
Implication of Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases


Abstract
:1. Introduction
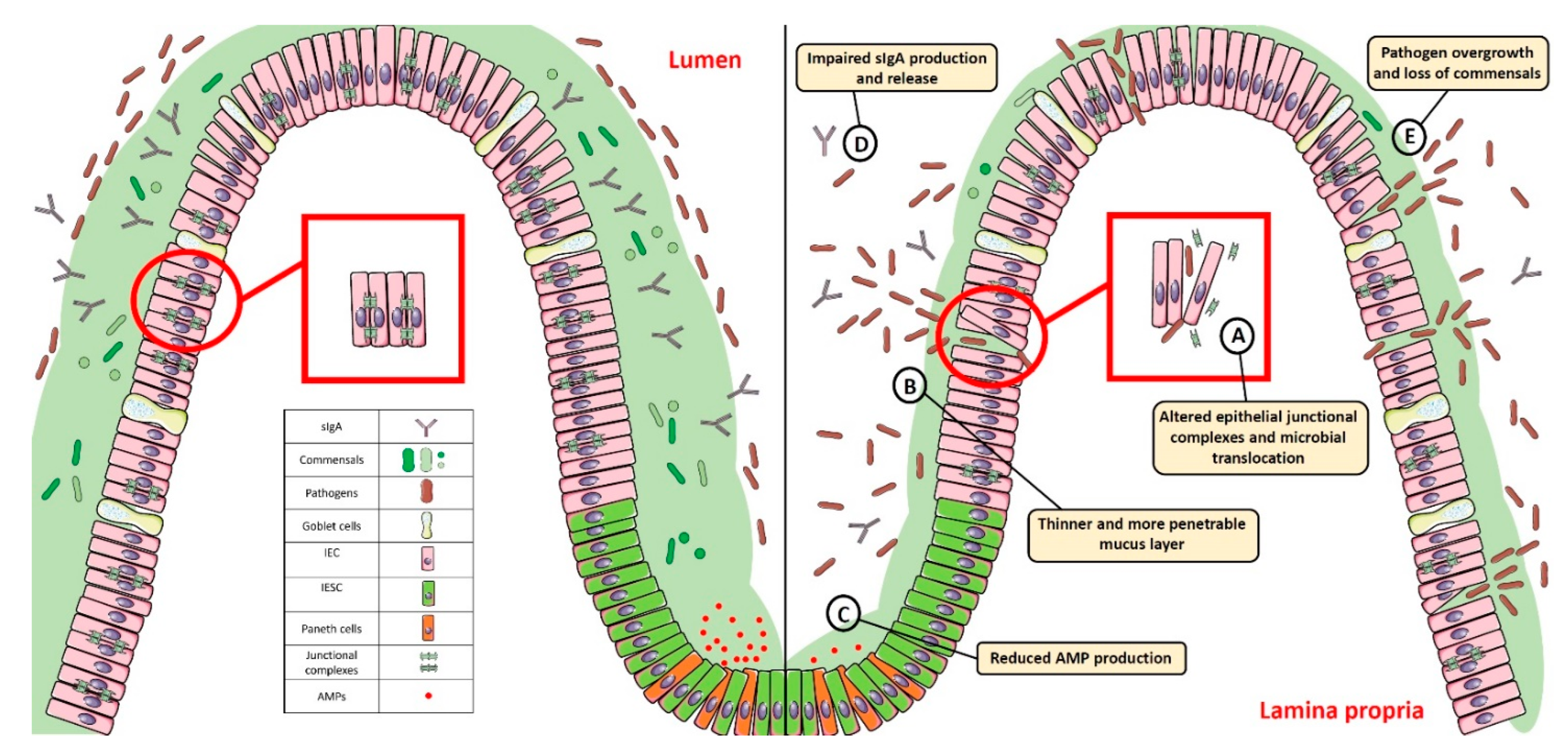
2. Breaking the Balance: Intestinal Barrier Dysfunction and Gut Dysbiosis
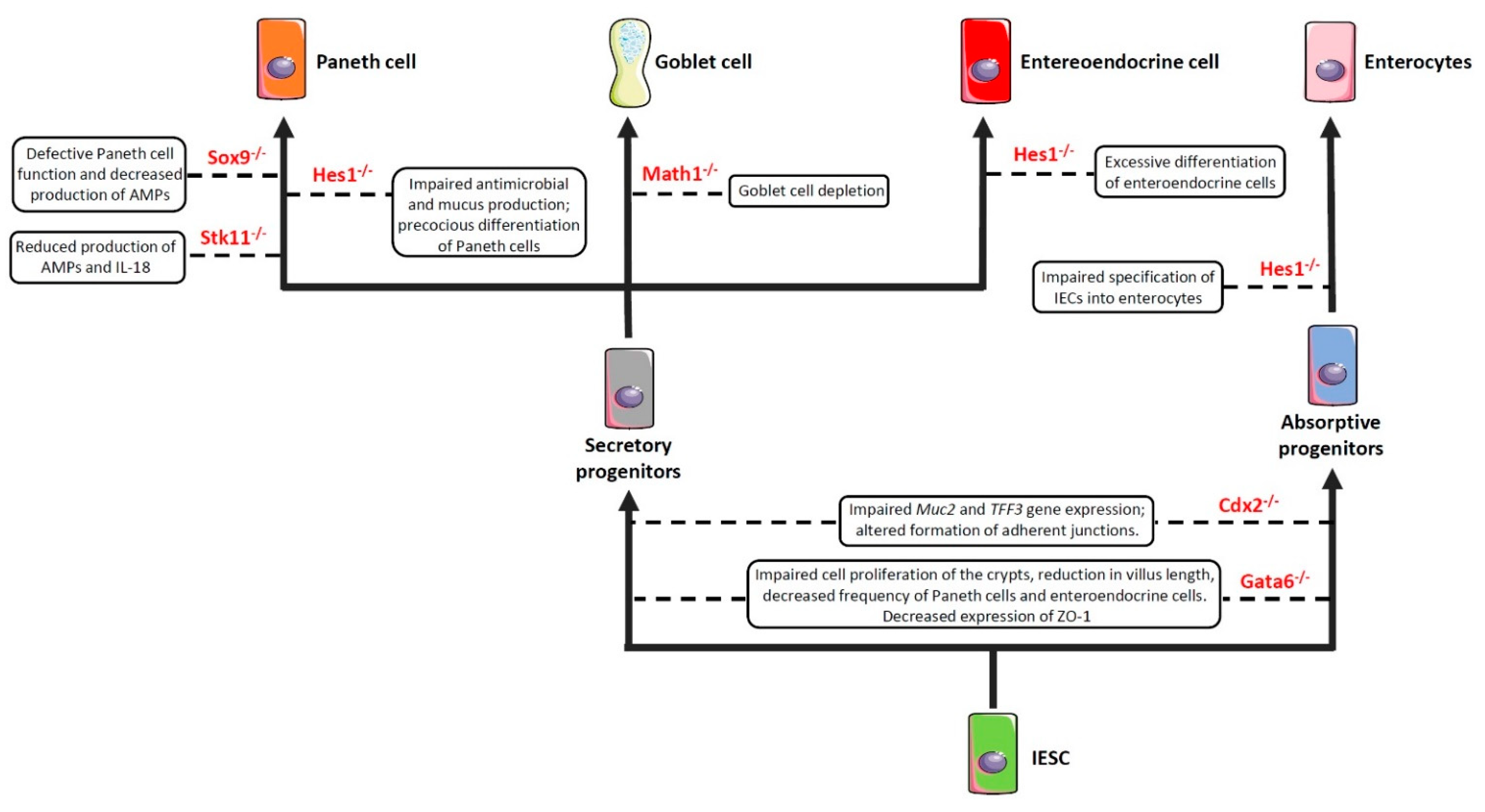
2.1. Impairment of Cell Commitment
2.2. Impairment of Epithelial Junctional Complexes
2.3. Thinning/Depletion of the Mucus Layer
2.4. Paneth Cell Dysfunction
2.5. Impairment of Microbial Sensing by Pattern Recognition Receptors (PRRs)
2.6. Modulation of Epithelial Oxidative Burst
2.7. Modulation of Xenobiotic Receptors
2.8. Impairment of Secretory IgA
3. Discussion and Therapeutic Perspectives
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Kiyono, H. Epithelial barrier: An interface for the cross-communication between gut flora and immune system. Immunol. Rev. 2012, 245, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, e338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, L.V.; Macpherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Martens, E.C.; Neumann, M.; Desai, M.S. Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nat. Rev. Microbiol. 2018, 16, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Nunez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Hayes, C.L.; Dong, J.; Galipeau, H.J.; Jury, J.; McCarville, J.; Huang, X.; Wang, X.Y.; Naidoo, A.; Anbazhagan, A.N.; Libertucci, J.; et al. Commensal microbiota induces colonic barrier structure and functions that contribute to homeostasis. Sci. Rep. 2018, 8, 14184. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Whitley, C.S.; Haribabu, B.; Jala, V.R. Regulation of Intestinal Barrier Function by Microbial Metabolites. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1463–1482. [Google Scholar] [CrossRef]
- Su, L.; Shen, L.; Clayburgh, D.R.; Nalle, S.C.; Sullivan, E.A.; Meddings, J.B.; Abraham, C.; Turner, J.R. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology 2009, 136, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankertz, J.; Schulzke, J.D. Altered permeability in inflammatory bowel disease: Pathophysiology and clinical implications. Curr. Opin. Gastroenterol. 2007, 23, 379–383. [Google Scholar] [CrossRef]
- Martini, E.; Krug, S.M.; Siegmund, B.; Neurath, M.F.; Becker, C. Mend Your Fences: The Epithelial Barrier and its Relationship with Mucosal Immunity in Inflammatory Bowel Disease. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitter, A.H.; Wullstein, F.; Fromm, M.; Schulzke, J.D. Epithelial barrier defects in ulcerative colitis: Characterization and quantification by electrophysiological imaging. Gastroenterology 2001, 121, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van Goudoever, J.B.; Buller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.; Price, G.R.; Tauro, S.B.; Taupin, D.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008, 5, e54. [Google Scholar] [CrossRef] [Green Version]
- Braun, A.; Treede, I.; Gotthardt, D.; Tietje, A.; Zahn, A.; Ruhwald, R.; Schoenfeld, U.; Welsch, T.; Kienle, P.; Erben, G.; et al. Alterations of phospholipid concentration and species composition of the intestinal mucus barrier in ulcerative colitis: A clue to pathogenesis. Inflamm. Bowel Dis. 2009, 15, 1705–1720. [Google Scholar] [CrossRef]
- Larsson, J.M.; Karlsson, H.; Crespo, J.G.; Johansson, M.E.; Eklund, L.; Sjovall, H.; Hansson, G.C. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm. Bowel Dis. 2011, 17, 2299–2307. [Google Scholar] [CrossRef]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Burgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef]
- Lammers, K.M.; Lu, R.; Brownley, J.; Lu, B.; Gerard, C.; Thomas, K.; Rallabhandi, P.; Shea-Donohue, T.; Tamiz, A.; Alkan, S.; et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology 2008, 135, 194–204.e3. [Google Scholar] [CrossRef] [Green Version]
- Fasano, A.; Not, T.; Wang, W.; Uzzau, S.; Berti, I.; Tommasini, A.; Goldblum, S.E. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet 2000, 355, 1518–1519. [Google Scholar] [CrossRef]
- El Asmar, R.; Panigrahi, P.; Bamford, P.; Berti, I.; Not, T.; Coppa, G.V.; Catassi, C.; Fasano, A. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure. Gastroenterology 2002, 123, 1607–1615. [Google Scholar] [CrossRef]
- Szakal, D.N.; Gyorffy, H.; Arato, A.; Cseh, A.; Molnar, K.; Papp, M.; Dezsofi, A.; Veres, G. Mucosal expression of claudins 2, 3 and 4 in proximal and distal part of duodenum in children with coeliac disease. Virchows Arch. Int. J. Pathol. 2010, 456, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Ciccocioppo, R.; Finamore, A.; Ara, C.; Di Sabatino, A.; Mengheri, E.; Corazza, G.R. Altered expression, localization, and phosphorylation of epithelial junctional proteins in celiac disease. Am. J. Clin. Pathol. 2006, 125, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E., Jr.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Li, J.Z.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 2014, 5, 3114. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Young, V.B. Microbial and metabolic interactions between the gastrointestinal tract and Clostridium difficile infection. Gut Microbes 2014, 5, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kachrimanidou, M.; Tsintarakis, E. Insights into the Role of Human Gut Microbiota in Clostridioides difficile Infection. Microorganisms 2020, 8, 200. [Google Scholar] [CrossRef] [Green Version]
- Hanning, N.; Edwinson, A.L.; Ceuleers, H.; Peters, S.A.; De Man, J.G.; Hassett, L.C.; De Winter, B.Y.; Grover, M. Intestinal barrier dysfunction in irritable bowel syndrome: A systematic review. Ther. Adv. Gastroenterol. 2021, 14, 1756284821993586. [Google Scholar] [CrossRef]
- Martinez, C.; Vicario, M.; Ramos, L.; Lobo, B.; Mosquera, J.L.; Alonso, C.; Sanchez, A.; Guilarte, M.; Antolin, M.; de Torres, I.; et al. The jejunum of diarrhea-predominant irritable bowel syndrome shows molecular alterations in the tight junction signaling pathway that are associated with mucosal pathobiology and clinical manifestations. Am. J. Gastroenterol. 2012, 107, 736–746. [Google Scholar] [CrossRef]
- Martinez, C.; Lobo, B.; Pigrau, M.; Ramos, L.; Gonzalez-Castro, A.M.; Alonso, C.; Guilarte, M.; Guila, M.; de Torres, I.; Azpiroz, F.; et al. Diarrhoea-predominant irritable bowel syndrome: An organic disorder with structural abnormalities in the jejunal epithelial barrier. Gut 2013, 62, 1160–1168. [Google Scholar] [CrossRef]
- Martinez, C.; Rodino-Janeiro, B.K.; Lobo, B.; Stanifer, M.L.; Klaus, B.; Granzow, M.; Gonzalez-Castro, A.M.; Salvo-Romero, E.; Alonso-Cotoner, C.; Pigrau, M.; et al. miR-16 and miR-125b are involved in barrier function dysregulation through the modulation of claudin-2 and cingulin expression in the jejunum in IBS with diarrhoea. Gut 2017, 66, 1537–1538. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, S.P.; Hebden, J.; Campbell, E.; Naesdal, J.; Olbe, L.; Perkins, A.C.; Spiller, R.C. Abnormal intestinal permeability in subgroups of diarrhea-predominant irritable bowel syndromes. Am. J. Gastroenterol. 2006, 101, 1288–1294. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Du, L.; Kim, J.J.; Chen, B.; Zhu, Y.; Zhang, Y.; Yao, S.; He, H.; Zheng, X.; Huang, Z.; et al. MLCK-mediated intestinal permeability promotes immune activation and visceral hypersensitivity in PI-IBS mice. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2018, 30, e13348. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Verne, M.L.; Fields, J.Z.; Lefante, J.J.; Basra, S.; Salameh, H.; Verne, G.N. Randomised placebo-controlled trial of dietary glutamine supplements for postinfectious irritable bowel syndrome. Gut 2019, 68, 996–1002. [Google Scholar] [CrossRef]
- Dhawan, P.; Singh, A.B.; Deane, N.G.; No, Y.; Shiou, S.R.; Schmidt, C.; Neff, J.; Washington, M.K.; Beauchamp, R.D. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J. Clin. Investig. 2005, 115, 1765–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.R.; Nalle, S.C.; Tretiakova, M.; Rubin, D.T.; Turner, J.R. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab. Investig. A J. Tech. Methods Pathol. 2008, 88, 1110–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE 2013, 8, e56964. [Google Scholar] [CrossRef] [Green Version]
- Boleij, A.; Hechenbleikner, E.M.; Goodwin, A.C.; Badani, R.; Stein, E.M.; Lazarev, M.G.; Ellis, B.; Carroll, K.C.; Albesiano, E.; Wick, E.C.; et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2015, 60, 208–215. [Google Scholar] [CrossRef]
- Biarc, J.; Nguyen, I.S.; Pini, A.; Gosse, F.; Richert, S.; Thierse, D.; Van Dorsselaer, A.; Leize-Wagner, E.; Raul, F.; Klein, J.P.; et al. Carcinogenic properties of proteins with pro-inflammatory activity from Streptococcus infantarius (formerly S.bovis). Carcinogenesis 2004, 25, 1477–1484. [Google Scholar] [CrossRef]
- Genser, L.; Aguanno, D.; Soula, H.A.; Dong, L.; Trystram, L.; Assmann, K.; Salem, J.E.; Vaillant, J.C.; Oppert, J.M.; Laugerette, F.; et al. Increased jejunal permeability in human obesity is revealed by a lipid challenge and is linked to inflammation and type 2 diabetes. J. Pathol. 2018, 246, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef] [Green Version]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meddings, J.B.; Jarand, J.; Urbanski, S.J.; Hardin, J.; Gall, D.G. Increased gastrointestinal permeability is an early lesion in the spontaneously diabetic BB rat. Am. J. Physiol. 1999, 276, G951–G957. [Google Scholar] [CrossRef]
- Neu, J.; Reverte, C.M.; Mackey, A.D.; Liboni, K.; Tuhacek-Tenace, L.M.; Hatch, M.; Li, N.; Caicedo, R.A.; Schatz, D.A.; Atkinson, M. Changes in intestinal morphology and permeability in the biobreeding rat before the onset of type 1 diabetes. J. Pediatric Gastroenterol. Nutr. 2005, 40, 589–595. [Google Scholar] [CrossRef]
- Bosi, E.; Molteni, L.; Radaelli, M.G.; Folini, L.; Fermo, I.; Bazzigaluppi, E.; Piemonti, L.; Pastore, M.R.; Paroni, R. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia 2006, 49, 2824–2827. [Google Scholar] [CrossRef] [Green Version]
- Watts, T.; Berti, I.; Sapone, A.; Gerarduzzi, T.; Not, T.; Zielke, R.; Fasano, A. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc. Natl. Acad. Sci. USA 2005, 102, 2916–2921. [Google Scholar] [CrossRef] [Green Version]
- Sapone, A.; de Magistris, L.; Pietzak, M.; Clemente, M.G.; Tripathi, A.; Cucca, F.; Lampis, R.; Kryszak, D.; Carteni, M.; Generoso, M.; et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives. Diabetes 2006, 55, 1443–1449. [Google Scholar] [CrossRef] [Green Version]
- Visser, J.T.; Lammers, K.; Hoogendijk, A.; Boer, M.W.; Brugman, S.; Beijer-Liefers, S.; Zandvoort, A.; Harmsen, H.; Welling, G.; Stellaard, F.; et al. Restoration of impaired intestinal barrier function by the hydrolysed casein diet contributes to the prevention of type 1 diabetes in the diabetes-prone BioBreeding rat. Diabetologia 2010, 53, 2621–2628. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.K.; Ou, J.; Liang, S.; Zhou, X.; Hu, X. Epithelial Hes1 maintains gut homeostasis by preventing microbial dysbiosis. Mucosal Immunol. 2018, 11, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.; Pedersen, E.E.; Galante, P.; Hald, J.; Heller, R.S.; Ishibashi, M.; Kageyama, R.; Guillemot, F.; Serup, P.; Madsen, O.D. Control of endodermal endocrine development by Hes-1. Nat. Genet. 2000, 24, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Fukui, H.; Kayahara, T.; Sawada, M.; Seno, H.; Hiai, H.; Kageyama, R.; Okano, H.; Chiba, T. Hes1-deficient mice show precocious differentiation of Paneth cells in the small intestine. Biochem. Biophys. Res. Commun. 2005, 328, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Bermingham, N.A.; Finegold, M.J.; Zoghbi, H.Y. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 2001, 294, 2155–2158. [Google Scholar] [CrossRef]
- Liu, X.; Lu, J.; Liu, Z.; Zhao, J.; Sun, H.; Wu, N.; Liu, H.; Liu, W.; Hu, Z.; Meng, G.; et al. Intestinal Epithelial Cell-Derived LKB1 Suppresses Colitogenic Microbiota. J. Immunol. 2018, 200, 1889–1900. [Google Scholar] [CrossRef]
- Shimada, T.; Koike, T.; Yamagata, M.; Yoneda, M.; Hiraishi, H. Regulation of TFF3 expression by homeodomain protein CDX2. Regul. Pept. 2007, 140, 81–87. [Google Scholar] [CrossRef]
- Beuling, E.; Baffour-Awuah, N.Y.; Stapleton, K.A.; Aronson, B.E.; Noah, T.K.; Shroyer, N.F.; Duncan, S.A.; Fleet, J.C.; Krasinski, S.D. GATA factors regulate proliferation, differentiation, and gene expression in small intestine of mature mice. Gastroenterology 2011, 140, 1219–1229.e2. [Google Scholar] [CrossRef] [Green Version]
- Beuling, E.; Aronson, B.E.; Tran, L.M.; Stapleton, K.A.; ter Horst, E.N.; Vissers, L.A.; Verzi, M.P.; Krasinski, S.D. GATA6 is required for proliferation, migration, secretory cell maturation, and gene expression in the mature mouse colon. Mol. Cell. Biol. 2012, 32, 3392–3402. [Google Scholar] [CrossRef] [Green Version]
- Laudisi, F.; Stolfi, C.; Bevivino, G.; Maresca, C.; Franze, E.; Troncone, E.; Lolli, E.; Marafini, I.; Pietrucci, D.; Teofani, A.; et al. GATA6 deficiency leads to epithelial barrier dysfunction and enhances susceptibility to gut inflammation. J. Crohn’s Colitis 2021, jjab145. [Google Scholar] [CrossRef]
- Mori-Akiyama, Y.; van den Born, M.; van Es, J.H.; Hamilton, S.R.; Adams, H.P.; Zhang, J.; Clevers, H.; de Crombrugghe, B. SOX9 is required for the differentiation of paneth cells in the intestinal epithelium. Gastroenterology 2007, 133, 539–546. [Google Scholar] [CrossRef]
- Riba, A.; Olier, M.; Lacroix-Lamande, S.; Lencina, C.; Bacquie, V.; Harkat, C.; Gillet, M.; Baron, M.; Sommer, C.; Mallet, V.; et al. Paneth Cell Defects Induce Microbiota Dysbiosis in Mice and Promote Visceral Hypersensitivity. Gastroenterology 2017, 153, 1594–1606.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laukoetter, M.G.; Nava, P.; Lee, W.Y.; Severson, E.A.; Capaldo, C.T.; Babbin, B.A.; Williams, I.R.; Koval, M.; Peatman, E.; Campbell, J.A.; et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 2007, 204, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Wada, M.; Tamura, A.; Takahashi, N.; Tsukita, S. Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology 2013, 144, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Choi, W.; Kuo, W.T.; Singh, G.; Sailer, A.; Wang, Y.; Shen, L.; Fanning, A.S.; Turner, J.R. The scaffolding protein ZO-1 coordinates actomyosin and epithelial apical specializations in vitro and in vivo. J. Biol. Chem. 2018, 293, 17317–17335. [Google Scholar] [CrossRef] [Green Version]
- Kuo, W.T.; Zuo, L.; Odenwald, M.A.; Madha, S.; Singh, G.; Gurniak, C.B.; Abraham, C.; Turner, J.R. The Tight Junction Protein ZO-1 Is Dispensable for Barrier Function but Critical for Effective Mucosal Repair. Gastroenterology 2021, 161, 1924–1939. [Google Scholar] [CrossRef]
- Marchelletta, R.R.; Krishnan, M.; Spalinger, M.R.; Placone, T.W.; Alvarez, R.; Sayoc-Becerra, A.; Canale, V.; Shawki, A.; Park, Y.S.; Bernts, L.H.; et al. T cell protein tyrosine phosphatase protects intestinal barrier function by restricting epithelial tight junction remodeling. J. Clin. Investig. 2021, 131, e138230. [Google Scholar] [CrossRef]
- Liso, M.; De Santis, S.; Verna, G.; Dicarlo, M.; Calasso, M.; Santino, A.; Gigante, I.; Eri, R.; Raveenthiraraj, S.; Sobolewski, A.; et al. A Specific Mutation in Muc2 Determines Early Dysbiosis in Colitis-Prone Winnie Mice. Inflamm. Bowel Dis. 2020, 26, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Eri, R.D.; Adams, R.J.; Tran, T.V.; Tong, H.; Das, I.; Roche, D.K.; Oancea, I.; Png, C.W.; Jeffery, P.L.; Radford-Smith, G.L.; et al. An intestinal epithelial defect conferring ER stress results in inflammation involving both innate and adaptive immunity. Mucosal Immunol. 2011, 4, 354–364. [Google Scholar] [CrossRef]
- Shroyer, N.F.; Wallis, D.; Venken, K.J.; Bellen, H.J.; Zoghbi, H.Y. Gfi1 functions downstream of Math1 to control intestinal secretory cell subtype allocation and differentiation. Genes Dev. 2005, 19, 2412–2417. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Luo, J.; Li, J.; Kim, G.; Chen, E.S.; Xiao, S.; Snapper, S.B.; Bao, B.; An, D.; Blumberg, R.S.; et al. Foxo1 controls gut homeostasis and commensalism by regulating mucus secretion. J. Exp. Med. 2021, 218, e20210324. [Google Scholar] [CrossRef]
- Simms, L.A.; Doecke, J.D.; Walsh, M.D.; Huang, N.; Fowler, E.V.; Radford-Smith, G.L. Reduced alpha-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn’s disease. Gut 2008, 57, 903–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehkamp, J.; Harder, J.; Weichenthal, M.; Schwab, M.; Schaffeler, E.; Schlee, M.; Herrlinger, K.R.; Stallmach, A.; Noack, F.; Fritz, P.; et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 2004, 53, 1658–1664. [Google Scholar] [CrossRef] [Green Version]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadwell, K.; Patel, K.K.; Maloney, N.S.; Liu, T.C.; Ng, A.C.; Storer, C.E.; Head, R.D.; Xavier, R.; Stappenbeck, T.S.; Virgin, H.W. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 2010, 141, 1135–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Bock, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Koslowski, M.J.; Kubler, I.; Chamaillard, M.; Schaeffeler, E.; Reinisch, W.; Wang, G.; Beisner, J.; Teml, A.; Peyrin-Biroulet, L.; Winter, S.; et al. Genetic variants of Wnt transcription factor TCF-4 (TCF7L2) putative promoter region are associated with small intestinal Crohn’s disease. PLoS ONE 2009, 4, e4496. [Google Scholar] [CrossRef]
- Wehkamp, J.; Wang, G.; Kubler, I.; Nuding, S.; Gregorieff, A.; Schnabel, A.; Kays, R.J.; Fellermann, K.; Burk, O.; Schwab, M.; et al. The Paneth cell alpha-defensin deficiency of ileal Crohn’s disease is linked to Wnt/Tcf-4. J. Immunol. 2007, 179, 3109–3118. [Google Scholar] [CrossRef] [Green Version]
- Neal, M.D.; Sodhi, C.P.; Jia, H.; Dyer, M.; Egan, C.E.; Yazji, I.; Good, M.; Afrazi, A.; Marino, R.; Slagle, D.; et al. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. J. Biol. Chem. 2012, 287, 37296–37308. [Google Scholar] [CrossRef] [Green Version]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cezard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef]
- Cooney, R.; Baker, J.; Brain, O.; Danis, B.; Pichulik, T.; Allan, P.; Ferguson, D.J.; Campbell, B.J.; Jewell, D.; Simmons, A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010, 16, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Petnicki-Ocwieja, T.; Hrncir, T.; Liu, Y.J.; Biswas, A.; Hudcovic, T.; Tlaskalova-Hogenova, H.; Kobayashi, K.S. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 15813–15818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, G.; Rossi, R.; Commere, P.H.; Jay, P.; Sansonetti, P.J. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe 2014, 15, 792–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasberger, H.; Magis, A.T.; Sheng, E.; Conomos, M.P.; Zhang, M.; Garzotto, L.S.; Hou, G.; Bishu, S.; Nagao-Kitamoto, H.; El-Zaatari, M.; et al. DUOX2 variants associate with preclinical disturbances in microbiota-immune homeostasis and increased inflammatory bowel disease risk. J. Clin. Investig. 2021, 131, e141676. [Google Scholar] [CrossRef] [PubMed]
- Pircalabioru, G.; Aviello, G.; Kubica, M.; Zhdanov, A.; Paclet, M.H.; Brennan, L.; Hertzberger, R.; Papkovsky, D.; Bourke, B.; Knaus, U.G. Defensive Mutualism Rescues NADPH Oxidase Inactivation in Gut Infection. Cell Host Microbe 2016, 19, 651–663. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef] [Green Version]
- Dring, M.M.; Goulding, C.A.; Trimble, V.I.; Keegan, D.; Ryan, A.W.; Brophy, K.M.; Smyth, C.M.; Keeling, P.W.; O’Donoghue, D.; O’Sullivan, M.; et al. The pregnane X receptor locus is associated with susceptibility to inflammatory bowel disease. Gastroenterology 2006, 130, 341–348. [Google Scholar] [CrossRef]
- Martinez, A.; Marquez, A.; Mendoza, J.; Taxonera, C.; Fernandez-Arquero, M.; Diaz-Rubio, M.; de la Concha, E.G.; Urcelay, E. Role of the PXR gene locus in inflammatory bowel diseases. Inflamm. Bowel Dis. 2007, 13, 1484–1487. [Google Scholar] [CrossRef]
- Alvarado, D.M.; Chen, B.; Iticovici, M.; Thaker, A.I.; Dai, N.; VanDussen, K.L.; Shaikh, N.; Lim, C.K.; Guillemin, G.J.; Tarr, P.I.; et al. Epithelial Indoleamine 2,3-Dioxygenase 1 Modulates Aryl Hydrocarbon Receptor and Notch Signaling to Increase Differentiation of Secretory Cells and Alter Mucus-Associated Microbiota. Gastroenterology 2019, 157, 1093–1108.e11. [Google Scholar] [CrossRef]
- Metidji, A.; Omenetti, S.; Crotta, S.; Li, Y.; Nye, E.; Ross, E.; Li, V.; Maradana, M.R.; Schiering, C.; Stockinger, B. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2018, 49, 353–362.e5. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Wang, Q.; Ma, Y.; Li, L.; Yu, K.; Zhang, Z.; Chen, G.; Li, X.; Xiao, W.; Xu, P.; et al. Aryl Hydrocarbon Receptor Activation Modulates Intestinal Epithelial Barrier Function by Maintaining Tight Junction Integrity. Int. J. Biol. Sci. 2018, 14, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, J.R.; Strauss, J.D.; Bielecka, A.; Porto, A.F.; Lobo, F.M.; Urban, A.; Schofield, W.B.; Palm, N.W. IgA-deficient humans exhibit gut microbiota dysbiosis despite secretion of compensatory IgM. Sci. Rep. 2019, 9, 13574. [Google Scholar] [CrossRef] [PubMed]
- Johansen, F.E.; Pekna, M.; Norderhaug, I.N.; Haneberg, B.; Hietala, M.A.; Krajci, P.; Betsholtz, C.; Brandtzaeg, P. Absence of epithelial immunoglobulin A transport, with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory component-deficient mice. J. Exp. Med. 1999, 190, 915–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, S.; Kawaguchi-Miyashita, M.; Kushiro, A.; Sato, T.; Nanno, M.; Sako, T.; Matsuoka, Y.; Sudo, K.; Tagawa, Y.; Iwakura, Y.; et al. Generation of polymeric immunoglobulin receptor-deficient mouse with marked reduction of secretory IgA. J. Immunol. 1999, 163, 5367–5373. [Google Scholar] [PubMed]
- Menta, P.L.R.; Andrade, M.E.R.; Leocadio, P.C.L.; Fraga, J.R.; Dias, M.T.S.; Cara, D.C.; Cardoso, V.N.; Borges, L.F.; Capettini, L.S.A.; Aguilar, E.C.; et al. Wheat gluten intake increases the severity of experimental colitis and bacterial translocation by weakening of the proteins of the junctional complex. Br. J. Nutr. 2019, 121, 361–373. [Google Scholar] [CrossRef]
- Johnson, R.J.; Rivard, C.; Lanaspa, M.A.; Otabachian-Smith, S.; Ishimoto, T.; Cicerchi, C.; Cheeke, P.R.; Macintosh, B.; Hess, T. Fructokinase, Fructans, Intestinal Permeability, and Metabolic Syndrome: An Equine Connection? J. Equine Vet. Sci. 2013, 33, 120–126. [Google Scholar] [CrossRef] [Green Version]
- Gulhane, M.; Murray, L.; Lourie, R.; Tong, H.; Sheng, Y.H.; Wang, R.; Kang, A.; Schreiber, V.; Wong, K.Y.; Magor, G.; et al. High Fat Diets Induce Colonic Epithelial Cell Stress and Inflammation that is Reversed by IL-22. Sci. Rep. 2016, 6, 28990. [Google Scholar] [CrossRef] [Green Version]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.Y.; Nguyen, D.; Bui, V.; Nguyen, H.; Hoa, N. Ethanol modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. 1999, 276, G965–G974. [Google Scholar] [CrossRef]
- Elamin, E.; Jonkers, D.; Juuti-Uusitalo, K.; van Ijzendoorn, S.; Troost, F.; Duimel, H.; Broers, J.; Verheyen, F.; Dekker, J.; Masclee, A. Effects of ethanol and acetaldehyde on tight junction integrity: In vitro study in a three dimensional intestinal epithelial cell culture model. PLoS ONE 2012, 7, e35008. [Google Scholar] [CrossRef] [Green Version]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankaran-Walters, S.; Hart, R.; Dills, C. Guardians of the Gut: Enteric Defensins. Front. Microbiol. 2017, 8, 647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.; Nakamura, K.; Yoshii, A.; Yokoi, Y.; Kikuchi, M.; Shinozaki, R.; Nakamura, S.; Ohira, S.; Sugimoto, R.; Ayabe, T. Paneth cell alpha-defensin misfolding correlates with dysbiosis and ileitis in Crohn’s disease model mice. Life Sci. Alliance 2020, 3, e201900592. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhou, S.Y.; Gillilland, M., 3rd; Li, J.Y.; Lee, A.; Gao, J.; Zhang, G.; Xu, X.; Owyang, C. Bile acid toxicity in Paneth cells contributes to gut dysbiosis induced by high-fat feeding. JCI Insight 2020, 5, e138881. [Google Scholar] [CrossRef]
- Liu, T.C.; Kern, J.T.; Jain, U.; Sonnek, N.M.; Xiong, S.; Simpson, K.F.; VanDussen, K.L.; Winkler, E.S.; Haritunians, T.; Malique, A.; et al. Western diet induces Paneth cell defects through microbiome alterations and farnesoid X receptor and type I interferon activation. Cell Host Microbe 2021, 29, 988–1001.e6. [Google Scholar] [CrossRef]
- Brawner, K.M.; Yeramilli, V.A.; Duck, L.W.; Van Der Pol, W.; Smythies, L.E.; Morrow, C.D.; Elson, C.O.; Martin, C.A. Depletion of dietary aryl hydrocarbon receptor ligands alters microbiota composition and function. Sci. Rep. 2019, 9, 14724. [Google Scholar] [CrossRef] [Green Version]
- Schanz, O.; Chijiiwa, R.; Cengiz, S.C.; Majlesain, Y.; Weighardt, H.; Takeyama, H.; Forster, I. Dietary AhR Ligands Regulate AhRR Expression in Intestinal Immune Cells and Intestinal Microbiota Composition. Int. J. Mol. Sci. 2020, 21, 3189. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Wu, S.; Xia, Y.; Sun, J. Salmonella infection upregulates the leaky protein claudin-2 in intestinal epithelial cells. PLoS ONE 2013, 8, e58606. [Google Scholar] [CrossRef] [Green Version]
- Fasano, A.; Baudry, B.; Pumplin, D.W.; Wasserman, S.S.; Tall, B.D.; Ketley, J.M.; Kaper, J.B. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc. Natl. Acad. Sci. USA 1991, 88, 5242–5246. [Google Scholar] [CrossRef] [Green Version]
- Fasano, A.; Fiorentini, C.; Donelli, G.; Uzzau, S.; Kaper, J.B.; Margaretten, K.; Ding, X.; Guandalini, S.; Comstock, L.; Goldblum, S.E. Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J. Clin. Investig. 1995, 96, 710–720. [Google Scholar] [CrossRef]
- Jakobsson, H.E.; Rodriguez-Pineiro, A.M.; Schutte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Backhed, F.; Hansson, G.C.; Johansson, M.E. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015, 16, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Huang, Y.; Wang, Y.; Wang, P.; Song, H.; Wang, F. Antibiotics induced intestinal tight junction barrier dysfunction is associated with microbiota dysbiosis, activated NLRP3 inflammasome and autophagy. PLoS ONE 2019, 14, e0218384. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Young, C.; Neu, J. Molecular modulation of intestinal epithelial barrier: Contribution of microbiota. J. Biomed. Biotechnol. 2010, 2010, 305879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.; Clevers, H. Cell fate specification and differentiation in the adult mammalian intestine. Nat. Rev. Mol. Cell Biol. 2021, 22, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Fre, S.; Huyghe, M.; Mourikis, P.; Robine, S.; Louvard, D.; Artavanis-Tsakonas, S. Notch signals control the fate of immature progenitor cells in the intestine. Nature 2005, 435, 964–968. [Google Scholar] [CrossRef]
- Calon, A.; Gross, I.; Lhermitte, B.; Martin, E.; Beck, F.; Duclos, B.; Kedinger, M.; Duluc, I.; Domon-Dell, C.; Freund, J.N. Different effects of the Cdx1 and Cdx2 homeobox genes in a murine model of intestinal inflammation. Gut 2007, 56, 1688–1695. [Google Scholar] [CrossRef]
- Mesquita, P.; Jonckheere, N.; Almeida, R.; Ducourouble, M.P.; Serpa, J.; Silva, E.; Pigny, P.; Silva, F.S.; Reis, C.; Silberg, D.; et al. Human MUC2 mucin gene is transcriptionally regulated by Cdx homeodomain proteins in gastrointestinal carcinoma cell lines. J. Biol. Chem. 2003, 278, 51549–51556. [Google Scholar] [CrossRef] [Green Version]
- Lorentz, O.; Duluc, I.; Arcangelis, A.D.; Simon-Assmann, P.; Kedinger, M.; Freund, J.N. Key role of the Cdx2 homeobox gene in extracellular matrix-mediated intestinal cell differentiation. J. Cell Biol. 1997, 139, 1553–1565. [Google Scholar] [CrossRef] [Green Version]
- Hinoi, T.; Lucas, P.C.; Kuick, R.; Hanash, S.; Cho, K.R.; Fearon, E.R. CDX2 regulates liver intestine-cadherin expression in normal and malignant colon epithelium and intestinal metaplasia. Gastroenterology 2002, 123, 1565–1577. [Google Scholar] [CrossRef]
- Keller, M.S.; Ezaki, T.; Guo, R.J.; Lynch, J.P. Cdx1 or Cdx2 expression activates E-cadherin-mediated cell-cell adhesion and compaction in human COLO 205 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G104–G114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrisey, E.E.; Ip, H.S.; Lu, M.M.; Parmacek, M.S. GATA-6: A zinc finger transcription factor that is expressed in multiple cell lineages derived from lateral mesoderm. Dev. Biol. 1996, 177, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menard, S.; Cerf-Bensussan, N.; Heyman, M. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal Immunol. 2010, 3, 247–259. [Google Scholar] [CrossRef]
- Schneeberger, E.E.; Lynch, R.D. Structure, function, and regulation of cellular tight junctions. Am. J. Physiol. 1992, 262, L647–L661. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.M. Breaking through the tight junction barrier. J. Cell Biol. 1993, 123, 1631–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukita, S.; Furuse, M. The structure and function of claudins, cell adhesion molecules at tight junctions. Ann. N. Y. Acad. Sci. 2000, 915, 129–135. [Google Scholar] [CrossRef]
- Harhaj, N.S.; Antonetti, D.A. Regulation of tight junctions and loss of barrier function in pathophysiology. Int. J. Biochem. Cell Biol. 2004, 36, 1206–1237. [Google Scholar] [CrossRef]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Et Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Clemente, M.G.; De Virgiliis, S.; Kang, J.S.; Macatagney, R.; Musu, M.P.; Di Pierro, M.R.; Drago, S.; Congia, M.; Fasano, A. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut 2003, 52, 218–223. [Google Scholar] [CrossRef]
- Tripathi, A.; Lammers, K.M.; Goldblum, S.; Shea-Donohue, T.; Netzel-Arnett, S.; Buzza, M.S.; Antalis, T.M.; Vogel, S.N.; Zhao, A.; Yang, S.; et al. Identification of human zonulin, a physiological modulator of tight junctions, as prehaptoglobin-2. Proc. Natl. Acad. Sci. USA 2009, 106, 16799–16804. [Google Scholar] [CrossRef] [Green Version]
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Kucharzik, T.; Walsh, S.V.; Chen, J.; Parkos, C.A.; Nusrat, A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am. J. Pathol. 2001, 159, 2001–2009. [Google Scholar] [CrossRef] [Green Version]
- Dhawan, P.; Ahmad, R.; Chaturvedi, R.; Smith, J.J.; Midha, R.; Mittal, M.K.; Krishnan, M.; Chen, X.; Eschrich, S.; Yeatman, T.J.; et al. Claudin-2 expression increases tumorigenicity of colon cancer cells: Role of epidermal growth factor receptor activation. Oncogene 2011, 30, 3234–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damci, T.; Nuhoglu, I.; Devranoglu, G.; Osar, Z.; Demir, M.; Ilkova, H. Increased intestinal permeability as a cause of fluctuating postprandial blood glucose levels in Type 1 diabetic patients. Eur. J. Clin. Investig. 2003, 33, 397–401. [Google Scholar] [CrossRef]
- Carratu, R.; Secondulfo, M.; de Magistris, L.; Iafusco, D.; Urio, A.; Carbone, M.G.; Pontoni, G.; Carteni, M.; Prisco, F. Altered intestinal permeability to mannitol in diabetes mellitus type I. J. Pediatric Gastroenterol. Nutr. 1999, 28, 264–269. [Google Scholar] [CrossRef]
- Khoshbin, K.; Camilleri, M. Effects of dietary components on intestinal permeability in health and disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G589–G608. [Google Scholar] [CrossRef]
- Johansson, M.E.; Sjovall, H.; Hansson, G.C. The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.E.; Ambort, D.; Pelaseyed, T.; Schutte, A.; Gustafsson, J.K.; Ermund, A.; Subramani, D.B.; Holmen-Larsson, J.M.; Thomsson, K.A.; Bergstrom, J.H.; et al. Composition and functional role of the mucus layers in the intestine. Cell. Mol. Life Sci. CMLS 2011, 68, 3635–3641. [Google Scholar] [CrossRef]
- Arike, L.; Hansson, G.C. The Densely O-Glycosylated MUC2 Mucin Protects the Intestine and Provides Food for the Commensal Bacteria. J. Mol. Biol. 2016, 428, 3221–3229. [Google Scholar] [CrossRef] [Green Version]
- Ayabe, T.; Satchell, D.P.; Wilson, C.L.; Parks, W.C.; Selsted, M.E.; Ouellette, A.J. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat. Immunol. 2000, 1, 113–118. [Google Scholar] [CrossRef]
- Ouellette, A.J. Paneth cells and innate immunity in the crypt microenvironment. Gastroenterology 1997, 113, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.M.; Rossi, O.; Meijerink, M.; van Baarlen, P. Epithelial crosstalk at the microbiota-mucosal interface. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4607–4614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgueno, J.F.; Abreu, M.T. Epithelial Toll-like receptors and their role in gut homeostasis and disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Nighot, M.; Rawat, M.; Al-Sadi, R.; Castillo, E.F.; Nighot, P.; Ma, T.Y. Lipopolysaccharide-Induced Increase in Intestinal Permeability Is Mediated by TAK-1 Activation of IKK and MLCK/MYLK Gene. Am. J. Pathol. 2019, 189, 797–812. [Google Scholar] [CrossRef] [PubMed]
- Burgueno, J.F.; Fritsch, J.; Gonzalez, E.E.; Landau, K.S.; Santander, A.M.; Fernandez, I.; Hazime, H.; Davies, J.M.; Santaolalla, R.; Phillips, M.C.; et al. Epithelial TLR4 Signaling Activates DUOX2 to Induce Microbiota-Driven Tumorigenesis. Gastroenterology 2021, 160, 797–808.e6. [Google Scholar] [CrossRef]
- He, B.; Xu, W.; Santini, P.A.; Polydorides, A.D.; Chiu, A.; Estrella, J.; Shan, M.; Chadburn, A.; Villanacci, V.; Plebani, A.; et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity 2007, 26, 812–826. [Google Scholar] [CrossRef] [Green Version]
- Cario, E.; Gerken, G.; Podolsky, D.K. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology 2007, 132, 1359–1374. [Google Scholar] [CrossRef]
- Cario, E.; Gerken, G.; Podolsky, D.K. Toll-like receptor 2 enhances ZO-1-associated intestinal epithelial barrier integrity via protein kinase C. Gastroenterology 2004, 127, 224–238. [Google Scholar] [CrossRef]
- Podolsky, D.K.; Gerken, G.; Eyking, A.; Cario, E. Colitis-associated variant of TLR2 causes impaired mucosal repair because of TFF3 deficiency. Gastroenterology 2009, 137, 209–220. [Google Scholar] [CrossRef]
- Xiao, H.; Gulen, M.F.; Qin, J.; Yao, J.; Bulek, K.; Kish, D.; Altuntas, C.Z.; Wald, D.; Ma, C.; Zhou, H.; et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity 2007, 26, 461–475. [Google Scholar] [CrossRef] [Green Version]
- Werts, C.; Rubino, S.; Ling, A.; Girardin, S.E.; Philpott, D.J. Nod-like receptors in intestinal homeostasis, inflammation, and cancer. J. Leukoc. Biol. 2011, 90, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Claes, A.K.; Zhou, J.Y.; Philpott, D.J. NOD-Like Receptors: Guardians of Intestinal Mucosal Barriers. Physiology 2015, 30, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasberger, H.; Gao, J.; Nagao-Kitamoto, H.; Kitamoto, S.; Zhang, M.; Kamada, N.; Eaton, K.A.; El-Zaatari, M.; Shreiner, A.B.; Merchant, J.L.; et al. Increased Expression of DUOX2 Is an Epithelial Response to Mucosal Dysbiosis Required for Immune Homeostasis in Mouse Intestine. Gastroenterology 2015, 149, 1849–1859. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef]
- Lipinski, S.; Till, A.; Sina, C.; Arlt, A.; Grasberger, H.; Schreiber, S.; Rosenstiel, P. DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J. Cell Sci. 2009, 122, 3522–3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botteaux, A.; Hoste, C.; Dumont, J.E.; Van Sande, J.; Allaoui, A. Potential role of Noxes in the protection of mucosae: H2O2 as a bacterial repellent. Microbes Infect. 2009, 11, 537–544. [Google Scholar] [CrossRef]
- Corcionivoschi, N.; Alvarez, L.A.; Sharp, T.H.; Strengert, M.; Alemka, A.; Mantell, J.; Verkade, P.; Knaus, U.G.; Bourke, B. Mucosal reactive oxygen species decrease virulence by disrupting Campylobacter jejuni phosphotyrosine signaling. Cell Host Microbe 2012, 12, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Chu, F.F.; Esworthy, R.S.; Doroshow, J.H.; Grasberger, H.; Donko, A.; Leto, T.L.; Gao, Q.; Shen, B. Deficiency in Duox2 activity alleviates ileitis in GPx1- and GPx2-knockout mice without affecting apoptosis incidence in the crypt epithelium. Redox Biol. 2017, 11, 144–156. [Google Scholar] [CrossRef]
- MacFie, T.S.; Poulsom, R.; Parker, A.; Warnes, G.; Boitsova, T.; Nijhuis, A.; Suraweera, N.; Poehlmann, A.; Szary, J.; Feakins, R.; et al. DUOX2 and DUOXA2 form the predominant enzyme system capable of producing the reactive oxygen species H2O2 in active ulcerative colitis and are modulated by 5-aminosalicylic acid. Inflamm. Bowel Dis. 2014, 20, 514–524. [Google Scholar] [CrossRef]
- Haberman, Y.; Tickle, T.L.; Dexheimer, P.J.; Kim, M.O.; Tang, D.; Karns, R.; Baldassano, R.N.; Noe, J.D.; Rosh, J.; Markowitz, J.; et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Investig. 2014, 124, 3617–3633. [Google Scholar] [CrossRef] [Green Version]
- Hayes, P.; Dhillon, S.; O’Neill, K.; Thoeni, C.; Hui, K.Y.; Elkadri, A.; Guo, C.H.; Kovacic, L.; Aviello, G.; Alvarez, L.A.; et al. Defects in NADPH Oxidase Genes NOX1 and DUOX2 in Very Early Onset Inflammatory Bowel Disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Shah, Y.M.; Gonzalez, F.J. Pregnane X receptor as a target for treatment of inflammatory bowel disorders. Trends Pharmacol. Sci. 2012, 33, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Xie, W. Pregnane X receptor and constitutive androstane receptor at the crossroads of drug metabolism and energy metabolism. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 2091–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frericks, M.; Meissner, M.; Esser, C. Microarray analysis of the AHR system: Tissue-specific flexibility in signal and target genes. Toxicol. Appl. Pharmacol. 2007, 220, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Terc, J.; Hansen, A.; Alston, L.; Hirota, S.A. Pregnane X receptor agonists enhance intestinal epithelial wound healing and repair of the intestinal barrier following the induction of experimental colitis. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2014, 55, 12–19. [Google Scholar] [CrossRef]
- Stockinger, B.; Shah, K.; Wincent, E. AHR in the intestinal microenvironment: Safeguarding barrier function. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 559–570. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Y.; Guo, C.; Wang, J.; Boral, D.; Nie, D. Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem. Pharmacol. 2012, 83, 1112–1126. [Google Scholar] [CrossRef] [Green Version]
- Satsu, H.; Hiura, Y.; Mochizuki, K.; Hamada, M.; Shimizu, M. Activation of pregnane X receptor and induction of MDR1 by dietary phytochemicals. J. Agric. Food Chem. 2008, 56, 5366–5373. [Google Scholar] [CrossRef]
- Ren, Y.; Yue, B.; Ren, G.; Yu, Z.; Luo, X.; Sun, A.; Zhang, J.; Han, M.; Wang, Z.; Dou, W. Activation of PXR by alantolactone ameliorates DSS-induced experimental colitis via suppressing NF-kappaB signaling pathway. Sci. Rep. 2019, 9, 16636. [Google Scholar] [CrossRef]
- Shah, Y.M.; Ma, X.; Morimura, K.; Kim, I.; Gonzalez, F.J. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1114–G1122. [Google Scholar] [CrossRef]
- Garg, A.; Zhao, A.; Erickson, S.L.; Mukherjee, S.; Lau, A.J.; Alston, L.; Chang, T.K.; Mani, S.; Hirota, S.A. Pregnane X Receptor Activation Attenuates Inflammation-Associated Intestinal Epithelial Barrier Dysfunction by Inhibiting Cytokine-Induced Myosin Light-Chain Kinase Expression and c-Jun N-Terminal Kinase 1/2 Activation. J. Pharmacol. Exp. Ther. 2016, 359, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hankinson, O. The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 307–340. [Google Scholar] [CrossRef] [PubMed]
- Mulero-Navarro, S.; Fernandez-Salguero, P.M. New Trends in Aryl Hydrocarbon Receptor Biology. Front. Cell Dev. Biol. 2016, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Cella, M.; McDonald, K.G.; Garlanda, C.; Kennedy, G.D.; Nukaya, M.; Mantovani, A.; Kopan, R.; Bradfield, C.A.; Newberry, R.D.; et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 2011, 13, 144–151. [Google Scholar] [CrossRef]
- Schiering, C.; Wincent, E.; Metidji, A.; Iseppon, A.; Li, Y.; Potocnik, A.J.; Omenetti, S.; Henderson, C.J.; Wolf, C.R.; Nebert, D.W.; et al. Feedback control of AHR signalling regulates intestinal immunity. Nature 2017, 542, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, S.A.; Fu, J.; Chang, P.V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 19376–19387. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.; Martin-Gallausiaux, C.; Bourhis, J.M.; Beguet-Crespel, F.; Blottiere, H.M.; Lapaque, N. Identification of the novel role of butyrate as AhR ligand in human intestinal epithelial cells. Sci. Rep. 2019, 9, 643. [Google Scholar] [CrossRef] [PubMed]
- Fagarasan, S.; Honjo, T. Intestinal IgA synthesis: Regulation of front-line body defences. Nat. Rev. Immunol. 2003, 3, 63–72. [Google Scholar] [CrossRef]
- Corthesy, B. Multi-faceted functions of secretory IgA at mucosal surfaces. Front. Immunol. 2013, 4, 185. [Google Scholar] [CrossRef] [Green Version]
- Pabst, O.; Slack, E. IgA and the intestinal microbiota: The importance of being specific. Mucosal Immunol. 2020, 13, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K. Diversified IgA-Bacteria Interaction in Gut Homeostasis. Adv. Exp. Med. Biol. 2020, 1254, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Turula, H.; Wobus, C.E. The Role of the Polymeric Immunoglobulin Receptor and Secretory Immunoglobulins during Mucosal Infection and Immunity. Viruses 2018, 10, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, F.E.; Kaetzel, C.S. Regulation of the polymeric immunoglobulin receptor and IgA transport: New advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol. 2011, 4, 598–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantis, N.J.; Rol, N.; Corthesy, B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011, 4, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Diana, J.; Moura, I.C.; Vaugier, C.; Gestin, A.; Tissandie, E.; Beaudoin, L.; Corthesy, B.; Hocini, H.; Lehuen, A.; Monteiro, R.C. Secretory IgA induces tolerogenic dendritic cells through SIGNR1 dampening autoimmunity in mice. J. Immunol. 2013, 191, 2335–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, J.; Park, C.G.; Mantis, N.J. Recognition of secretory IgA by DC-SIGN: Implications for immune surveillance in the intestine. Immunol. Lett. 2010, 131, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludvigsson, J.F.; Neovius, M.; Hammarstrom, L. Association between IgA deficiency & other autoimmune conditions: A population-based matched cohort study. J. Clin. Immunol. 2014, 34, 444–451. [Google Scholar] [CrossRef]
- Aghamohammadi, A.; Cheraghi, T.; Gharagozlou, M.; Movahedi, M.; Rezaei, N.; Yeganeh, M.; Parvaneh, N.; Abolhassani, H.; Pourpak, Z.; Moin, M. IgA deficiency: Correlation between clinical and immunological phenotypes. J. Clin. Immunol. 2009, 29, 130–136. [Google Scholar] [CrossRef]
- Reikvam, D.H.; Derrien, M.; Islam, R.; Erofeev, A.; Grcic, V.; Sandvik, A.; Gaustad, P.; Meza-Zepeda, L.A.; Jahnsen, F.L.; Smidt, H.; et al. Epithelial-microbial crosstalk in polymeric Ig receptor deficient mice. Eur. J. Immunol. 2012, 42, 2959–2970. [Google Scholar] [CrossRef]
- Kato-Nagaoka, N.; Shimada, S.; Yamakawa, Y.; Tsujibe, S.; Naito, T.; Setoyama, H.; Watanabe, Y.; Shida, K.; Matsumoto, S.; Nanno, M. Enhanced differentiation of intraepithelial lymphocytes in the intestine of polymeric immunoglobulin receptor-deficient mice. Immunology 2015, 146, 59–69. [Google Scholar] [CrossRef]
- Yamazaki, K.; Shimada, S.; Kato-Nagaoka, N.; Soga, H.; Itoh, T.; Nanno, M. Accumulation of intestinal intraepithelial lymphocytes in association with lack of polymeric immunoglobulin receptor. Eur. J. Immunol. 2005, 35, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Uren, T.K.; Johansen, F.E.; Wijburg, O.L.; Koentgen, F.; Brandtzaeg, P.; Strugnell, R.A. Role of the polymeric Ig receptor in mucosal B cell homeostasis. J. Immunol. 2003, 170, 2531–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [Green Version]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vazquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Schirmer, M.; Franzosa, E.A.; Lloyd-Price, J.; McIver, L.J.; Schwager, R.; Poon, T.W.; Ananthakrishnan, A.N.; Andrews, E.; Barron, G.; Lake, K.; et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol. 2018, 3, 337–346. [Google Scholar] [CrossRef]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Erickson, A.R.; Cantarel, B.L.; Lamendella, R.; Darzi, Y.; Mongodin, E.F.; Pan, C.; Shah, M.; Halfvarson, J.; Tysk, C.; Henrissat, B.; et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS ONE 2012, 7, e49138. [Google Scholar] [CrossRef] [Green Version]
- Heintz-Buschart, A.; May, P.; Laczny, C.C.; Lebrun, L.A.; Bellora, C.; Krishna, A.; Wampach, L.; Schneider, J.G.; Hogan, A.; de Beaufort, C.; et al. Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nat. Microbiol. 2016, 2, 16180. [Google Scholar] [CrossRef]
- Martinez, C.; Antolin, M.; Santos, J.; Torrejon, A.; Casellas, F.; Borruel, N.; Guarner, F.; Malagelada, J.R. Unstable composition of the fecal microbiota in ulcerative colitis during clinical remission. Am. J. Gastroenterol. 2008, 103, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Rajca, S.; Grondin, V.; Louis, E.; Vernier-Massouille, G.; Grimaud, J.C.; Bouhnik, Y.; Laharie, D.; Dupas, J.L.; Pillant, H.; Picon, L.; et al. Alterations in the intestinal microbiome (dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 978–986. [Google Scholar] [CrossRef]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Liang, Q.Y.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef]
- Li, H.; Christman, L.M.; Li, R.; Gu, L. Synergic interactions between polyphenols and gut microbiota in mitigating inflammatory bowel diseases. Food Funct. 2020, 11, 4878–4891. [Google Scholar] [CrossRef] [PubMed]
- Vezza, T.; Rodriguez-Nogales, A.; Algieri, F.; Utrilla, M.P.; Rodriguez-Cabezas, M.E.; Galvez, J. Flavonoids in Inflammatory Bowel Disease: A Review. Nutrients 2016, 8, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Weigmann, B. A Novel Pathway of Flavonoids Protecting against Inflammatory Bowel Disease: Modulating Enteroendocrine System. Metabolites 2022, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Crawford, M.S.; McCole, D.F. JAK-STAT Pathway Regulation of Intestinal Permeability: Pathogenic Roles and Therapeutic Opportunities in Inflammatory Bowel Disease. Pharmaceuticals 2021, 14, 840. [Google Scholar] [CrossRef] [PubMed]
- Wagenaar, C.A.; van de Put, M.; Bisschops, M.; Walrabenstein, W.; de Jonge, C.S.; Herrema, H.; van Schaardenburg, D. The Effect of Dietary Interventions on Chronic Inflammatory Diseases in Relation to the Microbiome: A Systematic Review. Nutrients 2021, 13, 3208. [Google Scholar] [CrossRef]
- Wark, G.; Samocha-Bonet, D.; Ghaly, S.; Danta, M. The Role of Diet in the Pathogenesis and Management of Inflammatory Bowel Disease: A Review. Nutrients 2020, 13, 135. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- Tomova, A.; Bukovsky, I.; Rembert, E.; Yonas, W.; Alwarith, J.; Barnard, N.D.; Kahleova, H. The Effects of Vegetarian and Vegan Diets on Gut Microbiota. Front. Nutr. 2019, 6, 47. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016, 65, 1812–1821. [Google Scholar] [CrossRef]
- Sultan, S.; El-Mowafy, M.; Elgaml, A.; Ahmed, T.A.E.; Hassan, H.; Mottawea, W. Metabolic Influences of Gut Microbiota Dysbiosis on Inflammatory Bowel Disease. Front. Physiol. 2021, 12, 715506. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, J.W.; Lau, C.; Jacomino, M.; Finegold, M.; Henning, S.J. Improving access to intestinal stem cells as a step toward intestinal gene transfer. Hum. Gene Ther. 1994, 5, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Kitson, C.; Farley, R.; Steel, R.; Marriott, C.; Parkins, D.A.; Scarpa, M.; Wainwright, B.; Evans, M.J.; Colledge, W.H.; et al. Mucus altering agents as adjuncts for nonviral gene transfer to airway epithelium. Gene Ther. 2001, 8, 1380–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickles, R.J.; Fahrner, J.A.; Petrella, J.M.; Boucher, R.C.; Bergelson, J.M. Retargeting the coxsackievirus and adenovirus receptor to the apical surface of polarized epithelial cells reveals the glycocalyx as a barrier to adenovirus-mediated gene transfer. J. Virol. 2000, 74, 6050–6057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, H.; Kimura, T.; Haga, K.; Kasahara, N.; Anton, P.; McGowan, I. Effective in vivo and ex vivo gene transfer to intestinal mucosa by VSV-G-pseudotyped lentiviral vectors. BMC Gastroenterol. 2010, 10, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, H.; Haga, K.; Ohno, I.; Hiraoka, K.; Kimura, T.; Hermann, K.; Kasahara, N.; Anton, P.; McGowan, I. Mucosal gene therapy using a pseudotyped lentivirus vector encoding murine interleukin-10 (mIL-10) suppresses the development and relapse of experimental murine colitis. BMC Gastroenterol. 2014, 14, 68. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, J.; Van Montfrans, C.; Brennan, F.; Van Deventer, S.; Drillenburg, P.; Hodgson, H.; Te Velde, A.; Sol Rodriguez Pena, M. IL-10 gene therapy prevents TNBS-induced colitis. Gene Ther. 2002, 9, 1715–1721. [Google Scholar] [CrossRef] [Green Version]
- Buckinx, R.; Timmermans, J.P. Targeting the gastrointestinal tract with viral vectors: State of the art and possible applications in research and therapy. Histochem. Cell Biol. 2016, 146, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, S.; Galle, P.R.; Neurath, M.F. Efficient gene delivery to the inflamed colon by local administration of recombinant adenoviruses with normal or modified fibre structure. Gut 1999, 44, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, P.L.; Yuan, L.; Sun, K.; Kunovska, L.; Seregin, S.; Amalfitano, A.; Basson, M.D. Regulation of epithelial differentiation in rat intestine by intraluminal delivery of an adenoviral vector or silencing RNA coding for Schlafen 3. PLoS ONE 2013, 8, e79745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.R.; Thiele, D.L.; Silva, M.; Beutler, B. Adenoviral vectors given intravenously to immunocompromised mice yield stable transduction of the colonic epithelium. Gastroenterology 1997, 112, 1586–1594. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Observation | Ref. |
|---|---|---|
| IBD |
| [15,16,17,18,19] |
| Celiac disease |
| [20,21,22,23,24] |
| CDI |
| [25,26,27] |
| IBS |
| [28,29,30,31,32,33,34] |
| CRC |
| [35,36,37,38,39,40] |
| Obesity |
| [41,42,43,44] |
| Type 1 diabetes |
| [44,45,46,47,48,49,50] |
| Category | Gene | Effects on Intestinal Barrier | Ref. |
|---|---|---|---|
| Cell commitment | Hes1 | Reduced production of AMPs and mucus, gut dysbiosis, and inflammation. Precocious differentiation of Paneth cells. Impaired specification of IECs into enterocytes. | [51,52,53] |
| Math1 | Decreased frequency of goblet cells. | [54] | |
| Stk11 | Impaired released of AMPs and IL-18. Colitogenic bacteria overgrowth. | [55] | |
| Cdx2 | Altered mucus production and increased intestinal permeability and susceptibility to DSS-induced colitis. | [15,56] | |
| Gata6 | Impaired stem cell proliferation, reduction in villus length, Paneth cell, and enterocyte and enteroendocrine cell frequency. Increased number of goblet-like cells. Decreased levels of ZO-1 and increased intestinal permeability and susceptibility to experimental colitis and ileitis. Gut dysbiosis. | [57,58,59] | |
| Sox9 | Lack of differentiated Paneth cells, crypt enlargement, gut dysbiosis. | [60,61] | |
| Junctional Complexes | Jam-A/F11R | Increased intestinal permeability, low-grade intestinal inflammation, and increased susceptibility to DSS-induced colitis. | [62] |
| Cldn-2 and Cldn-15 double-KO | Impaired paracellular Na+ flow and malnutrition. | [63] | |
| Tjp1 | Apical surface brush border membrane and crevasses at intercellular junctions between enterocytes. Increased susceptibility to experimental colitis, delayed cell division, and mucosal healing. | [64,65] | |
| Ptpn2 | Increased claudin-2 expression, intestinal permeability, and inflammatory cytokine production. | [66] | |
| Mucus layer | Muc2 | ER stress and decreased frequency of goblet cells, altered mucus production, increased intestinal permeability, gut dysbiosis, and chronic intestinal inflammation. | [16,67,68] |
| Gfi1 | Accumulation of secretory progenitors, decrease in mucus and AMPs release. | [69] | |
| Foxo1 | Impaired mucus layer formation, overgrowth of mucin-degrading bacteria, and decrease of short-chain fatty acid-producing microbial species. Enhanced susceptibility to infection and inflammation. | [70] | |
| Paneth cells | Nod2 | Impaired α-defensins secretion. | [71,72] |
| Atg16l1 | Impaired autophagy in response to viral infection and decreased AMPs release. | [73,74] | |
| Xbp1 | Chronic ER stress in response to viral infection and decreased AMP release. | [75] | |
| Tcf4 | Reduced α-defensins secretion and CD development. | [76,77] | |
| PRRs | MyD88 | Increased stem cell proliferation. | [78] |
| Nod2 | Impaired pathogen sensing and clearance, and gut dysbiosis. No protection against oxidative stress-mediated cell death, and impaired epithelial regeneration. | [79,80,81,82,83] | |
| Oxidative Burst | Duox2 | Enhanced pathogen translocation to host lymphatic tissues. | [84] |
| Cyba | Decreased DUOX2 activity in response to Citrobacter rodentium. | [85] | |
| Xenobiotic Receptors | Pxr-Nr1I2 | Dysregulated TLR4-NF-κB signalling pathway, reduced ZO-1 and E-cadherin expression, increase in Claudin-2 levels. Higher susceptibility to IBD development. | [86,87,88] |
| AhR | Increased susceptibility to intestinal infection (Citrobacter rodentium), reduced mucus production, impaired tight junctions, and crypt hyperplasia. | [89,90,91] | |
| Secretory IgA | IgA | Gut dysbiosis. | [92] |
| pIgR | Impaired SIgA transcytosis across epithelial cells and gut dysbiosis. | [93,94] |
| Category | Diet | Effects on Intestinal Barrier | Ref. |
| Junctional Complexes | Gluten | Alterations in adherent junctions and desmosomes and increased intestinal permeability and susceptibility to experimental colitis. Disassembly of ZO-1 from the tight junctional complex. | [20,95] |
| Glucose/Fructose | TJ and AJ proteins dysfunction, increased susceptibility to pathogen infection, gut dysbiosis, metabolic syndrome, oxidative stress, and chronic inflammation | [44,96] | |
| High-fat diet | ER stress in IECs, impairment of Claudin-1 expression and mucus barrier, and increased endotoxin serum levels. Increased taurocholic bile acid production and gut dysbiosis. | [97,98] | |
| Ethanol | Altered ZO-1 and occludin localization and impaired paracellular permeability. | [99,100] | |
| Mucus Layer | Fiber-deprived diet | Thinner mucus layer, gut dysbiosis, and chronic intestinal inflammation. | [67,101] |
| Paneth cells | Vitamin D deficiency and exposure to high-fat diet | Impaired expression of α-defensins, MMP7, and tight junction-related proteins; increased intestinal permeability; gut dysbiosis; and metabolic syndrome. Indiction of ER stress and secretion of misfolded α-defensins. | [102,103] |
| High fat diet | Decreased AMP expression, ER stress and autophagy induction, and gut dysbiosis. | [104] | |
| Western diet (deoxycholic acid) | Excessive activation of the farnesoid X receptor and type I interferon signalling pathways. | [105] | |
| Xenobiotic receptors | AhR ligand-free diet | Higher susceptibility to experimental colitis and gut dysbiosis. | [106,107] |
| Category | Bacterial infection | Effects on intestinal barrier | Ref. |
| Junctional Complexes | Infection by Salmonella typhimurium | Increased claudin-2 expression and bacterial invasion. | [108] |
| Infection by Vibrio cholerae | Zonula occludes toxin production and altered paracellular permeability. | [109,110] | |
| PRRs | LPS | Increased intestinal permeability. | [111] |
| Category | Medication exposure | Effects on intestinal barrier | Ref. |
| Junctional Complexes | Antibiotic treatment | Decreased production of microbial-derived short-chain fatty acids. Gut dysbiosis. Reduced ZO-1, occludin, and claudin-1 expression, and increased intestinal permeability. Altered microvilli morphology and reduced rate of intestinal epithelial cell turnover. | [112,113] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stolfi, C.; Maresca, C.; Monteleone, G.; Laudisi, F. Implication of Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases. Biomedicines 2022, 10, 289. https://doi.org/10.3390/biomedicines10020289
Stolfi C, Maresca C, Monteleone G, Laudisi F. Implication of Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases. Biomedicines. 2022; 10(2):289. https://doi.org/10.3390/biomedicines10020289
Chicago/Turabian StyleStolfi, Carmine, Claudia Maresca, Giovanni Monteleone, and Federica Laudisi. 2022. "Implication of Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases" Biomedicines 10, no. 2: 289. https://doi.org/10.3390/biomedicines10020289