
Suppressive Role of ACVR1/ALK2 in Basal and TGFβ1-Induced Cell Migration in Pancreatic Ductal Adenocarcinoma Cells and Identification of a Self-Perpetuating Autoregulatory Loop Involving the Small GTPase RAC1b
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Transient Transfections
2.2. Immunoblotting
2.3. Quantitative PCR Analysis
2.4. xCELLigence-Based Cell Migration Assays
2.5. Statistical Analysis
3. Results
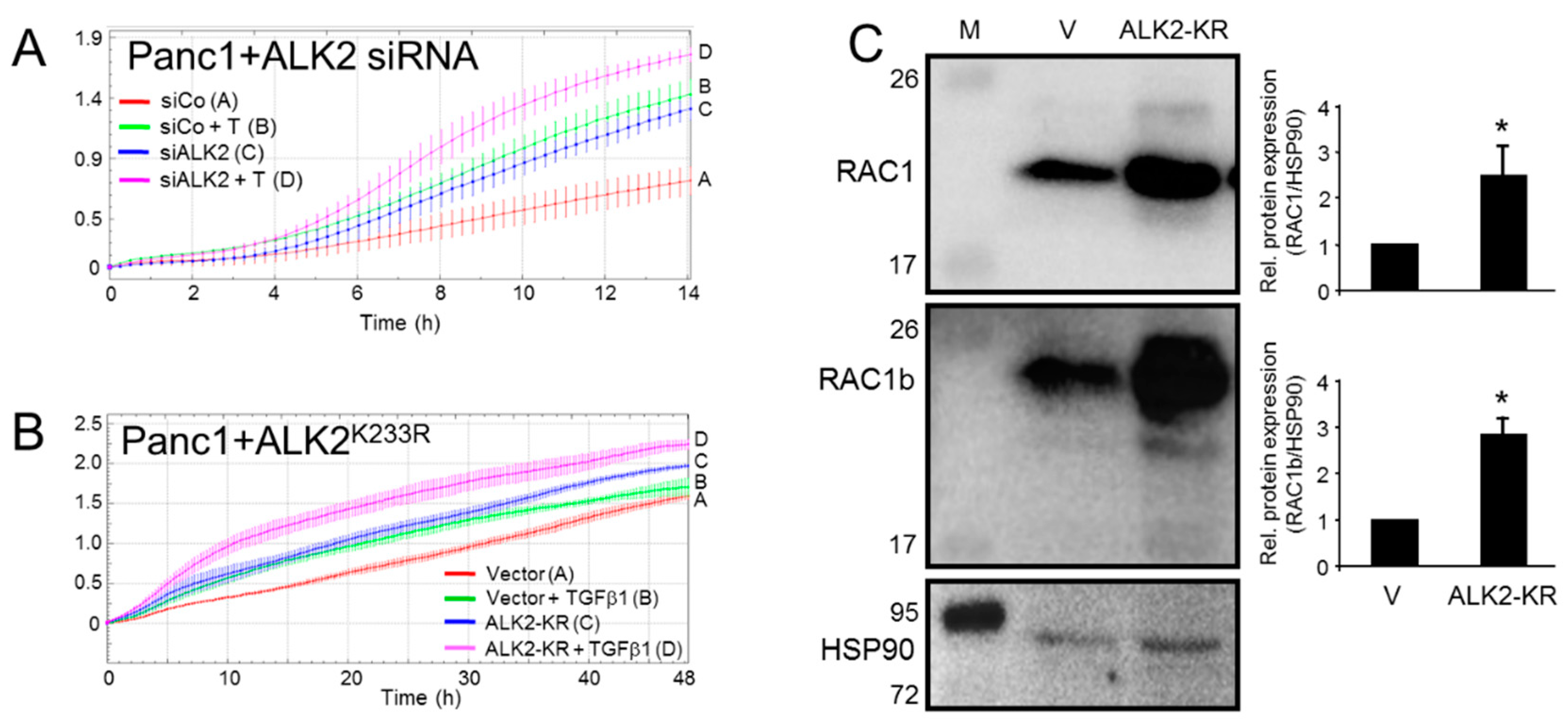
3.1. Inhibition of the BMP Type I Receptor ALK2 Enhances Basal and TGFβ1-Induced Cell Migration
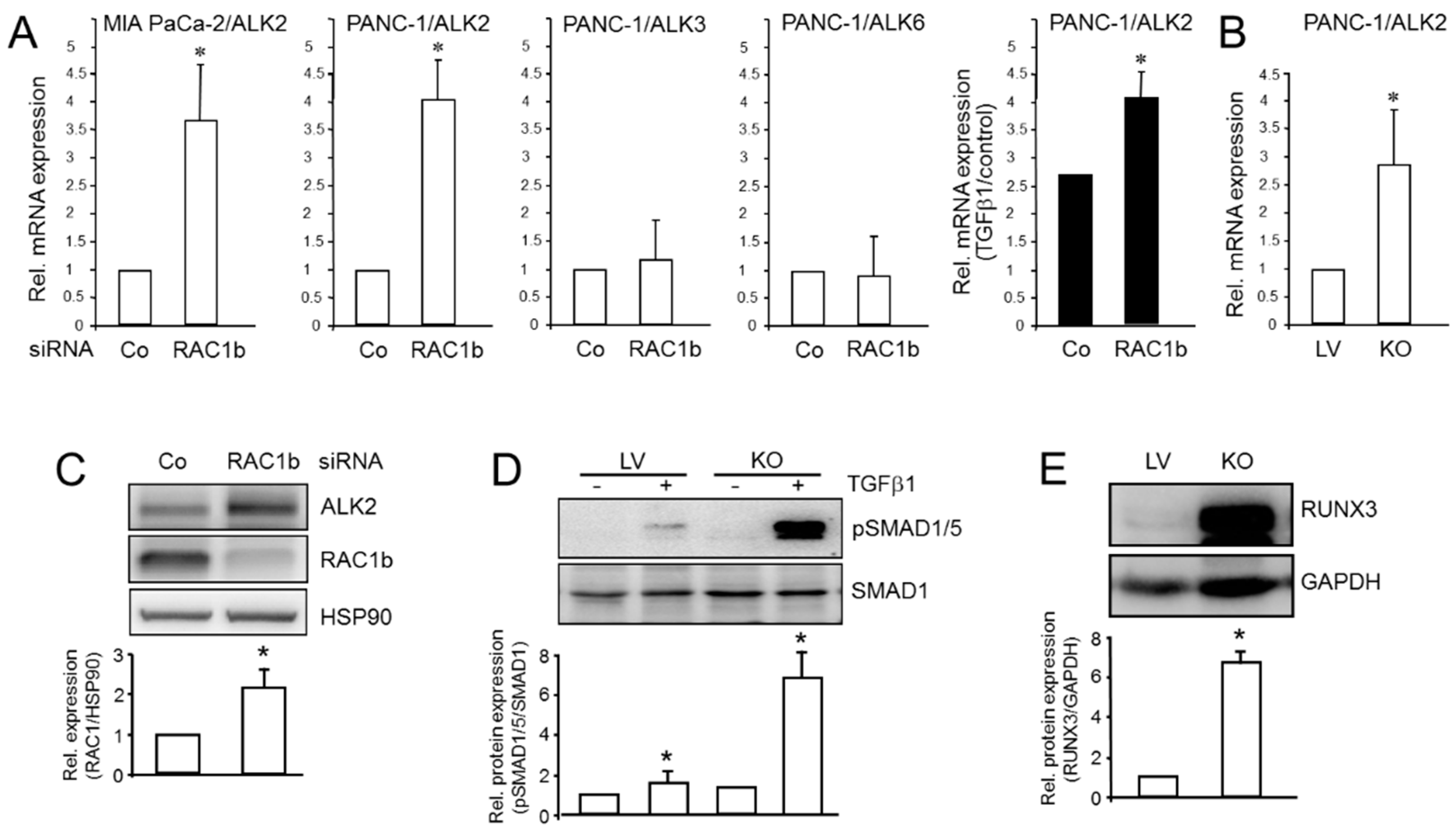
3.2. ALK2 Expression, SMAD1/5 Signaling, and Target Gene Expression Is Subject to Negative Regulation by RAC1b
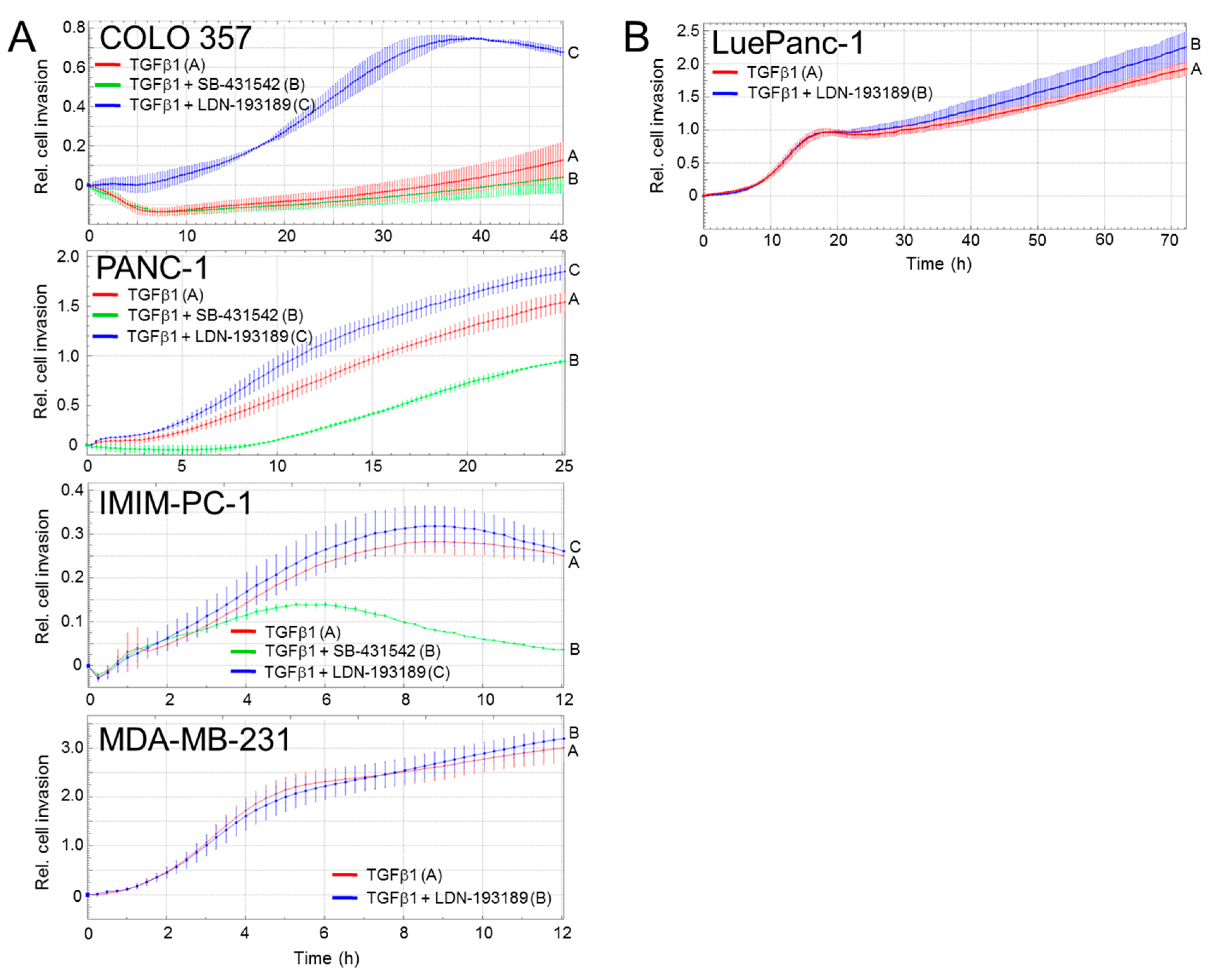
3.3. The Inhibitory Effect of ALK2 on TGFβ-Driven Cell Migration Depends on the PDAC Differentiation Subtype
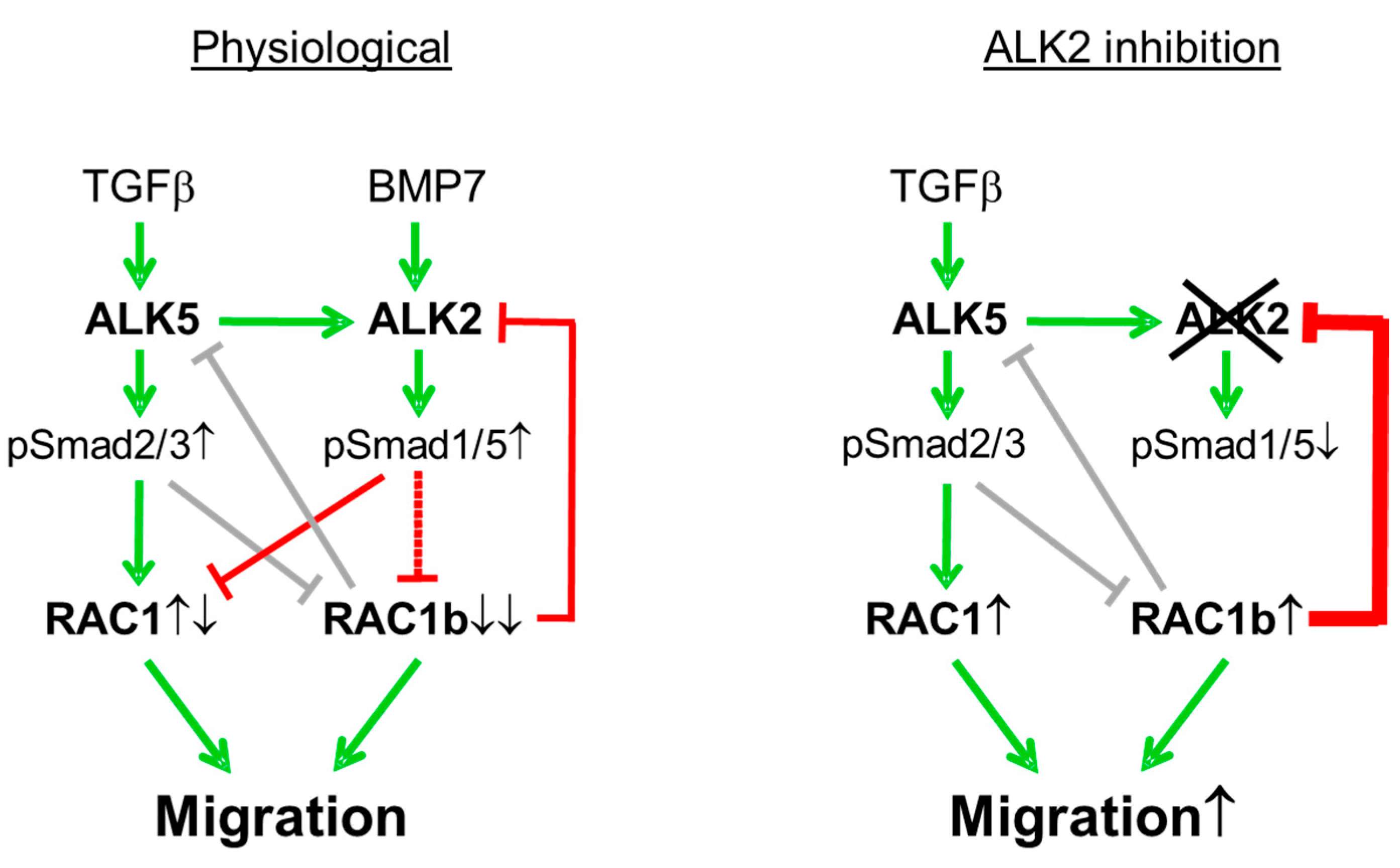
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thomas, S.K.; Lee, J.; Beatty, G.L. Paracrine and cell autonomous signalling in pancreatic cancer progression and metastasis. eBioMedicine 2020, 53, 102662. [Google Scholar] [CrossRef] [PubMed]
- Dardare, J.; Witz, A.; Merlin, J.L.; Bochnakian, A.; Toussaint, P.; Gilson, P.; Harlé, A. Epithelial to Mesenchymal Transition in Patients with Pancreatic Ductal Adenocarcinoma: State-of-the-Art and Therapeutic Opportunities. Pharmaceuticals 2021, 14, 740. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Zhao, Y.; Ye, Y.; Yu, J. Opposing roles and potential antagonistic mechanism between TGF-β and BMP pathways: Implications for cancer progression. eBioMedicine 2019, 41, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; Li, H.; Yang, S.; Li, M.; Zhao, C.; Zhang, J.; Xu, G.; Wang, F. Inhibitory Effect of Bone Morphogenetic Protein 4 in Retinal Pigment Epithelial-Mesenchymal Transition. Sci. Rep. 2016, 6, 32182. [Google Scholar] [CrossRef]
- Zeisberg, M.; Hanai, J.-i.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Meurer, S.K.; Esser, M.; Tihaa, L.; Weiskirchen, R. BMP-7/TGF-β1 signalling in myoblasts: Components involved in signalling and BMP-7-dependent blockage of TGF-β-mediated CTGF expression. Eur. J. Cell Biol. 2012, 91, 450–463. [Google Scholar] [CrossRef]
- Macías-Silva, M.; Hoodless, P.A.; Tang, S.J.; Buchwald, M.; Wrana, J.L. Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. J. Biol. Chem. 1998, 273, 25628–25636. [Google Scholar] [CrossRef] [Green Version]
- Na, Y.R.; Seok, S.H.; Kim, D.J.; Han, J.H.; Kim, T.H.; Jung, H.; Lee, B.H.; Park, J.H. Bone morphogenetic protein 7 induces mesenchymal-to-epithelial transition in melanoma cells, leading to inhibition of metastasis. Cancer Sci. 2009, 100, 2218–2225. [Google Scholar] [CrossRef]
- Zeisberg, M.; Shah, A.A.; Kalluri, R. Bone morphogenic protein-7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J. Biol. Chem. 2005, 280, 8094–8100. [Google Scholar] [CrossRef] [Green Version]
- Katoch, A.; Jamwal, V.L.; Faheem, M.M.; Kumar, S.; Senapati, S.; Yadav, G.; Gandhi, S.G.; Goswami, A. Overlapping targets exist between the Par-4 and miR-200c axis which regulate EMT and proliferation of pancreatic cancer cells. Transl. Oncol. 2021, 14, 100879. [Google Scholar] [CrossRef]
- Ramachandran, A.; Vizán, P.; Das, D.; Chakravarty, P.; Vogt, J.; Rogers, K.W.; Müller, P.; Hinck, A.P.; Sapkota, G.P.; Hill, C.S. TGF-β uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial-to-mesenchymal transition. elife 2018, 7, e31756. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.C.; Randall, R.A.; Hill, C.S. Transforming growth factor β-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol. Cell. Biol. 2008, 28, 6889–6902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H.; Otterbein, H.; Fiedler, C.; Mihara, K.; Hollenberg, M.D.; Gieseler, F.; Lehnert, H.; Witte, D. RAC1B Suppresses TGF-β1-Dependent Cell Migration in Pancreatic Carcinoma Cells through Inhibition of the TGF-β Type I Receptor ALK5. Cancers 2019, 11, 691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H.; Thürling, I.; Färber, B.; Kowalke, T.; Fischer, T.; De Assis, L.V.M.; Braun, R.; Castven, D.; Oster, H.; Konukiewitz, B.; et al. The Quasimesenchymal Pancreatic Ductal Epithelial Cell Line PANC-1-A Useful Model to Study Clonal Heterogeneity and EMT Subtype Shifting. Cancers 2022, 14, 2057. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Cho, K.; Kim, N.H.; Seo, S.H.; Song, S.H.; Jeong, C.H.; Kim, H.S.; Um, J.E.; Ku, M.; Yang, J.; Park, J.Y.; et al. A micellized bone morphogenetic protein-7 prodrug ameliorates liver fibrosis by suppressing transforming growth factor-β signaling. Am. J. Cancer Res 2022, 12, 763–778. [Google Scholar]
- Braun, R.; Lapshyna, O.; Watzelt, J.; Drenckhan, M.; Künstner, A.; Färber, B.; Hael, A.A.M.; Bolm, L.; Honselmann, K.; Konukiewitz, B.; et al. Establishment and molecular characterization of two patient-derived pancreatic ductal adenocarinoma cell lines as preclinical models for treatment response. Cells, 2022; accepted pending revision. [Google Scholar]
- Witte, D.; Otterbein, H.; Förster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-β1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, 17313. [Google Scholar] [CrossRef] [Green Version]
- Zinn, R.; Otterbein, H.; Lehnert, H.; Ungefroren, H. RAC1B: A guardian of the epithelial phenotype and protector against epithelial-mesenchymal transition. Cells 2019, 8, 1569. [Google Scholar] [CrossRef] [Green Version]
- Heid, I.; Lubeseder-Martellato, C.; Sipos, B.; Mazur, P.K.; Lesina, M.; Schmid, R.M.; Siveke, J.T. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology 2011, 141, 719–730. [Google Scholar] [CrossRef]
- Ungefroren, H.; Witte, D.; Lehnert, H. The role of small GTPases of the Rho/Rac family in TGF-β-induced EMT and cell motility in cancer. Dev. Dyn. 2018, 247, 451–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Zhang, X.; Shang, R.; Wang, J.; Yang, X.; Zhang, H.; Liu, Q.; Wang, D.; Wang, L.; Dou, K. Blockade of ARHGAP11A reverses malignant progress via inactivating Rac1B in hepatocellular carcinoma. Cell Commun. Signal. 2018, 16, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Tian, Y.; Jia, Y.; Shen, Q.; Jiang, W.; Chen, G.; Shang, B.; Shi, M.; Wang, Z.; Zhao, X. RUNX3 inhibits the invasion and migration of esophageal squamous cell carcinoma by reversing the epithelial-mesenchymal transition through TGF-β/Smad signaling. Oncol. Rep. 2020, 43, 1289–1299. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, Q.; Ji, C.; Liu, X.; Li, L.; Luo, J. RUNX3 plays an important role in mediating the BMP9-induced osteogenic differentiation of mesenchymal stem cells. Int. J. Mol. Med. 2017, 40, 1991–1999. [Google Scholar] [CrossRef] [Green Version]
- Roman, B.L.; Hinck, A.P. ALK1 signaling in development and disease: New paradigms. Cell. Mol. Life Sci. 2017, 74, 4539–4560. [Google Scholar] [CrossRef]
- Inman, G.J.; Nicolas, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Schmidtlein, P.M.; Volz, C.; Braun, R.; Thürling, I.; Lapshyna, O.; Wellner, U.F.; Konukiewitz, B.; Lehnert, H.; Marquardt, J.U.; Ungefroren, H. A comparative endocrine trans-differentiation approach to pancreatic ductal adenocarcinoma cells with different EMT phenotypes identifies quasi-mesenchymal tumor cells as those with highest plasticity. Cancers 2021, 13, 4663. [Google Scholar] [CrossRef]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-b induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungefroren, H.; Braun, R.; Lapshyna, O.; Konukiewitz, B.; Wellner, U.F.; Lehnert, H.; Marquardt, J.-U. Suppressive Role of ACVR1/ALK2 in Basal and TGFβ1-Induced Cell Migration in Pancreatic Ductal Adenocarcinoma Cells and Identification of a Self-Perpetuating Autoregulatory Loop Involving the Small GTPase RAC1b. Biomedicines 2022, 10, 2640. https://doi.org/10.3390/biomedicines10102640
Ungefroren H, Braun R, Lapshyna O, Konukiewitz B, Wellner UF, Lehnert H, Marquardt J-U. Suppressive Role of ACVR1/ALK2 in Basal and TGFβ1-Induced Cell Migration in Pancreatic Ductal Adenocarcinoma Cells and Identification of a Self-Perpetuating Autoregulatory Loop Involving the Small GTPase RAC1b. Biomedicines. 2022; 10(10):2640. https://doi.org/10.3390/biomedicines10102640
Chicago/Turabian StyleUngefroren, Hendrik, Rüdiger Braun, Olha Lapshyna, Björn Konukiewitz, Ulrich F. Wellner, Hendrik Lehnert, and Jens-Uwe Marquardt. 2022. "Suppressive Role of ACVR1/ALK2 in Basal and TGFβ1-Induced Cell Migration in Pancreatic Ductal Adenocarcinoma Cells and Identification of a Self-Perpetuating Autoregulatory Loop Involving the Small GTPase RAC1b" Biomedicines 10, no. 10: 2640. https://doi.org/10.3390/biomedicines10102640