, 61. [Google Scholar] [CrossRef] [Green Version]
- Llorente-Folch, I.; Rueda, C.; Pardo, B.; Szabadkai, G.; Duchen, M.R.; Satrustegui, J. The regulation of neuronal mitochondrial metabolism by calcium. J. Physiol. 2015, 593, 3447–3462. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Cho, J. Targeting chaperones, heat shock factor-1, and unfolded protein response: Promising therapeutic approaches for neurodegenerative disorders. Ageing Res. Rev. 2017, 35, 155–175. [Google Scholar] [CrossRef]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; De Snoo, M.; Gondard, E.; Neudorfer, C.; Chau, H.; Ngana, S.G.; O’Hara, D.M.; Brotchie, J.M.; Koprich, J.B.; Lozano, A.; et al. Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol. Commun. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Madureira, M.; Connor-Robson, N.; Wade-Martins, R. LRRK2: Autophagy and Lysosomal Activity. Front. Neurosci. 2020, 14, 498. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Ashkenazi, A.; Jimenez-Sanchez, M.; Rubinsztein, D.C. The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat. Commun. 2016, 7, 11803. [Google Scholar] [CrossRef]
- Sanyal, A.; Novis, H.S.; Gasser, E.; Lin, S.; Lavoie, M.J. LRRK2 Kinase Inhibition Rescues Deficits in Lysosome Function Due to Heterozygous GBA1 Expression in Human iPSC-Derived Neurons. Front. Neurosci. 2020, 14, 442. [Google Scholar] [CrossRef] [PubMed]
- Gorenberg, E.L.; Chandra, S.S. The Role of Co-chaperones in Synaptic Proteostasis and Neurodegenerative Disease. Front. Neurosci. 2017, 11, 248. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Nehru, B. Curcumin affords neuroprotection and inhibits α-synuclein aggregation in lipopolysaccharide-induced Parkinson’s disease model. Inflammopharmacology 2017, 26, 349–360. [Google Scholar] [CrossRef]
- Wang, M.S.; Boddapati, S.; Emadi, S.; Sierks, M.R. Curcumin reduces α-synuclein induced cytotoxicity in Parkinson’s disease cell model. BMC Neurosci. 2010, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018, 8, 16165. [Google Scholar] [CrossRef] [Green Version]
- Levenson, J.M.; Schroeter, S.; Carroll, J.C.; Cullen, V.; Asp, E.; Proschitsky, M.; Chung, C.H.-Y.; Gilead, S.; Nadeem, M.; Dodiya, H.B.; et al. NPT088 reduces both amyloid-β and tau pathologies in transgenic mice. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2016, 2, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Michelson, D.; Grundman, M.; Magnuson, K.; Fisher, R.; Levenson, J.M.; Aisen, P.; Marek, K.; Gray, M.; Hefti, F. Randomized, placebo controlled trial of NPT088, a phage-derived, amyloid-targeted treatment for Alzheimer’s disease. J. Prev. Alzheimer’s Dis. 2019, 6, 228–231. [Google Scholar] [CrossRef]
- Uehara, T.; Choong, C.-J.; Nakamori, M.; Hayakawa, H.; Nishiyama, K.; Kasahara, Y.; Baba, K.; Nagata, T.; Yokota, T.; Tsuda, H.; et al. Amido-bridged nucleic acid (AmNA)-modified antisense oligonucleotides targeting α-synuclein as a novel therapy for Parkinson’s disease. Sci. Rep. 2019, 9, 7567. [Google Scholar] [CrossRef]
- Alarcón-Arís, D.; Recasens, A.; Galofré, M.; Carballo-Carbajal, I.; Zacchi, N.; Ruiz-Bronchal, E.; Pavia-Collado, R.; Chica, R.; Ferrés-Coy, A.; Santos, M.; et al. Selective α-Synuclein Knockdown in Monoamine Neurons by Intranasal Oligonucleotide Delivery: Potential Therapy for Parkinson’s Disease. Mol. Ther. 2018, 26, 550–567. [Google Scholar] [CrossRef] [Green Version]
- Cole, T.A.; Zhao, H.; Collier, T.J.; Sandoval, I.; Sortwell, C.E.; Steece-Collier, K.; Daley, B.F.; Booms, A.; Lipton, J.; Welch, M.; et al. α-Synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson’s disease. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Chatterjee, D.; Bhatt, M.; Butler, D.; De Genst, E.; Dobson, C.M.; Messer, A.; Kordower, J.H. Proteasome-targeted nanobodies alleviate pathology and functional decline in an α-synuclein-based Parkinson’s disease model. NPJ Parkinsons Dis. 2018, 4, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jęśko, H.; Lenkiewicz, A.; Adamczyk, A. Treatments and compositions targeting α-synuclein: A patent review (2010–2016). Expert Opin. Ther. Patents 2016, 27, 427–438. [Google Scholar] [CrossRef]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hishida, R.; Kurahashi, K.; Narita, S.; Baba, T.; Matsunaga, M. “Wearing-off” and β2-adrenoceptor agonist in Parkinson’s disease. Lancet 1992, 339, 870. [Google Scholar] [CrossRef]
- Alexander, G.M.; Schwartzman, R.J.; Nukes, T.A.; Grothusen, J.R.; Hooker, M.D. β2-Adrenergic agonist as adjunct therapy to levodopa in Parkinson’s disease. Neurology 1994, 44, 1511. [Google Scholar] [CrossRef]
- Uc, E.Y.; Lambert, C.P.; Harik, S.I.; Rodnitzky, R.L.; Evans, W.J. Albuterol Improves Response to Levodopa and Increases Skeletal Muscle Mass in Patients with Fluctuating Parkinson Disease. Clin. Neuropharmacol. 2003, 26, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Thenganatt, M.A.; Jankovic, J. The relationship between essential tremor and Parkinson’s disease. Park. Relat. Disord. 2016, 22, S162–S165. [Google Scholar] [CrossRef]
- Gronich, N.; Abernethy, D.R.; Auriel, E.; Lavi, I.; Rennert, G.; Saliba, W. β2-adrenoceptor agonists and antagonists and risk of Parkinson’s disease. Mov. Disord. 2018, 33, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.S.; Gross, A.; Camacho-Soto, A.; Willis, A.W.; Racette, B.A. β2-adrenoreceptor medications and risk of Parkinson disease. Ann. Neurol. 2018, 84, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Laureys, G.; Clinckers, R.; Gerlo, S.; Spooren, A.; Wilczak, N.; Kooijman, R.; Smolders, I.; Michotte, Y.; De Keyser, J. Astrocytic β2-adrenergic receptors: From physiology to pathology. Prog. Neurobiol. 2010, 91, 189–199. [Google Scholar] [CrossRef]
- Scanzano, A.; Cosentino, M. Adrenergic regulation of innate immunity: A review. Front. Pharmacol. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Liu, Y.; Sorce, S.; Nuvolone, M.; Domange, J.; Aguzzi, A. Lymphocyte activation gene 3 (Lag3) expression is increased in prion infections but does not modify disease progression. Sci. Rep. 2018, 8, 14600. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Paudel, Y.N.; Villa, C.; Shaikh, M.F.; Piperi, C. Lymphocyte-Activation Gene 3 (LAG3) Protein as a Possible Therapeutic Target for Parkinson’s Disease: Molecular Mechanisms Connecting Neuroinflammation to α-Synuclein Spreading Pathology. Biology 2020, 9, 86. [Google Scholar] [CrossRef]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.-I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.-H.; et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353. [Google Scholar] [CrossRef] [Green Version]
- Cui, S.; Du, J.; Liu, S.; Meng, J.; Lin, Y.; Li, G.; He, Y.; Zhang, P.; Chen, S.; Wang, G. Serum soluble lymphocyte activation gene-3 as a diagnostic biomarker in Parkinson’s disease: A pilot multicenter study. Mov. Disord. 2018, 34, 138–141. [Google Scholar] [CrossRef]
- Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.C.-K.; Song, J.; Lu, J.; et al. Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Duan, C.; Gao, G.; Wang, X.; Yang, H. Alpha-synuclein overexpression negatively regulates insulin receptor substrate 1 by activating mTORC1/S6K1 signaling. Int. J. Biochem. Cell Biol. 2015, 64, 25–33. [Google Scholar] [CrossRef]
- Malagelada, C.; Ryu, E.J.; Biswas, S.C.; Jackson-Lewis, V.; Greene, L.A. RTP801 Is Elevated in Parkinson Brain Substantia Nigral Neurons and Mediates Death in Cellular Models of Parkinson’s Disease by a Mechanism Involving Mammalian Target of Rapamycin Inactivation. J. Neurosci. 2006, 26, 9996–10005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin Protects against Neuron Death in In Vitro and In Vivo Models of Parkinson’s Disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, M.; Huh, Y.-J.; Lee, Y.-I. The Impairments of α-Synuclein and Mechanistic Target of Rapamycin in Rotenone-Induced SH-SY5Y Cells and Mice Model of Parkinson’s Disease. Front. Neurosci. 2019, 13, 1028. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, V.; Di Maio, A.; Marino, G.; Cardinale, A.; Natale, G.; De Rosa, A.; Campanelli, F.; Mancini, M.; Napolitano, F.; Avallone, L.; et al. Rapamycin, by Inhibiting mTORC1 Signaling, Prevents the Loss of Striatal Bidirectional Synaptic Plasticity in a Rat Model of L-DOPA-Induced Dyskinesia. Front. Aging Neurosci. 2020, 12, 230. [Google Scholar] [CrossRef]
- Bové, J.; Martinez-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.-F.; Zhang, Y.-J.; Zhou, H.-Y.; Wang, H.-M.; Tian, L.-P.; Liu, J.; Ding, J.-Q.; Chen, S.-D. Curcumin Ameliorates the Neurodegenerative Pathology in A53T α-synuclein Cell Model of Parkinson’s Disease Through the Downregulation of mTOR/p70S6K Signaling and the Recovery of Macroautophagy. J. Neuroimmune Pharmacol. 2013, 8, 356–369. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Sureda, A.; Dehpour, A.R.; Shirooie, S.; Silva, A.S.; Devi, K.P.; Ahmed, T.; Ishaq, N.; Hashim, R.; Sobarzo-Sánchez, E.; et al. Regulation of autophagy by polyphenols: Paving the road for treatment of neurodegeneration. Biotechnol. Adv. 2018, 36, 1768–1778. [Google Scholar] [CrossRef]
- Liu, J.; Chen, M.; Wang, X.; Wang, Y.; Duan, C.; Gao, G.; Lu, L.; Wu, X.; Wang, X.; Yang, H. Piperine induces autophagy by enhancing protein phosphotase 2A activity in a rotenone-induced Parkinson’s disease model. Oncotarget 2016, 7, 60823–60843. [Google Scholar] [CrossRef] [Green Version]
- Hoeffer, C.A.; Klann, E. mTOR signaling: At the crossroads of plasticity, memory and disease. Trends Neurosci. 2010, 33, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Damri, O.; Shemesh, N.; Agam, G. Is There Justification to Treat Neurodegenerative Disorders by Repurposing Drugs? The Case of Alzheimer’s Disease, Lithium, and Autophagy. Int. J. Mol. Sci. 2020, 22, 189. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a Novel mTOR-Independent Autophagy Enhancer, Accelerates the Clearance of Mutant Huntingtin and α-Synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.-X.; Lu, J.-H.; Liu, L.-F.; Chen, L.-L.; Durairajan, S.S.K.; Yue, Z.; Zhang, H.-Q.; Li, M. HMGB1 is involved in autophagy inhibition caused by SNCA/α-synuclein overexpression. Autophagy 2013, 10, 144–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.-X.; Sun, Y.-R.; Peluso, I.; Zeng, Y.; Yu, X.; Lu, J.; Xu, Z.; Wang, M.-Z.; Liu, L.-F.; Huang, Y.-Y.; et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 2016, 12, 1372–1389. [Google Scholar] [CrossRef]
- Lazzara, C.A.; Kim, Y.-H. Potential application of lithium in Parkinson’s and other neurodegenerative diseases. Front. Neurosci. 2015, 9, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decressac, M.; Björklund, A. mTOR Inhibition Alleviates L-DOPA-Induced Dyskinesia in Parkinsonian Rats. J. Park. Dis. 2013, 3, 13–17. [Google Scholar] [CrossRef]
- González-Martín, A.; Moyano, T.; Gutiérrez, D.A.; Carvajal, F.J.; Cerpa, W.; Hanley, J.G.; Gutiérrez, R.A.; Álvarez, A.R. c-Abl regulates a synaptic plasticity-related transcriptional program involved in memory and learning. Prog. Neurobiol. 2021, 205, 102122. [Google Scholar] [CrossRef]
- Hantschel, O.; Superti-Furga, G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004, 5, 33–44. [Google Scholar] [CrossRef]
- Abushouk, A.I.; Negida, A.; Elshenawy, R.A.; Zein, H.; Hammad, A.; Menshawy, A.; Mohamed, W.M. C-Abl Inhibition; A Novel Therapeutic Target for Parkinson’s Disease. CNS Neurol. Disord.—Drug Targets 2018, 17, 14–21. [Google Scholar] [CrossRef]
- Shin, J.-H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.-I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) Repression of PGC-1α Contributes to Neurodegeneration in Parkinson’s Disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Mochly-Rosen, D. The PKCδ-Abl complex communicates ER stress to the mitochondria—An essential step in subsequent apoptosis. J. Cell Sci. 2008, 121, 804–813. [Google Scholar] [CrossRef] [Green Version]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E.-H. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of -synuclein in Parkinson’s disease models. Hum. Mol. Genet. 2013, 22, 3315–3328. [Google Scholar] [CrossRef] [Green Version]
- Brahmachari, S.; Ge, P.; Lee, S.H.; Kim, D.; Karuppagounder, S.; Kumar, M.; Mao, X.; Shin, J.H.; Lee, Y.; Pletnikova, O.; et al. Activation of tyrosine kinase c-Abl contributes to α-synuclein–induced neurodegeneration. J. Clin. Investig. 2016, 126, 2970–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breedveld, P.; Pluim, D.; Cipriani, G.; Wielinga, P.; van Tellingen, O.; Schinkel, A.H.; Schellens, J.H. The Effect of Bcrp1 (Abcg2) on the In vivo Pharmacokinetics and Brain Penetration of Imatinib Mesylate (Gleevec): Implications for the Use of Breast Cancer Resistance Protein and P-Glycoprotein Inhibitors to Enable the Brain Penetration of Imatinib in Patients. Cancer Res. 2005, 65, 2577–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeRemer, D.L.; Ustun, C.; Natarajan, K. Nilotinib: A second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. Clin. Ther. 2008, 30, 1956–1975. [Google Scholar] [CrossRef]
- Fowler, A.; Torres-Yaghi, Y.; Pagan, F.; Hebron, M.; Wilmarth, B.; Lawler, A.; Mundel, E.; Yusuf, N.; Starr, J.; Anjum, M.; et al. Nilotinib alters microRNAs that regulate specific autophagy and ubiquitination genes in the CSF of individuals with Parkinson’s disease (5357). Neurology 2020, 94, 5357. [Google Scholar]
- Simuni, T.; Fiske, B.; Merchant, K.; Coffey, C.S.; Klingner, E.; Caspell-Garcia, C.; Lafontant, D.-E.; Matthews, H.; Wyse, R.K.; Brundin, P.; et al. Efficacy of Nilotinib in Patients with Moderately Advanced Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2021, 78, 312–320. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Imam, S.Z.; Trickler, W.; Kimura, S.; Binienda, Z.K.; Paule, M.G.; Slikker, W., Jr.; Li, S.; Clark, R.A.; Ali, S.F. Neuroprotective Efficacy of a New Brain-Penetrating C-Abl Inhibitor in a Murine Parkinson’s Disease Model. PLoS ONE 2013, 8, e65129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giansanti, P.; Preisinger, C.; Huber, K.; Gridling, M.; Superti-Furga, G.; Bennett, K.L.; Heck, A.J.R. Evaluating the Promiscuous Nature of Tyrosine Kinase Inhibitors Assessed in A431 Epidermoid Carcinoma Cells by Both Chemical- and Phosphoproteomics. ACS Chem. Biol. 2014, 9, 1490–1498. [Google Scholar] [CrossRef]
- Rix, U.; Rix, L.L.R.; Terker, A.S.; Fernbach, N.V.; Hantschel, O.; Planyavsky, M.; Breitwieser, F.P.; Herrmann, H.; Colinge, J.; Bennett, K.L.; et al. A comprehensive target selectivity survey of the BCR-ABL kinase inhibitor INNO-406 by kinase profiling and chemical proteomics in chronic myeloid leukemia cells. Leukemia 2009, 24, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Villar-Cheda, B.; Meijide, A.D.; Joglar, B.; Perez, A.I.R.; Guerra, M.J.; Labandeira-Garcia, J.L. Involvement of microglial RhoA/Rho-Kinase pathway activation in the dopaminergic neuron death. Role of angiotensin via angiotensin type 1 receptors. Neurobiol. Dis. 2012, 47, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.; Subramaniam, M.D.; Venkatesan, D.; Cho, S.-G.; Ryding, M.; Meyer, M.; Vellingiri, B. Role of RhoA-ROCK signaling in Parkinson’s disease. Eur. J. Pharmacol. 2020, 894, 173815. [Google Scholar] [CrossRef] [PubMed]
- Roser, A.-E.; Tönges, L.; Lingor, P. Modulation of Microglial Activity by Rho-Kinase (ROCK) Inhibition as Therapeutic Strategy in Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2017, 9, 94. [Google Scholar] [CrossRef] [PubMed]
- Tilve, S.; Difato, F.; Chieregatti, E. Cofilin 1 activation prevents the defects in axon elongation and guidance induced by extracellular alpha-synuclein. Sci. Rep. 2015, 5, 16524. [Google Scholar] [CrossRef] [Green Version]
- Esposito, A.; Dohm, C.P.; Kermer, P.; Bähr, M.; Wouters, F.S. α-Synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol. Dis. 2007, 26, 521–531. [Google Scholar] [CrossRef]
- Gentry, E.G.; Henderson, B.W.; Arrant, A.E.; Gearing, M.; Feng, Y.; Riddle, N.C.; Herskowitz, J.H. Rho Kinase Inhibition as a Therapeutic for Progressive Supranuclear Palsy and Corticobasal Degeneration. J. Neurosci. 2016, 36, 1316–1323. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Lopez, A.; Labandeira, C.M.; Labandeira-Garcia, J.L.; Muñoz, A. Rho kinase inhibitor fasudil reduces l -DOPA-induced dyskinesia in a rat model of Parkinson’s disease. J. Cereb. Blood Flow Metab. 2020, 177, 5622–5641. [Google Scholar] [CrossRef]
- Tatenhorst, L.; Tönges, L.; Saal, K.-A.; Koch, J.C.; Szegő, .M.; Bähr, M.; Lingor, P. Rho Kinase Inhibition by Fasudil in the Striatal 6-Hydroxydopamine Lesion Mouse Model of Parkinson Disease. J. Neuropathol. Exp. Neurol. 2014, 73, 770–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskal, N.; Riccio, V.; Bashkurov, M.; Taddese, R.; Datti, A.; Lewis, P.N.; McQuibban, G.A. ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway. Nat. Commun. 2020, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Pahan, K. Prospects of Statins in Parkinson Disease. Neuroscientist 2011, 17, 244–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-On, P.; Crews, L.; Koob, A.O.; Mizuno, H.; Adame, A.; Spencer, B.; Masliah, E. Statins reduce neuronal α-synuclein aggregation in in vitro models of Parkinson’s disease. J. Neurochem. 2008, 105, 1656–1667. [Google Scholar] [CrossRef]
- Yang, Y.-J.; Bu, L.-L.; Shen, C.; Ge, J.-J.; He, S.-J.; Yu, H.-L.; Tang, Y.-L.; Jue, Z.; Sun, Y.-M.; Yu, W.-B.; et al. Fasudil Promotes α-Synuclein Clearance in an AAV-Mediated α-Synuclein Rat Model of Parkinson’s Disease by Autophagy Activation. J. Park. Dis. 2020, 10, 969–979. [Google Scholar] [CrossRef]
- Carroll, C.B.; Webb, D.; Stevens, K.N.; Vickery, J.; Eyre, V.; Ball, S.; Wyse, R.; Webber, M.; Foggo, A.; Zajicek, J.; et al. Simvastatin as a neuroprotective treatment for Parkinson’s disease (PD STAT): Protocol for a double-blind, randomised, placebo-controlled futility study. BMJ Open 2019, 9, e029740. [Google Scholar] [CrossRef] [PubMed]
- Fracassi, A.; Marangoni, M.; Rosso, P.; Pallottini, V.; Fioramonti, M.; Siteni, S.; Segatto, M. Statins and the Brain: More than Lipid Lowering Agents? Curr. Neuropharmacol. 2018, 17, 59–83. [Google Scholar] [CrossRef]
- Bienias, K.; Fiedorowicz, A.; Sadowska, A.; Prokopiuk, S.; Car, H. Regulation of sphingomyelin metabolism. Pharmacol. Rep. 2016, 68, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Shayman, J.A. Sphingolipid Catabolism. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Ozelius, L.J.; Bar-Shira, A.; Saunders-Pullman, R.; Mirelman, A.; Kornreich, R.; Gana-Weisz, M.; Raymond, D.; Rozenkrantz, L.; Deik, A.; et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 2013, 80, 1606–1610. [Google Scholar] [CrossRef] [Green Version]
- Alecu, I.; Bennett, S.A.L. Dysregulated Lipid Metabolism and Its Role in α-Synucleinopathy in Parkinson’s Disease. Front. Neurosci. 2019, 13, 328. [Google Scholar] [CrossRef]
- Leftin, A.; Job, C.; Beyer, K.; Brown, M.F. Solid-State 13C NMR Reveals Annealing of Raft-Like Membranes Containing Cholesterol by the Intrinsically Disordered Protein α-Synuclein. J. Mol. Biol. 2013, 425, 2973–2987. [Google Scholar] [CrossRef] [Green Version]
- Dagan, E.; Schlesinger, I.; Ayoub, M.; Mory, A.; Nassar, M.; Kurolap, A.; Peretz-Aharon, J.; Gershoni-Baruch, R. The contribution of Niemann-Pick SMPD1 mutations to Parkinson disease in Ashkenazi Jews. Park. Relat. Disord. 2015, 21, 1067–1071. [Google Scholar] [CrossRef]
- Deng, S.; Deng, X.; Song, Z.; Xiu, X.; Guo, Y.; Xiao, J.; Deng, H. Systematic Genetic Analysis of the SMPD1 Gene in Chinese Patients with Parkinson’s Disease. Mol. Neurobiol. 2015, 53, 5025–5029. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.-N.; Liany, H.; Bei, J.-X.; Yu, X.-Q.; Liu, J.; Au, W.-L.; Prakash, K.M.; Tan, L.C.; Tan, E.-K. A rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiol. Aging 2013, 34. [Google Scholar] [CrossRef]
- Robak, L.A.; Jansen, I.E.; Van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M.; Nalls, M.A.; Plagnol, V.; et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Ylönen, S.; Siitonen, A.; Nalls, M.A.; Ylikotila, P.; Autere, J.; Eerola-Rautio, J.; Gibbs, R.; Hiltunen, M.; Tienari, P.J.; Soininen, H.; et al. Genetic risk factors in Finnish patients with Parkinson’s disease. Park. Relat. Disord. 2017, 45, 39–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, L.N.; Chan, R.; Cheng, R.; Liu, X.; Park, N.; Parmalee, N.; Kisselev, S.; Cortes, E.; Torres, P.A.; Pastores, G.M.; et al. Gene-Wise Association of Variants in Four Lysosomal Storage Disorder Genes in Neuropathologically Confirmed Lewy Body Disease. PLoS ONE 2015, 10, e0125204. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, R.N.; Mallet, V.; Vanderperre, B.; Tavassoly, O.; Dauvilliers, Y.; Wu, R.Y.J.; Ruskey, J.A.; Leblond, C.S.; Ambalavanan, A.; Laurent, S.B.; et al. SMPD1 mutations, activity, and α-synuclein accumulation in Parkinson’s disease. Mov. Disord. 2019, 34, 526–535. [Google Scholar] [CrossRef]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.-P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E. Nuclear translocation of NF-B is increased in dopaminergic neurons of patients with Parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldi, S.; Arcuri, C.; Hunot, S.; Légeron, F.-P.; Mecca, C.; Garcia-Gil, M.; Lazzarini, A.; Codini, M.; Beccari, T.; Tasegian, A.; et al. Neutral Sphingomyelinase Behaviour in Hippocampus Neuroinflammation of MPTP-Induced Mouse Model of Parkinson’s Disease and in Embryonic Hippocampal Cells. Mediat. Inflamm. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Albi, E.; Cataldi, S.; Codini, M.; Mariucci, G.; Lazzarini, A.; Ceccarini, M.R.; Ferri, I.; Laurenti, M.E.; Arcuri, C.; Patria, F.; et al. Neutral sphingomyelinase increases and delocalizes in the absence of Toll-Like Receptor 4: A new insight for MPTP neurotoxicity. Prostaglandins Other Lipid Mediat. 2019, 142, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yue, P.; Lu, T.; Wang, Y.; Wei, Y.; Wei, X. Role of lysosomes in physiological activities, diseases, and therapy. J. Hematol. Oncol. 2021, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Schumacker, P.T.; Guzman, J.D.; Ilijic, E.; Yang, B.; Zampese, E. Calcium and Parkinson’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1013–1019. [Google Scholar] [CrossRef] [Green Version]
- Ilijic, E.; Guzman, J.; Surmeier, D. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol. Dis. 2011, 43, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Lautenschläger, J.; Stephens, A.D.; Fusco, G.; Stroehl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef] [Green Version]
- Hopp, S.C. Targeting microglia L-type voltage-dependent calcium channels for the treatment of central nervous system disorders. J. Neurosci. Res. 2020, 99, 141–162. [Google Scholar] [CrossRef]
- Surendranathan, A.; Su, L.; Mak, E.; Passamonti, L.; Hong, Y.T.; Arnold, R.; Rodríguez, P.V.; Bevan-Jones, W.R.; Brain, S.A.E.; Fryer, T.D.; et al. Early microglial activation and peripheral inflammation in dementia with Lewy bodies. Brain 2018, 141, 3415–3427. [Google Scholar] [CrossRef]
- Joers, V.; Tansey, M.G.; Mulas, G.; Carta, A.R. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog. Neurobiol. 2016, 155, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, X.; Liu, Y.; Bao, Y.; An, L. Nimodipine protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. Neuropharmacology 2009, 56, 580–589. [Google Scholar] [CrossRef]
- Hopp, S.; Royer, S.; D’Angelo, H.M.; Kaercher, R.M.; Fisher, D.A.; Wenk, G.L. Differential Neuroprotective and Anti-Inflammatory Effects of L-Type Voltage Dependent Calcium Channel and Ryanodine Receptor Antagonists in the Substantia Nigra and Locus Coeruleus. J. Neuroimmune Pharmacol. 2014, 10, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Saegusa, H.; Huntula, S.; Tanabe, T. Blockade of microglial Cav1.2 Ca2+ channel exacerbates the symptoms in a Parkinson’s disease model. Sci. Rep. 2019, 9, 9138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson Study Group STEADY-PD III Investigators. Isradipine Versus Placebo in Early Parkinson Disease. Ann. Intern. Med. 2020, 172, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, D.A.; D’Alessio, D.A. Physiology of Proglucagon Peptides: Role of Glucagon and GLP-1 in Health and Disease. Physiol. Rev. 2015, 95, 513–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Choi, H.-I.; Wang, Y.; Luo, Y.; Hoffer, B.J.; Greig, N.H. A New Treatment Strategy for Parkinson’s Disease through the Gut-Brain Axis: The Glucagon-Like Peptide-1 Receptor Pathway. Cell Transplant. 2017, 26, 1560–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athauda, D.; Foltynie, T. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: Mechanisms of action. Drug Discov. Today 2016, 21, 802–818. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-Q.; Zhang, W.; Li, T.; Yang, T.; Yuan, X.; Zhou, Y.; Zou, Q.; Yang, H.; Gao, F.; Tian, Y.; et al. GLP-1R activation ameliorated novel-object recognition memory dysfunction via regulating hippocampal AMPK/NF-κB pathway in neuropathic pain mice. Neurobiol. Learn. Mem. 2021, 182, 107463. [Google Scholar] [CrossRef]
- El Salam, R.A.; Safar, M.M. Neuroprotective effects of vildagliptin in rat rotenone Parkinson’s disease model: Role of RAGE-NFκB and Nrf2-antioxidant signaling pathways. J. Neurochem. 2015, 133, 700–707. [Google Scholar] [CrossRef]
- Li, Y.; Tweedie, D.; Mattson, M.P.; Holloway, H.W.; Greig, N.H. Enhancing the GLP-1 receptor signaling pathway leads to proliferation and neuroprotection in human neuroblastoma cells. J. Neurochem. 2010, 113, 1621–1631. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yin, F.; Zheng, X.; Jing, J.; Hu, Y. Geniposide, a novel agonist for GLP-1 receptor, prevents PC12 cells from oxidative damage via MAP kinase pathway. Neurochem. Int. 2007, 51, 361–369. [Google Scholar] [CrossRef]
- Li, Y.; Perry, T.; Kindy, M.S.; Harvey, B.K.; Tweedie, D.; Holloway, H.W.; Powers, K.; Shen, H.; Egan, J.M.; Sambamurti, K.; et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc. Natl. Acad. Sci. USA 2009, 106, 1285–1290. [Google Scholar] [CrossRef] [Green Version]
- Salcedo, I.; Tweedie, D.; Li, Y.; Greig, N.H. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: An emerging opportunity to treat neurodegenerative and cerebrovascular disorders. J. Cereb. Blood Flow Metab. 2012, 166, 1586–1599. [Google Scholar] [CrossRef] [Green Version]
- Perry, T.; Lahiri, D.K.; Chen, D.; Zhou, J.; Shaw, K.T.Y.; Egan, J.M.; Greig, N.H. A Novel Neurotrophic Property of Glucagon-Like Peptide 1: A Promoter of Nerve Growth Factor-Mediated Differentiation in PC12 Cells. J. Pharmacol. Exp. Ther. 2002, 300, 958–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Ell, P.; Soderlund, T.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Exenatide and the treatment of patients with Parkinson’s disease. J. Clin. Investig. 2013, 123, 2730–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athauda, D.; Maclagan, K.; Skene, S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Li, Y.; Wu, K.-J.; Yu, S.-J.; Tamargo, I.A.; Wang, Y.; Greig, N.H. Neurotrophic and neuroprotective effects of oxyntomodulin in neuronal cells and a rat model of stroke. Exp. Neurol. 2016, 288, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The Coactivator PGC-1 Cooperates with Peroxisome Proliferator-Activated Receptor α in Transcriptional Control of Nuclear Genes Encoding Mitochondrial Fatty Acid Oxidation Enzymes. Mol. Cell. Biol. 2000, 20, 1868–1876. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, R.K.; Beal, M.F. PPAR: A therapeutic target in Parkinson’s disease. J. Neurochem. 2008, 106, 506–518. [Google Scholar] [CrossRef]
- Cowell, R.M.; Blake, K.R.; Inoue, T.; Russell, J.W. Regulation of PGC-1α and PGC-1α-responsive genes with forskolin-induced Schwann cell differentiation. Neurosci. Lett. 2008, 439, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Ulusoy, G.K.; Celik, T.; Kayir, H.; Gürsoy, M.; Isik, A.T.; Uzbay, T.I. Effects of pioglitazone and retinoic acid in a rotenone model of Parkinson’s disease. Brain Res. Bull. 2011, 85, 380–384. [Google Scholar] [CrossRef]
- NINDS Exploratory Trials in Parkinson Disease (NET-PD) FS-ZONE Investigators Pioglitazone in early Parkinson’s disease: A phase 2, multicentre, double-blind, randomised trial. Lancet Neurol. 2015, 14, 795–803. [CrossRef] [Green Version]
- Sadeghian, M.; Marinova-Mutafchieva, L.; Broom, L.; Davis, J.; Virley, D.; Medhurst, A.; Dexter, D. Full and partial peroxisome proliferation-activated receptor-gamma agonists, but not delta agonist, rescue of dopaminergic neurons in the 6-OHDA Parkinsonian model is associated with inhibition of microglial activation and MMP expression. J. Neuroimmunol. 2012, 246, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Iwashita, A.; Muramatsu, Y.; Yamazaki, T.; Muramoto, M.; Kita, Y.; Yamazaki, S.; Mihara, K.; Moriguchi, A.; Matsuoka, N. Neuroprotective Efficacy of the Peroxisome Proliferator-Activated Receptor δ-Selective Agonists in Vitro and in Vivo. J. Pharmacol. Exp. Ther. 2006, 320, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xue, L.; Zheng, J.; Tian, X.; Zhang, Y.; Tong, Q. PPARß/δ agonist alleviates NLRP3 inflammasome-mediated neuroinflammation in the MPTP mouse model of Parkinson’s disease. Behav. Brain Res. 2019, 356, 483–489. [Google Scholar] [CrossRef]
- Kreisler, A.; Gelé, P.; Wiart, J.-F.; Lhermitte, M.; Destée, A.; Bordet, R. Lipid-lowering drugs in the MPTP mouse model of Parkinson’s disease: Fenofibrate has a neuroprotective effect, whereas bezafibrate and HMG-CoA reductase inhibitors do not. Brain Res. 2007, 1135, 77–84. [Google Scholar] [CrossRef]
- Brown, P.J.; Smith-Oliver, T.A.; Charifson, P.S.; Tomkinson, N.; Fivush, A.M.; Sternbach, D.D.; Wade, L.E.; Orband-Miller, L.; Parks, D.J.; Blanchard, S.G.; et al. Identification of peroxisome proliferator-activated receptor ligands from a biased chemical library. Chem. Biol. 1997, 4, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Heneka, M.; Landreth, G.E. The Role of Peroxisome Proliferator-Activated Receptor-γ (PPARγ) in Alzheimer’s Disease. CNS Drugs 2008, 22, 1–14. [Google Scholar] [CrossRef]
- Carrasco, E.; Casper, D.; Werner, P. Dopaminergic neurotoxicity by 6-OHDA and MPP+: Differential requirement for neuronal cyclooxygenase activity. J. Neurosci. Res. 2005, 81, 121–131. [Google Scholar] [CrossRef]
- Kurkowska-Jastrzębska, I.; Babiuch, M.; Joniec, I.; Przybyłkowski, A.; Członkowski, A. Indomethacin protects against neurodegeneration caused by MPTP intoxication in mice. Int. Immunopharmacol. 2002, 2, 1213–1218. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Bruscoli, S.; Mazzon, E.; Crisafulli, C.; Donato, V.; Di Paola, R.; Velardi, E.; Esposito, E.; Nocentini, G.; Riccardi, C. Peroxisome Proliferator-Activated Receptor-α Contributes to the Anti-Inflammatory Activity of Glucocorticoids. Mol. Pharmacol. 2007, 73, 323–337. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.-F.; Ku, H.-C.; Lin, H. PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef] [Green Version]
- Piccinin, E.; Sardanelli, A.; Seibel, P.; Moschetta, A.; Cocco, T.; Villani, G. PGC-1s in the Spotlight with Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 3487. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Kang, S.-U.; Zhang, S.; Karuppagounder, S.; Xu, J.; Lee, Y.-K.; Kang, B.-G.; Lee, Y.; Zhang, J.; Pletnikova, O.; et al. Adult Conditional Knockout of PGC-1α Leads to Loss of Dopamine Neurons. eNEURO 2016, 3, ENEURO.0183-16.2016. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.; Reddy, S.; Zheng, K.; Betensky, R.A.; Simon, D.K. Association of PGC-1alphapolymorphisms with age of onset and risk of Parkinson’s disease. BMC Med. Genet. 2011, 12, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Huang, W.; Li, D.; Si, E.; Wang, J.; Wang, Y.; Chen, C.; Chen, X. Overexpression of PGC-1α Influences Mitochondrial Signal Transduction of Dopaminergic Neurons. Mol. Neurobiol. 2016, 53, 3756–3770. [Google Scholar] [CrossRef]
- Mudò, G.; Mäkelä, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Mälkiä, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’Aquila, C.; De Mari, M.; et al. Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2014, 1842, 902–915. [Google Scholar] [CrossRef] [Green Version]
- Anis, E.; Zafeer, M.F.; Firdaus, F.; Islam, S.N.; Anees Khan, A.; Ali, A.; Hossain, M.M. Ferulic acid reinstates mitochondrial dynamics through PGC1α expression modulation in 6-hydroxydopamine lesioned rats. Phytother. Res. 2020, 34, 214–226. [Google Scholar] [CrossRef]
- Eschbach, J.; Von Einem, B.; Muller, K.; Bayer, H.; Scheffold, A.; Morrison, B.E.; Rudolph, K.L.; Thal, D.R.; Witting, A.; Weydt, P.; et al. Mutual exacerbation of peroxisome proliferator-activated receptor γ coactivator 1α deregulation and α-synuclein oligomerization. Ann. Neurol. 2014, 77, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Schapira, A.H. Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci. Rep. 2018, 8, 1385. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Kurian, M.A.; Hogarth, P. Neurodegeneration with brain iron accumulation. Handb. Clin. Neurol. 2018, 147, 293–305. [Google Scholar] [CrossRef]
- Lingor, P.; Carboni, E.; Koch, J.C. Alpha-synuclein and iron: Two keys unlocking Parkinson’s disease. J. Neural Transm. 2017, 124, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- Dexter, D.T.; Statton, S.A.; Whitmore, C.; Freinbichler, W.; Weinberger, P.; Tipton, K.F.; Della Corte, L.; Ward, R.J.; Crichton, R.R. Clinically available iron chelators induce neuroprotection in the 6-OHDA model of Parkinson’s disease after peripheral administration. J. Neural Transm. 2010, 118, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Pichler, I.; Del Greco, M.F.; Gögele, M.; Lill, C.M.; Bertram, L.; Do, C.B.; Eriksson, N.; Foroud, T.; Myers, R.H.; Nalls, M.; et al. Serum Iron Levels and the Risk of Parkinson Disease: A Mendelian Randomization Study. PLoS Med. 2013, 10, e1001462. [Google Scholar] [CrossRef]
- Guo, C.; Hao, L.-J.; Yang, Z.-H.; Chai, R.; Zhang, S.; Gu, Y.; Gao, H.-L.; Zhong, M.-L.; Wang, T.; Li, J.-Y.; et al. Deferoxamine-mediated up-regulation of HIF-1α prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp. Neurol. 2016, 280, 13–23. [Google Scholar] [CrossRef]
- Kaur, D.; Yantiri, F.; Rajagopalan, S.; Kumar, J.; Mo, J.Q.; Boonplueang, R.; Viswanath, V.; Jacobs, R.; Yang, L.; Beal, M.; et al. Genetic or Pharmacological Iron Chelation Prevents MPTP-Induced Neurotoxicity In Vivo: A Novel Therapy for Parkinson’s Disease. Neuron 2003, 37, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, D.I.; Hare, D.J.; Billings, J.L.; Sedjahtera, A.; Nurjono, M.; Arthofer, E.; George, S.; Culvenor, J.G.; Bush, A.I.; Adlard, P.A. Clioquinol Improves Cognitive, Motor Function, and Microanatomy of the Alpha-Synuclein hA53T Transgenic Mice. ACS Chem. Neurosci. 2015, 7, 119–129. [Google Scholar] [CrossRef]
- Ben Shachar, D.; Kahana, N.; Kampel, V.; Warshawsky, A.; Youdim, M.B. Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydroxydopamine lession in rats. Neuropharmacology 2003, 46, 254–263. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Gross, A.; Finberg, J.P.M. Rasagiline [N-propargyl-1R(+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. J. Cereb. Blood Flow Metab. 2001, 132, 500–506. [Google Scholar] [CrossRef]
- Gal, S.; Zheng, H.; Fridkin, M.; Youdim, M.B.H. Restoration of Nigrostriatal Dopamine Neurons in Post-MPTP Treatment by the Novel Multifunctional Brain-Permeable Iron Chelator-Monoamine Oxidase Inhibitor Drug, M30. Neurotox. Res. 2009, 17, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, P.; Mena, N.P.; Carrasco, C.M.; Muñoz, Y.; Pérez-Henríquez, P.; Morales, R.A.; Cassels, B.K.; Mendez-Galvez, C.; García-Beltrán, O.; Gonzalez-Billault, C.; et al. Iron Chelators and Antioxidants Regenerate Neuritic Tree and Nigrostriatal Fibers of MPP+/MPTP-Lesioned Dopaminergic Neurons. PLoS ONE 2015, 10, e0144848. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef]
- Charvin, D.; Medori, R.; Hauser, R.A.; Rascol, O. Therapeutic strategies for Parkinson disease: Beyond dopaminergic drugs. Nat. Rev. Drug Discov. 2018, 17, 804–822. [Google Scholar] [CrossRef] [PubMed]
- Atashrazm, F.; Dzamko, N. LRRK2 inhibitors and their potential in the treatment of Parkinson’s disease: Current perspectives. Clin. Pharmacol. Adv. Appl. 2016, 8, 177–189. [Google Scholar] [CrossRef] [Green Version]
- LaValley, N.J.; Slone, S.R.; Ding, H.; West, A.; Yacoubian, T.A. 14-3-3 Proteins regulate mutant LRRK2 kinase activity and neurite shortening. Hum. Mol. Genet. 2015, 25, 109–122. [Google Scholar] [CrossRef]
- Moehle, M.; Webber, P.J.; Tse, T.; Sukar, N.; Standaert, D.; DeSilva, T.M.; Cowell, R.; West, A.B. LRRK2 Inhibition Attenuates Microglial Inflammatory Responses. J. Neurosci. 2012, 32, 1602–1611. [Google Scholar] [CrossRef]
- Liu, G.-H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Howlett, E.H.; Jensen, N.; Belmonte, F.; Zafar, F.; Hu, X.; Kluss, J.; Schüle, B.; Kaufman, B.A.; Greenamyre, J.T.; Sanders, L.H. LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson’s disease. Hum. Mol. Genet. 2017, 26, 4340–4351. [Google Scholar] [CrossRef] [Green Version]
- Fell, M.J.; Mirescu, C.; Basu, K.; Cheewatrakoolpong, B.; DeMong, D.E.; Ellis, J.M.; Hyde, L.A.; Lin, Y.; Markgraf, C.G.; Mei, H.; et al. MLi-2, a Potent, Selective, and Centrally Active Compound for Exploring the Therapeutic Potential and Safety of LRRK2 Kinase Inhibition. J. Pharmacol. Exp. Ther. 2015, 355, 397–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Korzenik, J.R.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; et al. LRRK2 Is Involved in the IFN-γ Response and Host Response to Pathogens. J. Immunol. 2010, 185, 5577–5585. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Liu, X.; Li, Y.; Zhao, J.; Liu, Z.; Hu, Z.; Wang, Y.; Yao, Y.; Miller, A.W.; Su, B.; et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J. Exp. Med. 2017, 214, 3051–3066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Pan, Y.; Yan, R.; Zeng, B.; Wang, H.; Zhang, X.; Li, W.; Wei, H.; Liu, Z. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat. Immunol. 2015, 16, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Herzig, M.C.; Kolly, C.; Persohn, E.; Theil, D.; Schweizer, T.; Hafner, T.; Stemmelen, C.; Troxler, T.J.; Schmid, P.; Danner, S.; et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum. Mol. Genet. 2011, 20, 4209–4223. [Google Scholar] [CrossRef] [Green Version]
- Fuji, R.N.; Flagella, M.; Baca, M.; Baptista, M.A.S.; Brodbeck, J.; Chan, B.K.; Fiske, B.K.; Honigberg, L.; Jubb, A.M.; Katavolos, P.; et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl. Med. 2015, 7, 273ra15. [Google Scholar] [CrossRef]
- Tong, Y.; Yamaguchi, H.; Giaime, E.; Boyle, S.; Kopan, R.; Kelleher, R.J.; Shen, J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of -synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9879–9884. [Google Scholar] [CrossRef] [Green Version]
- Baptista, M.A.S.; Merchant, K.; Barrett, T.; Bhargava, S.; Bryce, D.K.; Ellis, J.M.; Estrada, A.A.; Fell, M.J.; Fiske, B.K.; Fuji, R.N.; et al. LRRK2 inhibitors induce reversible changes in nonhuman primate lungs without measurable pulmonary deficits. Sci. Transl. Med. 2020, 12, eaav0820. [Google Scholar] [CrossRef]
- Denali Therapeutics Announces Positive Clinical Results from LRRK2 Inhibitor Program for Parkinson’s Disease. 2018. Available online: https://denalitherapeutics.gcs-web.com/node/6741/pdf (accessed on 20 November 2021).
- Denali Therapeutics Announces First Patient Dosed in Phase 1b Study of DNL151 for Parkinson’s Disease and Launch of Its Engage Parkinson’s Website. Available online: https://www.globenewswire.com/news-release/2019/09/04/1910858/0/en/Denali-Therapeutics-Announces-First-Patient-Dosed-in-Phase-1b-Study-of-DNL151-for-Parkinson-s-Disease-and-Launch-of-Its-Engage-Parkinson-s-Website.html (accessed on 20 November 2021).
- Mazzulli, J.R.; Xu, Y.-H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Barkhuizen, M.; Anderson, D.G.; Grobler, A.F. Advances in GBA-associated Parkinson’s disease—Pathology, presentation and therapies. Neurochem. Int. 2016, 93, 6–25. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of -Glucocerebrosidase Reduces Pathological -Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- Sardi, S.P.; Cedarbaum, J.M.; Brundin, P. Targeted Therapies for Parkinson’s Disease: From Genetics to the Clinic. Mov. Disord. 2018, 33, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, D.L.; Gombash, S.E.; Kemp, C.J.; Manfredsson, F.P.; Polinski, N.K.; Duffy, M.F.; Sortwell, C.E. Viral Vector-Based Modeling of Neurodegenerative Disorders: Parkinson’s Disease. Hum. Press 2016, 1382, 367–382. [Google Scholar] [CrossRef]
- Lin, J.-Y.; Xie, C.-L.; Zhang, S.-F.; Yuan, W.; Liu, Z.-G. Current Experimental Studies of Gene Therapy in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maegawa, G.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.R.; et al. Identification and Characterization of Ambroxol as an Enzyme Enhancement Agent for Gaucher Disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the Treatment of Patients with Parkinson Disease with and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijer, J.M.D.; Kruithof, A.C.; van Amerongen, G.; de Kam, M.L.; Thijssen, E.; Grievink, H.W.; Moerland, M.; Walker, M.; Been, K.; Skerlj, R.; et al. A randomized single and multiple ascending dose study in healthy volunteers of LTI-291, a centrally penetrant glucocerebrosidase activator. Br. J. Clin. Pharmacol. 2021, 87, 3561–3573. [Google Scholar] [CrossRef]
- Fujishige, K.; Kotera, J.; Omori, K. Striatum- and testis-specific phosphodiesterase PDE10A isolation and characterization of a rat PDE10A. Eur. J. Biochem. 1999, 266, 1118–1127. [Google Scholar] [CrossRef] [Green Version]
- Girault, J.-A. Integrating Neurotransmission in Striatal Medium Spiny Neurons. Adv. Exp. Med. Biol. 2012, 970, 407–429. [Google Scholar] [CrossRef]
- Giorgi, M.; D’Angelo, V.; Esposito, Z.; Nuccetelli, V.; Sorge, R.; Martorana, A.; Stefani, A.; Bernardi, G.; Sancesario, G. Lowered cAMP and cGMP signalling in the brain during levodopa-induced dyskinesias in hemiparkinsonian rats: New aspects in the pathogenetic mechanisms. Eur. J. Neurosci. 2008, 28, 941–950. [Google Scholar] [CrossRef]
- Lee, Y.-Y.; Park, J.-S.; Leem, Y.-H.; Park, J.-E.; Kim, D.-Y.; Choi, Y.-H.; Park, E.-M.; Kang, J.L.; Kim, H.-S. The phosphodiesterase 10 inhibitor papaverine exerts anti-inflammatory and neuroprotective effects via the PKA signaling pathway in neuroinflammation and Parkinson’s disease mouse models. J. Neuroinflamm. 2019, 16, 246. [Google Scholar] [CrossRef] [PubMed]
- Niccolini, F.; Foltynie, T.; Marques, T.R.; Muhlert, N.; Tziortzi, A.C.; Searle, G.E.; Natesan, S.; Kapur, S.; Rabiner, E.A.; Gunn, R.N.; et al. Loss of phosphodiesterase 10A expression is associated with progression and severity in Parkinson’s disease. Brain 2015, 138, 3003–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Rockenstein, E.; Spencer, B.; Kim, H.-K.; Adame, A.; Trejo, M.; Stafa, K.; Lee, H.-J.; Lee, S.-J.; Masliah, E. Antagonizing Neuronal Toll-like Receptor 2 Prevents Synucleinopathy by Activating Autophagy. Cell Rep. 2015, 13, 771–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzamko, N.; Gysbers, A.; Perera, G.; Bahar, A.; Shankar, A.; Gao, J.; Fu, Y.; Halliday, G.M. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2016, 133, 303–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Spencer, B.; Rockenstein, E.; Yamakado, H.; Mante, M.; Adame, A.; Fields, J.A.; Masliah, D.; Iba, M.; Lee, H.-J.; et al. Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating α-synuclein transmission and neuroinflammation. Mol. Neurodegener. 2018, 13, 43. [Google Scholar] [CrossRef]
- Leitner, G.R.; Wenzel, T.; Marshall, N.; Gates, E.J.; Klegeris, A. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther. Targets 2019, 23, 865–882. [Google Scholar] [CrossRef]
- Kumar, V. Toll-like receptors in the pathogenesis of neuroinflammation. J. Neuroimmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated α-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Kanda, T.; Jenner, P. Can adenosine A2A receptor antagonists modify motor behavior and dyskinesia in experimental models of Parkinson’s disease? Park. Relat. Disord. 2020, 80, S21–S27. [Google Scholar] [CrossRef]
- Schiffmann, S.; Libert, F.; Vassart, G.; Vanderhaeghen, J.-J. Distribution of adenosine A2 receptor mRNA in the human brain. Neurosci. Lett. 1991, 130, 177–181. [Google Scholar] [CrossRef]
- Pollack, A.; Fink, J. Adenosine antagonists potentiate D2 dopamine-dependent activation of Fos in the striatopallidal pathway. Neuroscience 1995, 68, 721–728. [Google Scholar] [CrossRef]
- Ferreira, D.G.; Batalha, V.L.; Miranda, H.V.; Coelho, J.; Gomes, R.; Gonçalves, F.Q.; Real, J.I.; Rino, J.; Albino-Teixeira, A.; Cunha, R.; et al. Adenosine A2A Receptors Modulate α-Synuclein Aggregation and Toxicity. Cereb. Cortex 2015, bhv268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, S.; Croft, N.R.; Jenner, P. The novel adenosine A2a antagonist ST1535 potentiates the effects of a threshold dose of l-dopa in unilaterally 6-OHDA-lesioned rats. Brain Res. 2007, 1133, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Jackson, M.J.; Smith, L.A.; Stockwell, K.; Johnson, L.; Carminati, P.; Jenner, P. The novel adenosine A2a receptor antagonist ST1535 potentiates the effects of a threshold dose of l-DOPA in MPTP treated common marmosets. Eur. J. Pharmacol. 2006, 546, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Bleickardt, C.; Mullins, D.; Parker, E.; Hodgson, R. A2A receptor antagonists do not induce dyskinesias in drug-naive or l-dopa sensitized rats. Brain Res. Bull. 2013, 98, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Kieburtz, K.; Olanow, C.W.; Krishnaswami, J.; Resburg, C.; Kerwood, F.; Glass, A.; Kenney, C. A Phase 3 Study of Tozadenant (TOZ-PD) as a Maintenance Therapy for Patients with Parkinson’s Disease Experiencing Motor Fluctuations: Characterization of Study Population (P2.045). Neurology 2018, 90, P2.045. [Google Scholar]
- Hauser, R.A.; Olanow, C.W.; Kieburtz, K.D.; Pourcher, E.; Docu-Axelerad, A.; Lew, M.; Koziolkin, O.; Neale, A.; Resburg, C.; Meya, U.; et al. Tozadenant (SYN115) in patients with Parkinson’s disease who have motor fluctuations on levodopa: A phase 2b, double-blind, randomised trial. Lancet Neurol. 2014, 13, 767–776. [Google Scholar] [CrossRef]
- Hauser, R.A.; Cantillon, M.; Pourcher, E.; Micheli, F.; Mok, V.; Onofrj, M.; Huyck, S.; Wolski, K. Preladenant in patients with Parkinson’s disease and motor fluctuations: A phase 2, double-blind, randomised trial. Lancet Neurol. 2011, 10, 221–229. [Google Scholar] [CrossRef]
- Chen, J.-F.; Cunha, R. The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal. 2020, 16, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Bibbiani, F.; Oh, J.D.; Chase, T.N. Serotonin 5-HT1A agonist improves motor complications in rodent and primate parkinsonian models. Neurology 2001, 57, 1829–1834. [Google Scholar] [CrossRef]
- Goetz, C.G.; Laska, E.; Hicking, C.; Damier, P.; Müller, T.; Nutt, J.; Olanow, C.W.; Rascol, O.; Russ, H. Placebo influences on dyskinesia in Parkinson’s disease. Mov. Disord. 2008, 23, 700–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, C.G.; Damier, P.; Hicking, C.; Laska, E.; Müller, T.; Olanow, C.W.; Rascol, O.; Russ, H. Sarizotan as a treatment for dyskinesias in Parkinson’s disease: A double-blind placebo-controlled trial. Mov. Disord. 2006, 22, 179–186. [Google Scholar] [CrossRef]
- Ludwig, C.L.; Weinberger, D.R.; Bruno, G.; Gillespie, M.; Bakker, K.; LeWitt, P.A.; Chase, T.N. Buspirone, Parkinson’s Disease, and the Locus Ceruleus. Clin. Neuropharmacol. 1986, 9, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, P.; Rosenblad, C.; Arvidsson, K.A.E.; Wictorin, K.; Keywood, C.; Shankar, B.; Lowe, D.A.; Björklund, A.; Widner, H. Eltoprazine counteracts l-DOPA-induced dyskinesias in Parkinson’s disease: A dose-finding study. Brain 2015, 138, 963–973. [Google Scholar] [CrossRef]
- Bezard, E.; Tronci, E.; Pioli, E.Y.; Li, Q.; Porras, G.; Björklund, A.; Carta, M. Study of the antidyskinetic effect of eltoprazine in animal models of levodopa-induced dyskinesia. Mov. Disord. 2013, 28, 1088–1096. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Budnick, I.; Singh, M.; Thiruppathi, M.; Alharshawi, K.; Elshabrawy, H.; Holterman, M.J.; Prabhakar, B.S. Dual Role of GM-CSF as a Pro-Inflammatory and a Regulatory Cytokine: Implications for Immune Therapy. J. Interf. Cytokine Res. 2015, 35, 585–599. [Google Scholar] [CrossRef] [Green Version]
- Kosloski, L.M.; Kosmacek, E.A.; Olson, K.E.; Mosley, R.L.; Gendelman, H.E. GM-CSF induces neuroprotective and anti-inflammatory responses in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated mice. J. Neuroimmunol. 2013, 265, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gendelman, H.E.; Zhang, Y.; Santamaria, P.; Olson, K.E.; Schutt, C.; Bhatti, D.; Shetty, B.L.D.; LuAnn, L.; Estes, K.A.; Standaert, D.G.; et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson’s disease trial. NPJ Parkinson’s Dis. 2017, 3, 1–12. [Google Scholar] [CrossRef]
- Hovgaard, D.; Mortensen, B.T.; Schifter, S.; Nissen, N.I. Clinical pharmacokinetic studies of a human haemopoietic growth factor, GM-CSF. Eur. J. Clin. Investig. 1992, 22, 45–49. [Google Scholar] [CrossRef]
- Olson, K.E.; Namminga, K.L.; Lu, Y.; Thurston, M.J.; Schwab, A.D.; de Picciotto, S.; Tse, S.-W.; Walker, W.; Iacovelli, J.; Small, C.; et al. Granulocyte-macrophage colony-stimulating factor mRNA and Neuroprotective Immunity in Parkinson’s disease. Biomaterials 2021, 272, 120786. [Google Scholar] [CrossRef]
- Reynolds, A.D.; Stone, D.; Hutter, J.A.L.; Benner, E.J.; Mosley, R.L.; Gendelman, H.E. Regulatory T Cells Attenuate Th17 Cell-Mediated Nigrostriatal Dopaminergic Neurodegeneration in a Model of Parkinson’s Disease. J. Immunol. 2010, 184, 2261–2271. [Google Scholar] [CrossRef] [Green Version]
- Mosley, R.L.; Lu, Y.; Olson, K.E.; Machhi, J.; Yan, W.; Namminga, K.L.; Smith, J.R.; Shandler, S.J.; Gendelman, H.E. A Synthetic Agonist to Vasoactive Intestinal Peptide Receptor-2 Induces Regulatory T Cell Neuroprotective Activities in Models of Parkinson’s Disease. Front. Cell. Neurosci. 2019, 13, 421. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.E.; Bade, A.N.; Schutt, C.; Dong, J.; Shandler, S.J.; Boska, M.D.; Mosley, R.L.; Gendelman, H.E.; Liu, Y. Manganese-Enhanced Magnetic Resonance Imaging for Detection of Vasoactive Intestinal Peptide Receptor 2 Agonist Therapy in a Model of Parkinson’s Disease. Neurotherapeutics 2016, 13, 635–646. [Google Scholar] [CrossRef]
- Tarakad, A.; Jankovic, J. Recent advances in understanding and treatment of Parkinson’s disease. Fac. Rev. 2020, 9, 6. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, Y.; Yang, X.; Zhou, C.; Li, F.; Lei, P.; Zhong, L.; Jin, X.; Peng, G. Immune effects of optimized DNA vaccine and protective effects in a MPTP model of Parkinson’s disease. Neurol. Sci. 2013, 34, 1559–1570. [Google Scholar] [CrossRef]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.A.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2016, 32, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti-alpha-Synuclein Monoclonal Antibody, in Patients with Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef]
- Pagano, G.; Boess, F.G.; Taylor, K.I.; Ricci, B.; Mollenhauer, B.; Poewe, W.; Boulay, A.; Anzures-Cabrera, J.; Vogt, A.; Marchesi, M.; et al. A Phase II Study to Evaluate the Safety and Efficacy of Prasinezumab in Early Parkinson’s Disease (PASADENA): Rationale, Design, and Baseline Data. Front. Neurol. 2021, 12, 705407. [Google Scholar] [CrossRef] [PubMed]
- Weihofen, A.; Liu, Y.; Arndt, J.W.; Huy, C.; Quan, C.; Smith, B.A.; Baeriswyl, J.-L.; Cavegn, N.; Senn, L.; Su, L.; et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 2018, 124, 276–288. [Google Scholar] [CrossRef]
- Brys, M.; Fanning, L.; Hung, S.; Ellenbogen, A.; Penner, N.; Yang, M.; Welch, M.; Koenig, E.; David, E.; Fox, T.; et al. Randomized phase I clinical trial of anti–α-synuclein antibody BIIB054. Mov. Disord. 2019, 34, 1154–1163. [Google Scholar] [CrossRef]
- Kallab, M.; Herrera-Vaquero, M.; Johannesson, M.; Eriksson, F.; Sigvardson, J.; Poewe, W.; Wenning, G.K.; Nordström, E.; Stefanova, N. Region-Specific Effects of Immunotherapy with Antibodies Targeting α-synuclein in a Transgenic Model of Synucleinopathy. Front. Neurosci. 2018, 12, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, D.; Kordower, J.H. Immunotherapy in Parkinson’s disease: Current status and future directions. Neurobiol. Dis. 2019, 132, 104587. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Barker, R.A.; de Beaufort, I. Scientific and ethical issues related to stem cell research and interventions in neurodegenerative disorders of the brain. Prog. Neurobiol. 2013, 110, 63–73. [Google Scholar] [CrossRef]
- Barker, R.A.; Götz, M.; Parmar, M. New approaches for brain repair—from rescue to reprogramming. Nature 2018, 557, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Parmar, M.; Kirkeby, A.; Björklund, A.; Thompson, L.; Brundin, P. Are Stem Cell-Based Therapies for Parkinson’s Disease Ready for the Clinic in 2016? J. Park. Dis. 2016, 6, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriks, S.; Shim, J.-W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef]
- Brederlau, A.; Correia, A.S.; Anisimov, S.V.; Elmi, M.; Paul, G.; Roybon, L.; Morizane, A.; Bergquist, F.; Riebe, I.; Nannmark, U.; et al. Transplantation of Human Embryonic Stem Cell-Derived Cells to a Rat Model of Parkinson’s Disease: Effect of In Vitro Differentiation on Graft Survival and Teratoma Formation. Stem Cells 2006, 24, 1433–1440. [Google Scholar] [CrossRef] [Green Version]
- Scheiner, Z.S.; Talib, S.; Feigal, E.G. The Potential for Immunogenicity of Autologous Induced Pluripotent Stem Cell-derived Therapies. J. Biol. Chem. 2014, 289, 4571–4577. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Gan, S.U.; Lin, G.; Lim, Y.T.; Masilamani, J.; Mustafa, F.B.; Phua, M.L.; Rivino, L.; Phan, T.T.; Lee, K.O.; et al. Characterization of Human Umbilical Cord Lining-Derived Epithelial Cells and Transplantation Potential. Cell Transplant. 2011, 20, 1827–1841. [Google Scholar] [CrossRef] [PubMed]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.; Marek, K.; Ottman, R.; Meng, C.; Comyns, K.; Chan, P.; Ma, J.; Marras, C.; Langston, J.W.; Ross, G.W.; et al. Concordance for Parkinson’s disease in twins: A 20-year update. Ann. Neurol. 2019, 85, 600–605. [Google Scholar] [CrossRef]
- Barker, R.A. Designing stem-cell-based dopamine cell replacement trials for Parkinson’s disease. Nat. Med. 2019, 25, 1045–1053. [Google Scholar] [CrossRef]
- Díaz, M.L. Regenerative medicine: Could Parkinson’s be the first neurodegenerative disease to be cured? Future Sci. OA 2019, 5, FSO418. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.-Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef]
- Garitaonandia, I.; Gonzalez, R.; Sherman, G.; Semechkin, A.; Evans, A.; Kern, R. Novel Approach to Stem Cell Therapy in Parkinson’s Disease. Stem Cells Dev. 2018, 27, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Okun, M.S.; Kordower, J.H. Stem Cells: Scientific and Ethical Quandaries of a Personalized Approach to Parkinson’s Disease. Mov. Disord. 2020, 35, 1312–1314. [Google Scholar] [CrossRef]
- Parmar, M.; Björklund, A. From Skin to Brain: A Parkinson’s Disease Patient Transplanted with His Own Cells. Cell Stem Cell 2020, 27, 8–10. [Google Scholar] [CrossRef]
- Kimura, Y.; Shofuda, T.; Higuchi, Y.; Nagamori, I.; Oda, M.; Nakamori, M.; Onodera, M.; Kanematsu, D.; Yamamoto, A.; Katsuma, A.; et al. Human Genomic Safe Harbors and the Suicide Gene-Based Safeguard System for iPSC-Based Cell Therapy. Stem Cells Transl. Med. 2019, 8, 627–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reubinoff, B.; Pera, M.F.; Fong, C.-Y.; Trounson, A.; Bongso, A. Embryonic stem cell lines from human blastocysts: Somatic differentiation in vitro. Nat. Biotechnol. 2000, 18, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Hiller, B.M.; Marmion, D.J.; Gross, R.M.; Thompson, C.A.; Chavez, C.A.; Brundin, P.; Wakeman, D.R.; McMahon, C.W.; Kordower, J.H. Mitomycin-C treatment during differentiation of induced pluripotent stem cell-derived dopamine neurons reduces proliferation without compromising survival or function in vivo. Stem Cells Transl. Med. 2020, 10, 278–290. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; Garcia-Martin, E.; Agúndez, J.A. Cerebrospinal fluid biochemical studies in patients with Parkinson’s disease: Toward a potential search for biomarkers for this disease. Front. Cell. Neurosci. 2014, 8, 369. [Google Scholar] [CrossRef] [PubMed]
- Plog, B.A.; Dashnaw, M.L.; Hitomi, E.; Peng, W.; Liao, Y.; Lou, N.; Deane, R.; Nedergaard, M. Biomarkers of Traumatic Injury Are Transported from Brain to Blood via the Glymphatic System. J. Neurosci. 2015, 35, 518–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, C.A.; Brunkhorst, R.; Niessner, M.; Pfeilschifter, W.; Steinmetz, H.; Foerch, C. Blood Levels of Glial Fibrillary Acidic Protein (GFAP) in Patients with Neurological Diseases. PLoS ONE 2013, 8, e62101. [Google Scholar] [CrossRef]
- Santini, E.; Heiman, M.; Greengard, P.; Valjent, E.; Fisone, G. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA–induced dyskinesia. Sci. Signal. 2009, 2, ra36. [Google Scholar] [CrossRef] [Green Version]
- Foltynie, T.; Aviles-Olmos, I. Exenatide as a potential treatment for patients with Parkinson’s disease: First steps into the clinic. Alzheimer’s Dement. 2014, 10, S38–S46. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y. Oxidized DJ-1 as a possible biomarker of Parkinson’s disease. J. Clin. Biochem. Nutr. 2014, 54, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Ecosta, A.; Epeppe, A.; Carlesimo, G.A.; Ezabberoni, S.; Escalici, F.; Ecaltagirone, C.; Eangelucci, F. Brain-derived neurotrophic factor serum levels correlate with cognitive performance in Parkinson’s disease patients with mild cognitive impairment. Front. Behav. Neurosci. 2015, 9, 253. [Google Scholar] [CrossRef]
- Alirezaei, Z.; Pourhanifeh, M.H.; Borran, S.; Nejati, M.; Mirzaei, H.; Hamblin, M.R. Neurofilament Light Chain as a Biomarker, and Correlation with Magnetic Resonance Imaging in Diagnosis of CNS-Related Disorders. Mol. Neurobiol. 2019, 57, 469–491. [Google Scholar] [CrossRef]
- Fedorow, H.; Tribl, F.; Halliday, G.; Gerlach, M.; Riederer, P.; Double, K.L. Neuromelanin in human dopamine neurons: Comparison with peripheral melanins and relevance to Parkinson’s disease. Prog. Neurobiol. 2005, 75, 109–124. [Google Scholar] [CrossRef]
- Magdalinou, N.; Lees, A.J.; Zetterberg, H. Cerebrospinal fluid biomarkers in parkinsonian conditions: An update and future directions. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Pan, X.; Zhang, J.; Ma, A.; Yang, S.; Ma, J.; Xie, A. Plasma levels of miR-137 and miR-124 are associated with Parkinson’s disease but not with Parkinson’s disease with depression. Neurol. Sci. 2017, 38, 761–767. [Google Scholar] [CrossRef]
- Godau, J.; Knauel, K.; Weber, K.; Brockmann, K.; Maetzler, W.; Binder, G.; Berg, D. Serum Insulinlike Growth Factor 1 as Possible Marker for Risk and Early Diagnosis of Parkinson Disease. Arch. Neurol. 2011, 68, 925–931. [Google Scholar] [CrossRef] [Green Version]
- Desforges, N.M.; Hebron, M.L.; Algarzae, N.K.; Lonskaya, I.; Moussa, C.E.-H. Fractalkine Mediates Communication between Pathogenic Proteins and Microglia: Implications of Anti-Inflammatory Treatments in Different Stages of Neurodegenerative Diseases. Int. J. Alzheimer’s Dis. 2012, 2012, 345472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Školoudík, D.; Jelínková, M.; Blahuta, J.; Čermák, P.; Soukup, T.; Bártová, P.; Langová, K.; Herzig, R. Transcranial Sonography of the Substantia Nigra: Digital Image Analysis. Am. J. Neuroradiol. 2014, 35, 2273–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wennström, M.; Surova, Y.; Hall, S.; Nilsson, C.; Minthon, L.; Boström, F.; Hansson, O.; Nielsen, H.M. Low CSF Levels of Both α-Synuclein and the α-Synuclein Cleaving Enzyme Neurosin in Patients with Synucleinopathy. PLoS ONE 2013, 8, e53250. [Google Scholar] [CrossRef] [Green Version]
- Yuan, N.; Chen, Y.; Xia, Y.; Dai, J.; Liu, C. Inflammation-related biomarkers in major psychiatric disorders: A cross-disorder assessment of reproducibility and specificity in 43 meta-analyses. Transl. Psychiatry 2019, 9, 233. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.J.; Pervaiz, N.; Abbasi, A.A. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Sci. Rep. 2016, 6, 24475. [Google Scholar] [CrossRef] [PubMed]
- Oczkowska, A.; Kozubski, W.; Lianeri, M.; Dorszewska, J. Mutations in PRKN and SNCA Genes Important for the Progress of Parkinson’s Disease. Curr. Genom. 2014, 14, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Cramm, M.; Llorens, F.; Müller-Cramm, D.; Collins, S.; Atarashi, R.; Satoh, K.; Orrù, C.D.; Groveman, B.R.; Zafar, S.; et al. The real-time quaking-induced conversion assay for detection of human prion disease and study of other protein misfolding diseases. Nat. Protoc. 2016, 11, 2233–2242. [Google Scholar] [CrossRef] [PubMed]


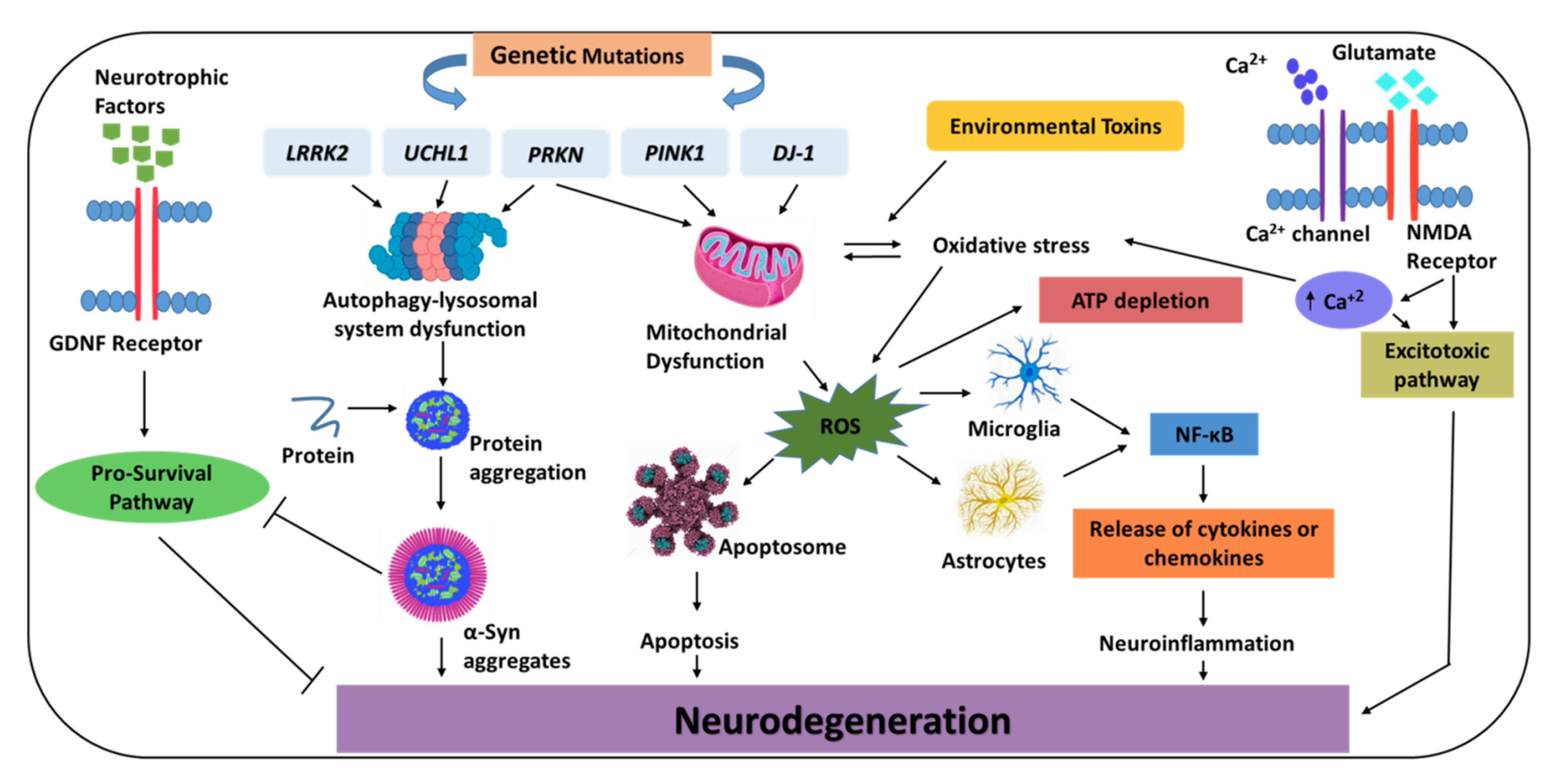{kind=link}
| Symptoms | Mode of Action | Therapeutics | References |
|---|---|---|---|
| Motor | Dopamimetic drugs | L-DOPA Dopamine agonists (ropinirole, pramipexole) | [17] |
| Prevention of L-DOPA/dopamine breakdown | Decarboxylase inhibitors (carbidopa, benserazide); MAO-B inhibitors (selegiline, rasagiline, safinamide); COMT inhibitors (entacapone, opicapone) | [8] | |
| Glutamate inhibition | Amantadine | [18] | |
| Restoration of the balance between dopamine and acetylcholine | Anticholinergics (benztropine, procyclidine, trihexyphenidyl) | [13] | |
| Surgery | Deep brain stimulation; MRgFUS | [14,15] | |
| Non-motor | Depending on the symptoms, antidepressants, cholinesterase inhibitors, or sedative agents may be used | [16] |
| Therapeutic Class | Agent | Target/Mode of Action | Clinical Trial ID | Stage | Status |
|---|---|---|---|---|---|
| Immunomodulatory | GM-CSF | Enhances Treg quantity and functionality, with no impact on the levels of effector T (Teff) cells | NCT03790670 | Phase Ib | Recruiting |
| LBT36 | Increases Treg abundance and performance, decreases microglial activation by altering Th1/Th17 cytokine responses | Not available | Not available | Not available | |
| Active immunization | PD01A | C-terminal α-Syn mimicking peptide vaccination | NCT02618941 | Phase I | Completed |
| PD03A | C-terminal α-Syn mimicking peptide vaccination | NCT02267434 | Phase I | Completed | |
| pVAX1-IL-4/ SYN-B | A nucleic acid vaccine that increases antibody titers against α-Syn | Not available | Not available | Not available | |
| Passive immunotherapy | Prasinezumab | A humanized IgG1 monoclonal antibody targeted towards C-terminal epitopes on α-Syn | NCT04777331 | Phase IIb | Recruiting |
| BIIB054 | A fully human monoclonal antibody directed against N-terminal α-Syn aggregate species | NCT03318523 | Phase II | Terminated | |
| Lu AF82422 | A humanized monoclonal IgG1 antibody targeting the C-terminal of α-Syn | NCT03611569 | Phase I | Completed | |
| ABBV-0805 | A humanized monoclonal antibody targeting α-Syn oligomeric and protofibrillar α-Syn species | NCT04127695 | Phase I | Withdrawn |
| Sponsor | Therapeutic/Type of Cell | Stage | Clinical Trial (ID) |
|---|---|---|---|
| TRANSEURO | Human fetal ventral mesencephalic tissue | Phase I | NCT01898390 |
| Kyoto University Hospital | Allogeneic iPSC-derived DA progenitors | Phase I/II | R000038278 |
| National Institutes of Health | Autologous iPSC-derived dopamine progenitor cells | Case report | |
| ISCO | ISCO human parthenogenetic neural stem cells (ISC-hpNSC) | Phase I | NCT02452723 |
| Therapeutic Class | Approaches | Drugs Penetrating BBB | Ref |
|---|---|---|---|
| α-Syn aggregation inhibitors (NPT200-11, NPT088); Antisense oligonucleotides; β2AR agonists (salbutamol); LAG3 receptor | Targeting α-Syn | NPT200-11; NPT088; Anle138b; Salbutamol | [49,50] [53,57] [61] [69] |
| mTOR signaling pathway (rapamycin); Inhibitors of c-Abl (nilotinib and bafetinib); ROCK inhibitors (simvastatin and lovastatin); ASMase | Enhancing autophagy | Rapamycin; Nilotinib; Simvastatin; Lovastatin | [290] [101] [116,117] [129,130] |
| L-VDCC (isradipine); GLP-1 agonist (exenatide and oxyntomodulin); PPAR agonists (pioglitazone); PGC-1α activators (ambroxol); Iron chelators (DFO) | Promoting neuroprotection | Isradipine; Exenatide; Oxyntomodulin; Pioglitazone; Ambroxol; DFO | [143] [156,291] [162,166] [182] [195] |
| LRRK2 kinase inhibitors (MLi-2, PF-06685360, and DNL201); GCase agonists (isofagomine and ambroxol) | Targeting mutated genes | MLi-2; PF-06685360; DNL201; Isofagomine | [202,211] [220] |
| PDE10A inhibitors (papaverine); TLRs (anti-TLR2 antibodies) | Targeting neuroinflammation | Papaverine | [224] [228,229] |
| A2A receptor antagonists (istradefylline and tozadenant); 5-HT1A receptor agonist (buspirone and eltoprazine) | Others | Istradefylline; Tozadenant | [240,241,242,243] [248] |
| Immunomodulatory (sargramostim); Active immunization (PD01A and PD03A); Passive immunotherapy (prasinezumab and cinpanemab) | Immunization | Sargramostim; PD01A, PD03A; Prasinezumab; Cinpanemab | [253] [258] [260] |
| Human parthenogenetic neural stem cells (ISC-hpNSC); Human fetal ventral mesencephalic tissue (hfVM); Allogeneic/autologous iPSC-derived DA progenitors | Stem cell therapy | NSCs can be delivered trans-cranially | [281] [278] [279,280] |
| Type | Name | Role/Significance | Measurement | References |
|---|---|---|---|---|
| Biochemical biomarkers that can be investigated either in CSF or blood | Glial fibrillary acidic protein (GFAP) | This brain-specific protein and its breakdown products can serve as possible early biomarkers for PD | Detected via an in-house ELISA kit based on antibodies, with a sensitivity of concentration 70 ng/L and intra- and inter-assay coefficients of variation of 4% and 8%, respectively | [288,289] |
| DJ-1 | DJ-1 expression is implicated during oxidative stress | During measuring DJ-1 content in plasma and CSF, an assessment of contamination or hemolysis of erythrocyte is required | [292] | |
| Brain-derived neurotrophic factor (BDNF) | Reduced expression of BDNF within the SN serves as a potential biomarker for PD | Can be determined by ELISA | [293] | |
| Neurofilament light chain protein (NFL) | Recognized in PD associated with LBs | Measuring NFL increasing levels is a very sensitive method to assess aggressive neuronal death | [294] | |
| Neuromelanin | Released from dying neurons | Can be measured by MRI techniques | [295] | |
| CSF α-Syn with lysosomal enzymes | The combination of CSF lysosomal enzymes, such as cathepsin and GCase in the CSF with CSF α-Syn aggregates facilitate an improved accuracy of diagnosis | Detected by protein-misfolding cyclic amplification and real-time quaking-induced conversion | [296,305] | |
| miR-124 | Plasma levels of miR-124 were markedly reduced in PD patients. | Detected in plasma | [297] | |
| Insulin-like growth factor 1 (IGF-1) | Potential marker for idiopathic PD in early disease stage | Detected in serum | [298] | |
| Inflammatory | Fractalkine | Low levels of fractalkine associated with neuroinflammation | Detected in serum | [299] |
| Neurosin | Can cleave and degrade α-Syn; decrease in neurosin levels have been reported in PD | Identified by Northern blotting/investigated in vivo using a commercial sandwich ELISA kit or a direct ELISA | [301] | |
| Genetic | Mutations in SCNA, PRKN, PINK1, DJ-1, LRRK2 and GBA | Triplication of the SCNA causes a two-fold increase in α-Syn expression. | Analysis of global gene expression with DNA microarrays has been performed in the peripheral blood of PD patients | [303,304] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gouda, N.A.; Elkamhawy, A.; Cho, J. Emerging Therapeutic Strategies for Parkinson’s Disease and Future Prospects: A 2021 Update. Biomedicines 2022, 10, 371. https://doi.org/10.3390/biomedicines10020371
Gouda NA, Elkamhawy A, Cho J. Emerging Therapeutic Strategies for Parkinson’s Disease and Future Prospects: A 2021 Update. Biomedicines. 2022; 10(2):371. https://doi.org/10.3390/biomedicines10020371
Chicago/Turabian StyleGouda, Noha A., Ahmed Elkamhawy, and Jungsook Cho. 2022. "Emerging Therapeutic Strategies for Parkinson’s Disease and Future Prospects: A 2021 Update" Biomedicines 10, no. 2: 371. https://doi.org/10.3390/biomedicines10020371