
Mitochondrial Ca2+ Signaling and Bioenergetics in Alzheimer’s Disease
 , and
, and 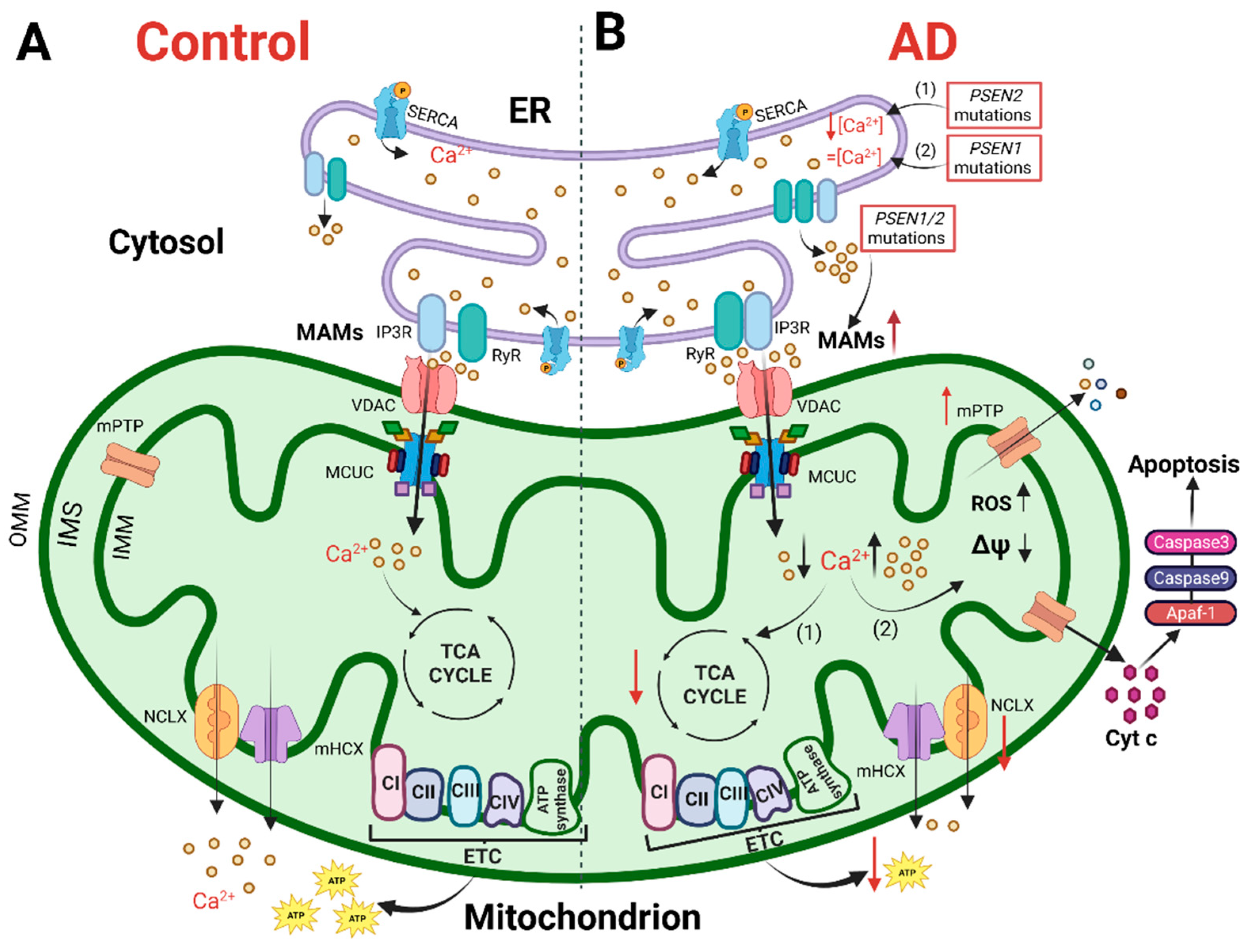{kind=link}
{kind=link}
Abstract
:1. Introduction
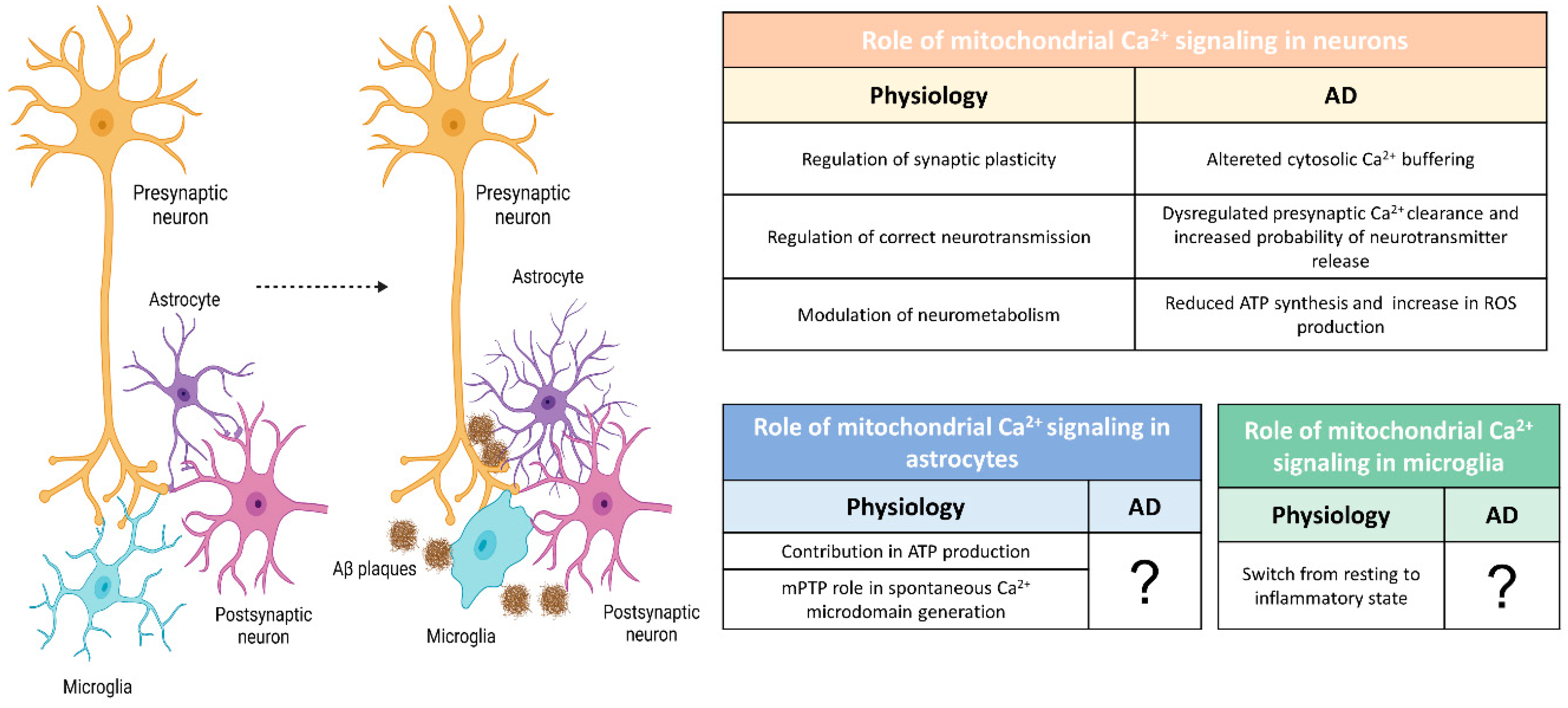
2. The Physiology of Brain Mitochondria: Ca2+ and Bioenergetics
2.1. Intracellular Ca2+ Handling
2.2. Mitochondrial Ca2+ Handling
2.3. Mitochondria, Ca2+ Hotspots and Ca2+ Microdomains
2.4. Mitochondrial Ca2+ Signaling and Bioenergetics: The Energy Match
3. Mitochondria in Alzheimer’s Disease: Ca2+ Signaling and Bioenergetics
3.1. Intracellular Ca2+ Signaling Alterations in AD
3.2. Mitochondrial Ca2+ Signaling Alterations in AD
3.3. Mitochondrial Bioenergetics in AD
4. Mitochondria Ca2+ Signaling and Bioenergetics as Therapeutic Targets
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sokoloff, L. Energetics of functional activation in neural tissues. Neurochem. Res. 1999, 24, 321–329. [Google Scholar] [CrossRef]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Pizzo, P. Highlighting the endoplasmic reticulum-mitochondria connection: Focus on Mitofusin 2. Pharmacol. Res. 2018, 128, 42–51. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Álvarez-Álvarez, I.; Guillén-Grima, F.; Aguinaga-Ontoso, I. Prevalence and incidence of Alzheimer’s disease in Europe: A meta-analysis. Neurologia 2017, 32, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 2016, 15, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Altomari, N.; Bruno, F.; Laganà, V.; Smirne, N.; Colao, R.; Curcio, S.; Di Lorenzo, R.; Frangipane, F.; Maletta, R.; Puccio, G.; et al. A Comparison of Behavioral and Psychological Symptoms of Dementia (BPSD) and BPSD Sub-Syndromes in Early-Onset and Late-Onset Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 85, 691–699. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Corbett, A.; Ballard, C. Emerging treatments for Alzheimer’s disease for non-amyloid and non-tau targets. Expert Rev. Neurother. 2017, 17, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Poudel, P.; Park, S. Recent Advances in the Treatment of Alzheimer’s Disease Using Nanoparticle-Based Drug Delivery Systems. Pharmaceutics 2022, 14, 835. [Google Scholar] [CrossRef]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, F.; Malvaso, A.; Canterini, S.; Bruni, A.C. Antimicrobial Peptides (AMPs) in the Pathogenesis of Alzheimer’s Disease: Implications for Diagnosis and Treatment. Antibiotics 2022, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020, 330, 113321. [Google Scholar] [CrossRef]
- Callens, M.; Loncke, J.; Bultynck, G. Dysregulated Ca2+ Homeostasis as a Central Theme in Neurodegeneration: Lessons from Alzheimer’s Disease and Wolfram Syndrome. Cells 2022, 11, 1963. [Google Scholar] [CrossRef] [PubMed]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Stevens, B. The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 2013, 61, 24–36. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Sidorenko, J.; Couvy-Duchesne, B.; Marioni, R.E.; Wright, M.J.; Goate, A.M.; Marcora, E.; Huang, K.L.; Porter, T.; Laws, S.M.; et al. Risk prediction of late-onset Alzheimer’s disease implies an oligogenic architecture. Nat. Commun. 2020, 11, 4799. [Google Scholar] [CrossRef]
- Wightman, D.P.; Jansen, I.E.; Savage, J.E.; Shadrin, A.A.; Bahrami, S.; Holland, D.; Rongve, A.; Børte, S.; Winsvold, B.S.; Drange, O.K.; et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 2021, 53, 1276–1282. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Cavalcante, G.C.; Brito, L.M.; Schaan, A.P.; Ribeiro-Dos-santos, Â.; de Araújo, G.S. Mitochondrial Genetics Reinforces Multiple Layers of Interaction in Alzheimer’s Disease. Biomedicines 2022, 10, 880. [Google Scholar] [CrossRef]
- Flaquer, A.; Baumbach, C.; Kriebel, J.; Meitinger, T.; Peters, A.; Waldenberger, M.; Grallert, H.; Strauch, K. Mitochondrial genetic variants identified to be associated with BMI in adults. PLoS ONE 2014, 9, e105116. [Google Scholar] [CrossRef] [PubMed]
- Rumshisky, A.; Ghassemi, M.; Naumann, T.; Szolovits, P.; Castro, V.M.; McCoy, T.H.; Perlis, R.H. Predicting early psychiatric readmission with natural language processing of narrative discharge summaries. Transl. Psychiatry 2016, 6, e921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, H.; Moser, N.; Huggenberger, S. Neuroanatomy of the Mouse; Springer International Publishing: Cham, Switzerland, 2020. [Google Scholar]
- de Rus Jacquet, A.; Denis, H.L.; Cicchetti, F.; Alpaugh, M. Current and future applications of induced pluripotent stem cell-based models to study pathological proteins in neurodegenerative disorders. Mol. Psychiatry 2021, 26, 2685–2706. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. VDAC isoforms in mammals. Biochim. Biophys. Acta 2012, 1818, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Perrone, M.; Caroccia, N.; Genovese, I.; Missiroli, S.; Modesti, L.; Pedriali, G.; Vezzani, B.; Vitto, V.A.M.; Antenori, M.; Lebiedzinska-Arciszewska, M.; et al. The role of mitochondria-associated membranes in cellular homeostasis and diseases. In International Review of Cell and Molecular Biology; Elsevier Publighing: Amsterdam, The Netherlands, 2020; ISBN 9780128197448. [Google Scholar]
- Hirabayashi, Y.; Kwon, S.-K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Ooi, K.; Hu, L.; Feng, Y.; Han, C.; Ren, X.; Qian, X.; Huang, H.; Chen, S.; Shi, Q.; Lin, H.; et al. Sigma-1 Receptor Activation Suppresses Microglia M1 Polarization via Regulating Endoplasmic Reticulum–Mitochondria Contact and Mitochondrial Functions in Stress-Induced Hypertension Rats. Mol. Neurobiol. 2021, 58, 6625–6646. [Google Scholar] [CrossRef]
- Göbel, J.; Engelhardt, E.; Pelzer, P.; Sakthivelu, V.; Jahn, H.M.; Jevtic, M.; Folz-Donahue, K.; Kukat, C.; Schauss, A.; Frese, C.K.; et al. Mitochondria-Endoplasmic Reticulum Contacts in Reactive Astrocytes Promote Vascular Remodeling. Cell Metab. 2020, 31, 791–808.e8. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.; Robitaille, R.; Volterra, A. Gliotransmitters Travel in Time and Space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, K.; Garaschuk, O. The role of intracellular calcium-store-mediated calcium signals in in vivo sensor and effector functions of microglia. J. Physiol. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Pizzo, P. Intracellular organelles in the saga of Ca2+ homeostasis: Different molecules for different purposes? Cell Mol. Life Sci. 2012, 69, 1077–1104. [Google Scholar] [CrossRef] [PubMed]
- Haydon, P.G.; Carmignoto, G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 2006, 86, 1009–1031. [Google Scholar] [CrossRef] [Green Version]
- Shigetomi, E.; Tong, X.; Kwan, K.Y.; Corey, D.P.; Khakh, B.S. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat. Neurosci. 2012, 15, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Färber, K.; Kettenmann, H. Functional role of calcium signals for microglial function. Glia 2006, 54, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.S.; Tang, Y.; Illes, P.; Verkhratsky, A. The Safeguarding Microglia: Central Role for P2Y12 Receptors. Front. Pharmacol. 2021, 11, 627760. [Google Scholar] [CrossRef] [PubMed]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef]
- Seo, M.D.; Enomoto, M.; Ishiyama, N.; Stathopulos, P.B.; Ikura, M. Structural insights into endoplasmic reticulum stored calcium regulation by inositol 1,4,5-trisphosphate and ryanodine receptors. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1853, 1980–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britzolaki, A.; Saurine, J.; Klocke, B.; Pitychoutis, P.M. A Role for SERCA Pumps in the Neurobiology of Neuropsychiatric and Neurodegenerative Disorders. In Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020. [Google Scholar]
- Lewis, R.S. Store-operated calcium channels: From function to structure and back again. Cold Spring Harb. Perspect. Biol. 2020, 12, a035055. [Google Scholar] [CrossRef]
- Wang, Q.C.; Zheng, Q.; Tan, H.; Zhang, B.; Li, X.; Yang, Y.; Yu, J.; Liu, Y.; Chai, H.; Wang, X.; et al. TMCO1 is an ER Ca2+ load-activated Ca2+ channel. Cell 2016, 165, 1454–1466. [Google Scholar] [CrossRef] [Green Version]
- Stafford, N.; Wilson, C.; Oceandy, D.; Neyses, L.; Cartwright, E.J. The plasma membrane calcium ATPases and their role as major new players in human disease. Physiol. Rev. 2017, 97, 1089–1125. [Google Scholar] [CrossRef] [Green Version]
- Schwaller, B. Cytosolic Ca2+ buffers are inherently Ca2+ signal modulators. Cold Spring Harb. Perspect. Biol. 2020, 12, a035543. [Google Scholar] [CrossRef]
- Elíes, J.; Yáñez, M.; Pereira, T.M.C.; Gil-Longo, J.; MacDougall, D.A.; Campos-Toimil, M. An Update to Calcium Binding Proteins. In Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020. [Google Scholar]
- Michalak, M.; Parker, J.M.R.; Opas, M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 2002, 32, 269–278. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef] [PubMed]
- Nemani, N.; Shanmughapriya, S.; Madesh, M. Molecular regulation of MCU: Implications in physiology and disease. Cell Calcium. 2018, 74, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; de Juan-Sanz, J.; Farrell, R.J.; Ryan, T.A. Molecular Tuning of the Axonal Mitochondrial Ca2+ Uniporter Ensures Metabolic Flexibility of Neurotransmission. Neuron 2020, 105, 678–687.e5. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Austin, S.; Mekis, R.; Mohammed, S.E.M.; Scalise, M.; Wang, W.-A.; Galluccio, M.; Pfeiffer, C.; Borovec, T.; Parapatics, K.; Vitko, D.; et al. TMBIM5 is the Ca2+/H+ antiporter of mammalian mitochondria. EMBO Rep. 2022, e54978. [Google Scholar] [CrossRef]
- Palty, R.; Sekler, I. The mitochondrial Na+/Ca2+ exchanger. Cell Calcium 2012, 52, 9–15. [Google Scholar] [CrossRef]
- Groten, C.J.; MacVicar, B.A. Mitochondrial Ca2+ uptake by the MCU facilitates pyramidal neuron excitability and metabolism during action potential firing. Commun. Biol. 2022, 5, 900. [Google Scholar] [CrossRef] [PubMed]
- Bas-Orth, C.; Schneider, J.; Lewen, A.; McQueen, J.; Hasenpusch-Theil, K.; Theil, T.; Hardingham, G.E.; Bading, H.; Kann, O. The mitochondrial calcium uniporter is crucial for the generation of fast cortical network rhythms. J. Cereb. Blood Flow Metab. 2020, 40, 2225–2239. [Google Scholar] [CrossRef]
- Lin, Y.; Li, L.-L.; Nie, W.; Liu, X.; Adler, A.; Xiao, C.; Lu, F.; Wang, L.; Han, H.; Wang, X.; et al. Brain activity regulates loose coupling between mitochondrial and cytosolic Ca2+ transients. Nat. Commun. 2019, 10, 5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, J.G.; Robinson, M.B. Regulation of mitochondrial dynamics in astrocytes: Mechanisms, consequences, and unknowns. Glia 2018, 66, 1213–1234. [Google Scholar] [CrossRef]
- Jackson, J.G.; Robinson, M.B. Reciprocal Regulation of Mitochondrial Dynamics and Calcium Signaling in Astrocyte Processes. J. Neurosci. 2015, 35, 15199–15213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brawek, B.; Garaschuk, O. Microglial calcium signaling in the adult, aged and diseased brain. Cell Calcium. 2013, 53, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, I.; Jha, S. Mitochondrial Dysfunction and Alzheimer’s Disease: Role of Microglia. Front. Aging Neurosci. 2020, 12, 252. [Google Scholar] [CrossRef]
- Filadi, R.; Pozzan, T. Generation and functions of second messengers microdomains. Cell Calcium 2015, 58, 405–414. [Google Scholar] [CrossRef]
- Shutov, L.P.; Kim, M.S.; Houlihan, P.R.; Medvedeva, Y.V.; Usachev, Y.M. Mitochondria and plasma membrane Ca2+-ATPase control presynaptic Ca2+ clearance in capsaicin-sensitive rat sensory neurons. J. Physiol. 2013, 591, 2443–2462. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium microdomains: Organization and function. Cell Calcium 2006, 40, 405–412. [Google Scholar] [CrossRef]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Kwon, S.-K.; Sando, R.; Lewis, T.L.; Hirabayashi, Y.; Maximov, A.; Polleux, F. LKB1 Regulates Mitochondria-Dependent Presynaptic Calcium Clearance and Neurotransmitter Release Properties at Excitatory Synapses along Cortical Axons. PLoS Biol. 2016, 14, e1002516. [Google Scholar] [CrossRef] [Green Version]
- Vaccaro, V.; Devine, M.J.; Higgs, N.F.; Kittler, J.T. Miro1-dependent mitochondrial positioning drives the rescaling of presynaptic Ca2+ signals during homeostatic plasticity. EMBO Rep. 2017, 18, 231–240. [Google Scholar] [CrossRef]
- Devine, M.J.; Szulc, B.R.; Howden, J.H.; López-Doménech, G.; Ruiz, A.; Kittler, J.T. Mitochondrial Ca2+ Uniporter haploinsufficiency enhances long-term potentiation at hippocampal mossy fibre synapses. J. Cell Sci. 2022. [Google Scholar] [CrossRef] [PubMed]
- Serrat, R.; Covelo, A.; Kouskoff, V.; Delcasso, S.; Ruiz-Calvo, A.; Chenouard, N.; Stella, C.; Blancard, C.; Salin, B.; Julio-Kalajzić, F.; et al. Astroglial ER-mitochondria calcium transfer mediates endocannabinoid-dependent synaptic integration. Cell Rep. 2022, 41, 111499. [Google Scholar] [CrossRef]
- Grosche, J.; Matyash, V.; Moller, T.; Verkhratsky, A.; Reichenbach, A.; Kettenmann, H. Microdomains for neuron-glia interaction: Parallel fiber signaling to Bergmann glial cells. Nat. Neurosci. 1999, 2, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Lia, A.; Henriques, V.J.; Zonta, M.; Chiavegato, A.; Carmignoto, G.; Gómez-Gonzalo, M.; Losi, G. Calcium Signals in Astrocyte Microdomains, a Decade of Great Advances. Front. Cell Neurosci. 2021, 15, 177. [Google Scholar] [CrossRef]
- Semyanov, A.; Henneberger, C.; Agarwal, A. Making sense of astrocytic calcium signals—From acquisition to interpretation. Nat. Rev. Neurosci. 2020, 21, 551–564. [Google Scholar] [CrossRef]
- Denizot, A.; Arizono, M.; Valentin Nägerl, U.; Soula, H.; Berry, H. Simulation of calcium signaling in fine astrocytic processes: Effect of spatial properties on spontaneous activity. PLoS Comput. Biol. 2019, 15, e1006795. [Google Scholar] [CrossRef]
- Agarwal, A.; Wu, P.-H.; Hughes, E.G.; Fukaya, M.; Tischfield, M.A.; Langseth, A.J.; Wirtz, D.; Bergles, D.E. Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 2017, 93, 587.e7–605.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin Kacerovsky, J.; Murai, K.K. Stargazing: Monitoring subcellular dynamics of brain astrocytes. Neuroscience 2016, 323, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Umpierre, A.D.; Bystrom, L.L.; Ying, Y.; Liu, Y.U.; Worrell, G.; Wu, L.J. Microglial calcium signaling is attuned to neuronal activity in awake mice. Elife 2020, 9, e56502. [Google Scholar] [CrossRef]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Greotti, E. The yin and yang of mitochondrial Ca2+ signaling in cell physiology and pathology. Cell Calcium 2021, 93, 102321. [Google Scholar] [CrossRef]
- Llorente-Folch, I.; Rueda, C.B.; Pardo, B.; Szabadkai, G.; Duchen, M.R.; Satrustegui, J. The regulation of neuronal mitochondrial metabolism by calcium. J. Physiol. 2015, 593, 3447–3462. [Google Scholar] [CrossRef] [Green Version]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Zampese, E.; Wokosin, D.L.; Gonzalez-Rodriguez, P.; Guzman, J.N.; Tkatch, T.; Kondapalli, J.; Surmeier, W.C.; D’Alessandro, K.B.; De Stefani, D.; Rizzuto, R.; et al. Ca2+ channels couple spiking to mitochondrial metabolism in substantia nigra dopaminergic neurons. Sci. Adv. 2022, 8, 8701. [Google Scholar] [CrossRef]
- Pérez-Liébana, I.; Juaristi, I.; González-Sánchez, P.; González-Moreno, L.; Rial, E.; Podunavac, M.; Zakarian, A.; Molgó, J.; Vallejo-Illarramendi, A.; Mosqueira-Martín, L.; et al. A Ca2+-Dependent Mechanism Boosting Glycolysis and OXPHOS by Activating Aralar-Malate-Aspartate Shuttle, upon Neuronal Stimulation. J. Neurosci. 2022, 42, 3879–3895. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertz, L.; Peng, L.; Dienel, G.A. Energy metabolism in astrocytes: High rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 2007, 27, 219–249. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, S.G.; Kegelman, T.P.; Su, Z.Z.; Das, S.K.; Dash, R.; Dasgupta, S.; Barral, P.M.; Hedvat, M.; Diaz, P.; et al. Role of Excitatory Amino Acid Transporter-2 (EAAT2) and glutamate in neurodegeneration: Opportunities for developing novel therapeutics. J. Cell Physiol. 2011, 226, 2484–2493. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genda, E.N.; Jackson, J.G.; Sheldon, A.L.; Locke, S.F.; Greco, T.M.; O’Donnell, J.C.; Spruce, L.A.; Xiao, R.; Guo, W.; Putt, M.; et al. Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J. Neurosci. 2011, 31, 18275–18288. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.E.; Jackson, J.G.; Genda, E.N.; Montoya, M.M.; Yudkoff, M.; Robinson, M.B. The glutamate transporter, GLAST, participates in a macromolecular complex that supports glutamate metabolism. Neurochem. Int. 2012, 61, 566–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Yan, Z.F.; Gao, J.H.; Sun, L.; Huang, X.Y.; Liu, Z.; Yu, S.Y.; Cao, C.J.; Zuo, L.J.; Chen, Z.J.; et al. Role and mechanism of microglial activation in iron-induced selective and progressive dopaminergic neurodegeneration. Mol. Neurobiol. 2014, 49, 1153–1165. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Mehta, M.M.; Weinberg, S.E.; Chandel, N.S. Mitochondrial control of immunity: Beyond ATP. Nat. Rev. Immunol. 2017, 17, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Palsson-Mcdermott, E.M.; O’Neill, L.A.J. The Warburg effect then and now: From cancer to inflammatory diseases. BioEssays 2013, 35, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.J.; Boshuizen, M.C.S.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Lopez-Collazo, E. Endotoxin tolerance: New mechanisms, molecules and clinical significance. Trends Immunol. 2009, 30, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Holland, R.; McIntosh, A.L.; Finucane, O.M.; Mela, V.; Rubio-Araiz, A.; Timmons, G.; McCarthy, S.A.; Gun’ko, Y.K.; Lynch, M.A. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain. Behav. Immun. 2018, 68, 183–196. [Google Scholar] [CrossRef]
- Harry, G.J.; Childers, G.; Giridharan, S.; Hernandes, I.L. An association between mitochondria and microglia effector function: What do we think we know? Neuroimmunol. Neuroinflamm. 2020, 2020, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Khachaturian, Z.S. Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol. Aging 1987, 8, 345–346. [Google Scholar] [CrossRef] [PubMed]
- Khachaturian, Z.S. Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann. N. Y. Acad. Sci. 1989, 568, 1–4. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Landfield, P.W.; Pitler, T.A. Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science 1984, 226, 1089–1092. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, W. Ca2+ homeostasis dysregulation in Alzheimer’s disease: A focus on plasma membrane and cell organelles. FASEB J. 2019, 33, 6697–6712. [Google Scholar] [CrossRef]
- Brunkan, A.L.; Goate, A.M. Presenilin function and γ-secretase activity. J. Neurochem. 2005, 93, 769–792. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, P.; Basso, E.; Filadi, R.; Greotti, E.; Leparulo, A.; Pendin, D.; Redolfi, N.; Rossini, M.; Vajente, N.; Pozzan, T.; et al. Presenilin-2 and Calcium Handling: Molecules, Organelles, Cells and Brain Networks. Cells 2020, 9, 2166. [Google Scholar] [CrossRef]
- Giacomello, M.; Barbiero, L.; Zatti, G.; Squitti, R.; Binetti, G.; Pozzan, T.; Fasolato, C.; Ghidoni, R.; Pizzo, P. Reduction of Ca2+ stores and capacitative Ca2+ entry is associated with the familial Alzheimer’s disease presenilin-2 T122R mutation and anticipates the onset of dementia. Neurobiol. Dis. 2005, 18, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Zatti, G.; Burgo, A.; Giacomello, M.; Barbiero, L.; Ghidoni, R.; Sinigaglia, G.; Florean, C.; Bagnoli, S.; Binetti, G.; Sorbi, S.; et al. Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 2006, 39, 539–550. [Google Scholar] [CrossRef]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Virumbrales, M.; Moreno, C.L.; Kruglikov, I.; Marazuela, P.; Sproul, A.; Jacob, S.; Zimmer, M.; Paull, D.; Zhang, B.; Schadt, E.E.; et al. CRISPR/Cas9-Correctable mutation-related molecular and physiological phenotypes in iPSC-derived Alzheimer’s PSEN2N141I neurons. Acta Neuropathol. Commun. 2017, 5, 77. [Google Scholar] [CrossRef]
- Moreno, C.L.; Della Guardia, L.; Shnyder, V.; Ortiz-Virumbrales, M.; Kruglikov, I.; Zhang, B.; Schadt, E.E.; Tanzi, R.E.; Noggle, S.; Buettner, C.; et al. IPSC-derived familial Alzheimer’s PSEN2 N141I cholinergic neurons exhibit mutation-dependent molecular pathology corrected by insulin signaling. Mol. Neurodegener. 2018, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.C.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Delekate, A.; Fuchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lines, J.; Baraibar, A.M.; Fang, C.; Martin, E.D.; Aguilar, J.; Lee, M.K.; Araque, A.; Kofuji, P. Astrocyte-neuronal network interplay is disrupted in Alzheimer’s disease mice. Glia 2022, 70, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Gsell, W.; Wahis, J.; Luckett, E.S.; Jamoulle, T.; Vermaercke, B.; Preman, P.; Moechars, D.; Hendrickx, V.; Jaspers, T.; et al. Astrocyte calcium dysfunction causes early network hyperactivity in Alzheimer’s disease. Cell Rep. 2022, 40. [Google Scholar] [CrossRef]
- Åbjørsbråten, K.S.; Skaaraas, G.H.E.S.; Cunen, C.; Bjørnstad, D.M.; Binder, K.M.G.; Bojarskaite, L.; Jensen, V.; Nilsson, L.N.G.; Rao, S.B.; Tang, W.; et al. Impaired astrocytic Ca2+ signaling in awake-behaving Alzheimer’s disease transgenic mice. Elife 2022, 11, e75055. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Gubert Olivé, M.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 Mutant iPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef] [Green Version]
- Itagaki, S.; McGeer, P.L.; Akiyama, H.; Zhu, S.; Selkoe, D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 1989, 24, 173–182. [Google Scholar] [CrossRef] [PubMed]
- McLarnon, J.G.; Choi, H.B.; Lue, L.F.; Walker, D.G.; Kim, S.U. Perturbations in calcium-mediated signal transduction in microglia from Alzheimer’s disease patients. J. Neurosci. Res. 2005, 81, 426–435. [Google Scholar] [CrossRef]
- McLarnon, J.G.; Ryu, J.K.; Walker, D.G.; Choi, H.B. Upregulated expression of purinergic P2X7 receptor in Alzheimer disease and amyloid-β peptide-treated microglia and in peptide-injected rat hippocampus. J. Neuropathol. Exp. Neurol. 2006, 65, 1090–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, M.; Xiaolong, Q.; Chaojuan, Y.; Ruiyuan, P.; Shukun, W.; Junbing, W.; Li, H.; Hong, C.; Jinbo, C.; Rong, W.; et al. Calhm2 governs astrocytic ATP releasing in the development of depression-like behaviors. Mol. Psychiatry 2018, 23, 883–891. [Google Scholar] [CrossRef]
- Cheng, J.; Dong, Y.; Ma, J.; Pan, R.; Liao, Y.; Kong, X.; Li, X.; Li, S.; Chen, P.; Wang, L.; et al. Microglial Calhm2 regulates neuroinflammation and contributes to Alzheimer’s disease pathology. Sci. Adv. 2021, 7, eabe3600. [Google Scholar] [CrossRef]
- Jairaman, A.; McQuade, A.; Granzotto, A.; Kang, Y.J.; Chadarevian, J.P.; Gandhi, S.; Parker, I.; Smith, I.; Cho, H.; Sensi, S.L.; et al. TREM2 regulates purinergic receptor-mediated calcium signaling and motility in human iPSC-derived microglia. Elife 2022, 11, e73021. [Google Scholar] [CrossRef]
- Brawek, B.; Schwendele, B.; Riester, K.; Kohsaka, S.; Lerdkrai, C.; Liang, Y.; Garaschuk, O. Impairment of in vivo calcium signaling in amyloid plaque-associated microglia. Acta Neuropathol. 2014, 127, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Contreras, L.; Drago, I.; Zampese, E.; Pozzan, T. Mitochondria: The calcium connection. Biochim. Biophys. Acta 2010, 1797, 607–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Rossa, A.; Laporte, M.H.; Astori, S.; Marissal, T.; Montessuit, S.; Sheshadri, P.; Ramos-Fernández, E.; Mendez, P.; Khani, A.; Quairiaux, C.; et al. Paradoxical neuronal hyperexcitability in a mouse model of mitochondrial pyruvate import deficiency. Elife 2022, 11, e72595. [Google Scholar] [CrossRef]
- Sanz-Blasco, S.; Valero, R.A.; Rodríguez-Crespo, I.; Villalobos, C.; Núñez, L. Mitochondrial Ca2+ overload underlies Aβ oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE 2008, 3, e2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, P.; Carraro, M.; Lippe, G. The mitochondrial permeability transition: Recent progress and open questions. FEBS J. 2021, 289, 7051–7074. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Zhang, W.; Rydzewska, M.; Yan, S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 2011, 32, 398–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Area-Gomez, E.; de Groof, A.J.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begley, J.G.; Duan, W.; Chan, S.; Duff, K.; Mattson, M.P. Altered calcium homeostasis and mitochondrial dysfunction in cortical synaptic compartments of presenilin-1 mutant mice. J. Neurochem. 1999, 72, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Korkotian, E.; Meshcheriakova, A.; Segal, M. Presenilin 1 Regulates [Ca2+]i and Mitochondria/ER Interaction in Cultured Rat Hippocampal Neurons. Oxid. Med. Cell Longev. 2019, 2019, 7284967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigotto, G.; Zentilin, L.; Pozzan, T.; Basso, E. Effects of mild excitotoxic stimulus on mitochondria Ca2+ handling in hippocampal cultures of a mouse model of alzheimer’s disease. Cells 2021, 10, 2046. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda-Falla, D.; Barrera-Ocampo, A.; Hagel, C.; Korwitz, A.; Vinueza-Veloz, M.F.; Zhou, K.; Schonewille, M.; Zhou, H.; Velazquez-Perez, L.; Rodriguez-Labrada, R.; et al. Familial Alzheimer’s disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J. Clin. Investig. 2014, 124, 1552–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, D.H.W.; Paillusson, S.; Hartopp, N.; Rupawala, H.; Mórotz, G.M.; Gomez-Suaga, P.; Greig, J.; Troakes, C.; Noble, W.; Miller, C.C.J. Disruption of endoplasmic reticulum-mitochondria tethering proteins in post-mortem Alzheimer’s disease brain. Neurobiol. Dis. 2020, 143, 105020. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [Green Version]
- Schreiner, B.; Hedskog, L.; Wiehager, B.; Ankarcrona, M. Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimer’s Dis. 2015, 43, 369–374. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Park, H.; Maharana, C.; Gwon, A.R.; Park, J.; Baek, S.H.; Bae, H.G.; Cho, Y.; Kim, H.K.; Sul, J.H.; et al. Alzheimer’s disease-causing presenilin-1 mutations have deleterious effects on mitochondrial function. Theranostics 2021, 11, 8855–8873. [Google Scholar] [CrossRef]
- Völgyi, K.; Badics, K.; Sialana, F.J.; Gulyássy, P.; Udvari, E.B.; Kis, V.; Drahos, L.; Lubec, G.; Kékesi, K.A.; Juhász, G. Early Presymptomatic Changes in the Proteome of Mitochondria-Associated Membrane in the APP/PS1 Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 7839–7857. [Google Scholar] [CrossRef]
- Fernandes, T.; Resende, R.; Silva, D.F.; Marques, A.P.; Santos, A.E.; Cardoso, S.M.; Rosário Domingues, M.; Moreira, P.I.; Pereira, C.F. Structural and functional alterations in mitochondria-associated membranes (Mams) and in mitochondria activate stress response mechanisms in an in vitro model of alzheimer’s disease. Biomedicines 2021, 9, 881. [Google Scholar] [CrossRef]
- Leal, N.S.; Dentoni, G.; Schreiner, B.; Kämäräinen, O.P.; Partanen, N.; Herukka, S.K.; Koivisto, A.M.; Hiltunen, M.; Rauramaa, T.; Leinonen, V.; et al. Alterations in mitochondria-endoplasmic reticulum connectivity in human brain biopsies from idiopathic normal pressure hydrocephalus patients. Acta Neuropathol. Commun. 2018, 6, 102. [Google Scholar] [CrossRef] [Green Version]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Montagna, E.; Crux, S.; Luckner, M.; Herber, J.; Colombo, A.V.; Marinkovic, P.; Tahirovic, S.; Lichtenthaler, S.F.; Wanner, G.; Muller, U.C.; et al. In vivo Ca2+ imaging of astrocytic microdomains reveals a critical role of the amyloid precursor protein for mitochondria. Glia 2019, 67, 985–998. [Google Scholar] [CrossRef]
- Blagov, A.V.; Grechko, A.V.; Nikiforov, N.G.; Borisov, E.E.; Sadykhov, N.K.; Orekhov, A.N. Role of Impaired Mitochondrial Dynamics Processes in the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 6954. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Jakkamsetti, V.; Marin-Valencia, I.; Ma, Q.; Good, L.B.; Terrill, T.; Rajasekaran, K.; Pichumani, K.; Khemtong, C.; Hooshyar, M.A.; Sundarrajan, C.; et al. Brain metabolism modulates neuronal excitability in a mouse model of pyruvate dehydrogenase deficiency. Sci. Transl. Med. 2019, 11, eaan0457. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Guan, J.S.; Borrelli, L.A.; Xu, J.; Serrano-Pozo, A.; Bacskai, B.J. Mitochondrial alterations near amyloid plaques in an Alzheimer’S disease mouse model. J. Neurosci. 2013, 33, 17042–17051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Rigotto, G.; Valente, G.; Giorgio, V.; Basso, E.; Filadi, R.; Pizzo, P. Defective Mitochondrial Pyruvate Flux Affects Cell Bioenergetics in Alzheimer’s Disease-Related Models. Cell Rep. 2020, 30, 2332–2348.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurey, P.; Connolly, N.M.C.; Fortunati, I.; Basso, E.; Lauwen, S.; Ferrante, C.; Moreira Pinho, C.; Joselin, A.; Gioran, A.; Bano, D.; et al. Systems biology identifies preserved integrity but impaired metabolism of mitochondria due to a glycolytic defect in Alzheimer’s disease neurons. Aging Cell 2019, 18, e12924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulland, T.K.; Song, W.M.; Huang, S.C.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- Pan, R.Y.; Ma, J.; Kong, X.X.; Wang, X.F.; Li, S.S.; Qi, X.L.; Yan, Y.H.; Cheng, J.; Liu, Q.; Jin, W.; et al. Sodium rutin ameliorates Alzheimer’s disease-like pathology by enhancing microglial amyloid-β clearance. Sci. Adv. 2020, 5, eaau6328. [Google Scholar] [CrossRef] [Green Version]
- Woods, J.J.; Nemani, N.; Shanmughapriya, S.; Kumar, A.; Zhang, M.; Nathan, S.R.; Thomas, M.; Carvalho, E.; Ramachandran, K.; Srikantan, S.; et al. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent. Sci. 2019, 5, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Arduino, D.M.; Wettmarshausen, J.; Vais, H.; Navas-Navarro, P.; Cheng, Y.; Leimpek, A.; Ma, Z.; Delrio-Lorenzo, A.; Giordano, A.; Garcia-Perez, C.; et al. Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening. Mol. Cell 2017, 67, 711–723 e7. [Google Scholar] [CrossRef] [PubMed]
- Kon, N.; Murakoshi, M.; Isobe, A.; Kagechika, K.; Miyoshi, N.; Nagayama, T. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 2017, 3, 17045. [Google Scholar] [CrossRef]
- Di Marco, G.; Vallese, F.; Jourde, B.; Bergsdorf, C.; Sturlese, M.; De Mario, A.; Techer-Etienne, V.; Haasen, D.; Oberhauser, B.; Schleeger, S.; et al. A High-Throughput Screening Identifies MICU1 Targeting Compounds. Cell Rep. 2020, 30, 2321–2331.e6. [Google Scholar] [CrossRef] [PubMed]
- De Mario, A.; Tosatto, A.; Hill, J.M.; Kriston-Vizi, J.; Ketteler, R.; Vecellio Reane, D.; Cortopassi, G.; Szabadkai, G.; Rizzuto, R.; Mammucari, C. Identification and functional validation of FDA-approved positive and negative modulators of the mitochondrial calcium uniporter. Cell Rep. 2021, 35, 109275. [Google Scholar] [CrossRef]
- Montero, M.; Lobaton, C.D.; Hernandez-Sanmiguel, E.; Santodomingo, J.; Vay, L.; Moreno, A.; Alvarez, J. Direct activation of the mitochondrial calcium uniporter by natural plant flavonoids. Biochem. J. 2004, 384, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermont, F.; Hermant, A.; Benninga, R.; Chabert, C.; Jacot, G.; Santo-Domingo, J.; Kraus, M.R.C.; Feige, J.N.; De Marchi, U. Targeting Mitochondrial Calcium Uptake with the Natural Flavonol Kaempferol, to Promote Metabolism/Secretion Coupling in Pancreatic β-cells. Nutrients 2020, 12, 538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, L.R.; Lapeña-Luzón, T.; Benetó, N.; Beltran-Beltran, V.; Pallardó, F.V.; Gonzalez-Cabo, P.; Navarro, J.A. Therapeutic Strategies Targeting Mitochondrial Calcium Signaling: A New Hope for Neurological Diseases? Antioxidants 2022, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.T.; Diaz-Sylvester, P.L.; Fleischer, S.; Copello, J.A. CGP-37157 inhibits the sarcoplasmic reticulum Ca2+ ATPase and activates ryanodine receptor channels in striated muscle. Mol. Pharmacol. 2011, 79, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Onyango, I.G. Modulation of mitochondrial bioenergetics as a therapeutic strategy in Alzheimer’s disease. Neural Regen. Res. 2018, 13, 19–25. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnst, N.; Redolfi, N.; Lia, A.; Bedetta, M.; Greotti, E.; Pizzo, P. Mitochondrial Ca2+ Signaling and Bioenergetics in Alzheimer’s Disease. Biomedicines 2022, 10, 3025. https://doi.org/10.3390/biomedicines10123025
Arnst N, Redolfi N, Lia A, Bedetta M, Greotti E, Pizzo P. Mitochondrial Ca2+ Signaling and Bioenergetics in Alzheimer’s Disease. Biomedicines. 2022; 10(12):3025. https://doi.org/10.3390/biomedicines10123025
Chicago/Turabian StyleArnst, Nikita, Nelly Redolfi, Annamaria Lia, Martina Bedetta, Elisa Greotti, and Paola Pizzo. 2022. "Mitochondrial Ca2+ Signaling and Bioenergetics in Alzheimer’s Disease" Biomedicines 10, no. 12: 3025. https://doi.org/10.3390/biomedicines10123025