
Immune Checkpoint Inhibitors and Other Immune Therapies in Breast Cancer: A New Paradigm for Prolonged Adjuvant Immunotherapy

Abstract
:1. Introduction
1.1. Conventional Immune Therapies in Breast Cancer
1.1.1. ICIs and ICB in Breast Cancer Therapy
1.1.2. PD-L1 Expression
1.1.3. PD-L1 Assay

{kind=link}
{kind=link}
| Parameter | Evaluation | Ref. | |
|---|---|---|---|
| Criterion | Analysis | ||
| Immune cells (iCs) | % | Percentage of tumor area involved by any intensity of PD-L1 IHC staining | [54] |
| Score | (Area of tumor infiltrated by PD-L1+ iCs/total tumor area) × 100% | [33,54,55] | |
| Tumor cells (TCs) | % | (Number of PD-L1+ iCs/total number of viable TCs) × 100% | [33,55,56] |
| Tumor cell score | The percentage of tumor area involved by PD-L1+ TCs related to the entire tumor area | [33,56] | |
| Tumor proportion score | (Number of viable PD-L1+ TCs/total number of viable TCs) × 100% | [33,55,57] | |
| iCs and TCs | Combined positive score | * (Number of PD-L1+ TCs plus iCs/total number of viable TCs) × 100% | [33,55,56] |
1.1.4. Pembrolizumab and Nivolumab (PD-1 Inhibitors) (Table 2)
1.1.5. Atezolizumab, Durvalumab, and Avelumab (PD-L1 Inhibitors)
1.1.6. Efficacy and Safety of Anti-PD1/PD-L1 Monotherapy in Metastatic Breast Cancer
1.1.7. Tremelimumab and Ipilimumab (CTLA-4 Inhibitors)
| A | ||||||||
|---|---|---|---|---|---|---|---|---|
| Trial | Phase | Drug | Mechanism of Action | Setting | Therapeutic Regimen | Pts (n) | Outcome | Ref. |
| Keynote-028 NCT 02054806 | Ib | Pembrolizumab (PE) | PD1 inhibitor | ABC PD-L1+ | Monotherapy for up to 2 years | 25 | ORR 20% SD 16% CB 20% MDR 12 mo. | [58] |
| Keynote-086 NCT 02447003 | II | mTNBC | Monotherapy, >1 prior systemic therapy | 170 | ORR 5.7% in PDL1+ pts PFS 2 mo. OS 9 mo. | [59] | ||
| Cohort B | Monotherapy, no prior systemic therapy | 84 | ORR 20% PFS 21 mo. OS 18 mo. | [60] | ||||
| Keynote-119 NCT 02555657 | III | mTNBC | PE vs. CT, prior systemic therapy | 312 vs. 310 | OS 12.7 vs. 11.6 mo. in PDL1+ pts | [61] | ||
| Keynote-355 NCT 02819518 | III | mTNBC | PE + CT vs. PB + CT, no prior CT | 566 vs. 281 | PFS 9.7 vs. 5.6 mo. in PDL1+ pts; PFS 7.6 vs. 5.6 mo. | [62] | ||
| Keynote-522 NCT 03036488 | III | Early, stage II-III TNBC | PE + PTX + CBD vs. PB + PTX + CBD | 401 vs. 201 | pCR 64.8% vs. 51.2% | [63] | ||
| Panacea-Keynote-014 | Ib-II | HER2+, Trastuzumab resistant ABC | PE + trastuzumab | 52 | ORR 15% in PDL1+ pts No response in PDL1− pts | [65] | ||
| TONIC | II | Nivolumab (NI) | mTNBC | (1) NI without induction; (2) NI with induction *; (3) RT; (4) CY; (5) CIS; (6) DOXO 3-4-5-6: followed by NI | 67 | ORR 20% for all pts CIS 23% DOXO 35% | [66] | |
| Institutional (open-label, single arm, single-center, closed early) | II | mTNBC | NI + cabozantinib | 18 | ORR 6% (1 PR in a PDL1− pt) | [12] | ||
| NCT03807765 | IB | BC with brain metastases | NI followed by SRS | 12 | 6 mo. control rate 55% 12 mo. control rate 22% | [67] | ||
| B | ||||||||
| Trial | Phase | Drug | Mechanism of Action | Setting | Therapeutic Regimen | Pts (n) | Outcome | Ref. |
| NCT01375842 | I | Atezolizumab (ATZ) | PD-L1 inhibitor | mTNBC unselected for PDL1 | Monotherapy | 116 | ORR 10–12% in PDL1+ Median PFS 1.4 mo. Median OS 8.9 mo. | [69] |
| iMpassion 130 NCT02425891 | III | mTNBC unselected for PDL1 | ATZ + Nab-PTX vs. PB + Nab-PTX | 451 vs. 451 | ORR 56–58.9% in PDL1+ Median PFS 7.5 mo. in PDL1+ Median OS 25 mo in PDL1+ | [70,73] | ||
| iMpassion 031 NCT03197935 | III | Stage II or III TNBC, no prior systemic therapy | ATZ + CT vs. PB + CT | 165 vs. 168 | pCR 58% vs. 41% p = 0.0044 | [74] | ||
| iMpassion 131 NCT03125902 | III | Locally advanced or mTNBC, no prior systemic therapy | ATZ + PTX vs. PB + PTX | 431 vs. 220 | Median PFS 6 vs. 5.7 mo. Median OS 22.1 vs. 28.3 mo in PDL1+ pts | [75] | ||
| NCT02484404 | I | Durvalumab (DRV) | Recurrent cancers including TNBC | DRV + cediranib + olaparib | 9 (1 TNBC) | CBR 67% PR 44% | [76] | |
| GeparNuevo NCT02685059 | II | NACT in early TNBC | DRV + Nab-PTX vs. PB + Nab-PTX followed by EC ** | 174 (117 the window cohort) | pCR 53.4% vs. 44.2% pCR 61% vs. 41.4% in window cohort | [77] | ||
| SAFIR 02 Breast Immuno NCT02299999 | II | HER2− mBC with prior CT | DRV vs. maintenance CT | 32 vs. 29 assessed | HR of death: 0.37 for PDL1+ pts vs. 0.49 PDL1− pts | [78] | ||
| Javelin NCT01772004 | Ib | Avelumab (AV) | Locally advanced or mTNBC, refractory or progressing | Monotherapy | 58 TNBC | ORR 22.2% in PDL1+ pts vs. 2.6% in PDL1− pts | [79] | |
| Institutional (open-label, single-arm, single-center, closed early) | I | Tremelimumab (TREMB) | CTLA-4 inhibitor | Incurable mBC | TREMB + RT | 6 (1 TNBC) | Median OS 50.8 mo. | [81] |
| ICON | Ib | Ipilimumab (IPI) | HR+ mBC | IPI + NI + PLD + CY vs. PLD + CY | 75 | Expected | [82] | |
1.1.8. PD-L1 Testing as Prognostic and/or Predictive Biomarker in Breast Cancer
1.2. ICIs Combined with DDRi
1.2.1. DNA Damage Repair Defects, Tumor Mutational Burden, and Neo-Antigens
1.2.2. Barriers to ICIs Response and Mechanisms by Which DDRi Synergizes with ICIs
1.2.3. Clinical Trials with ICIs and DDRi Combinatorial Therapy
1.3. Monoclonal Antibodies against Human Epidermal Growth Factor Receptor 2 (HER-2) Family in HER2+ Breast Cancer
Anti-HER2 mAbs and/or ADC Combined with ICIs
1.4. Other Investigational Immune-therapies
1.5. The Tumor Immunogenicity: A True or an Incomplete Understanding?
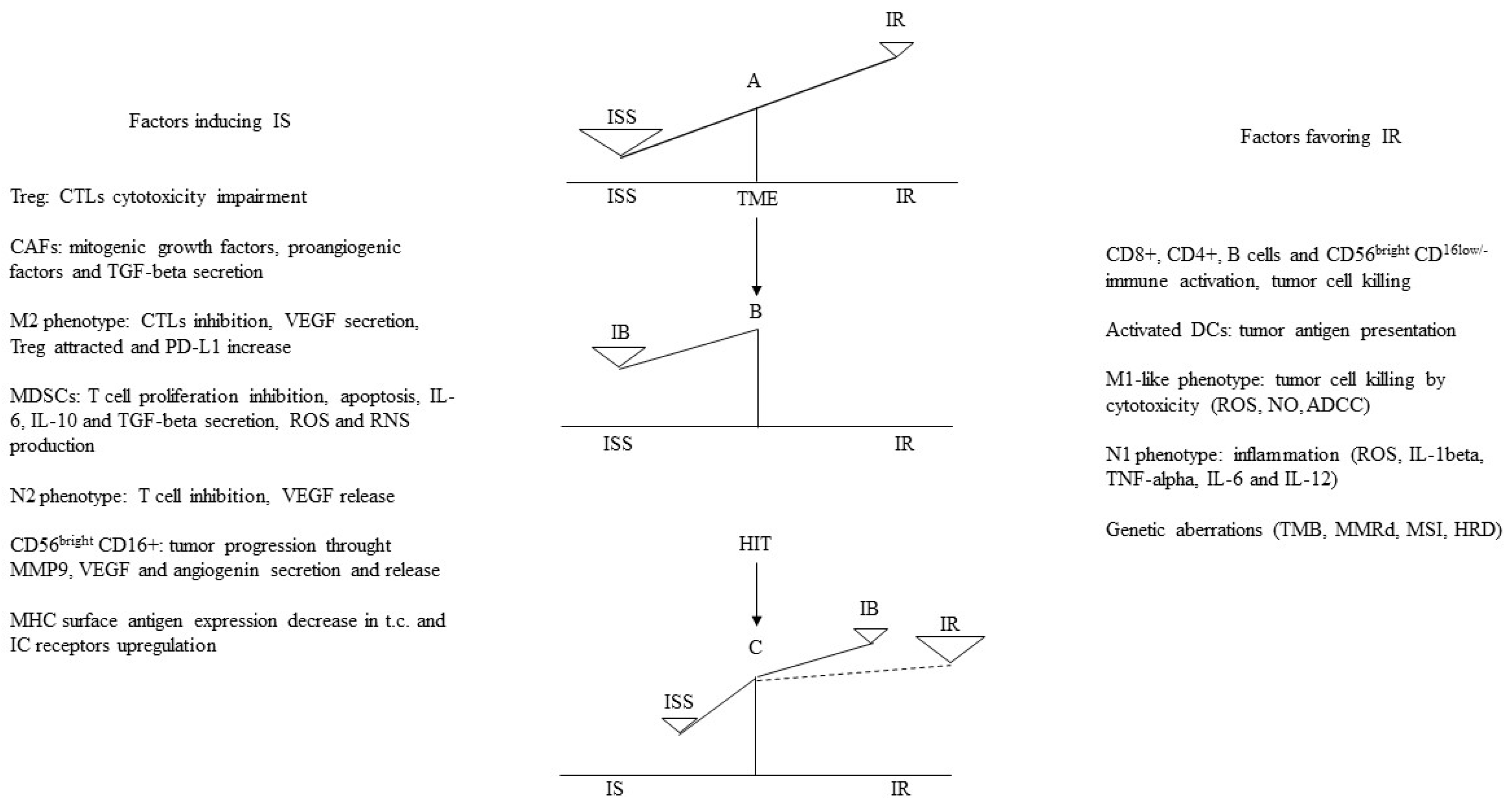
1.5.1. The Immunological Balance Resulting from the Interplay between Cancer Cells and TME: Mechanisms Inhibiting or Promoting an Antitumor Immune Response
1.5.2. Anti-Tumoral Immune Activities
1.5.3. Immune Activities Promoting Immune Inhibition and Tumor Progression
Tregs, TAMs, and MDSCs
N2 Phenotype, CD56bright CD16+ NK Subset, DCs, and Exosomes
1.5.4. Conditions Favoring a Successful TME Immune Manipulation
1.5.5. Metastatic Breast Cancer as an Incurable Disease
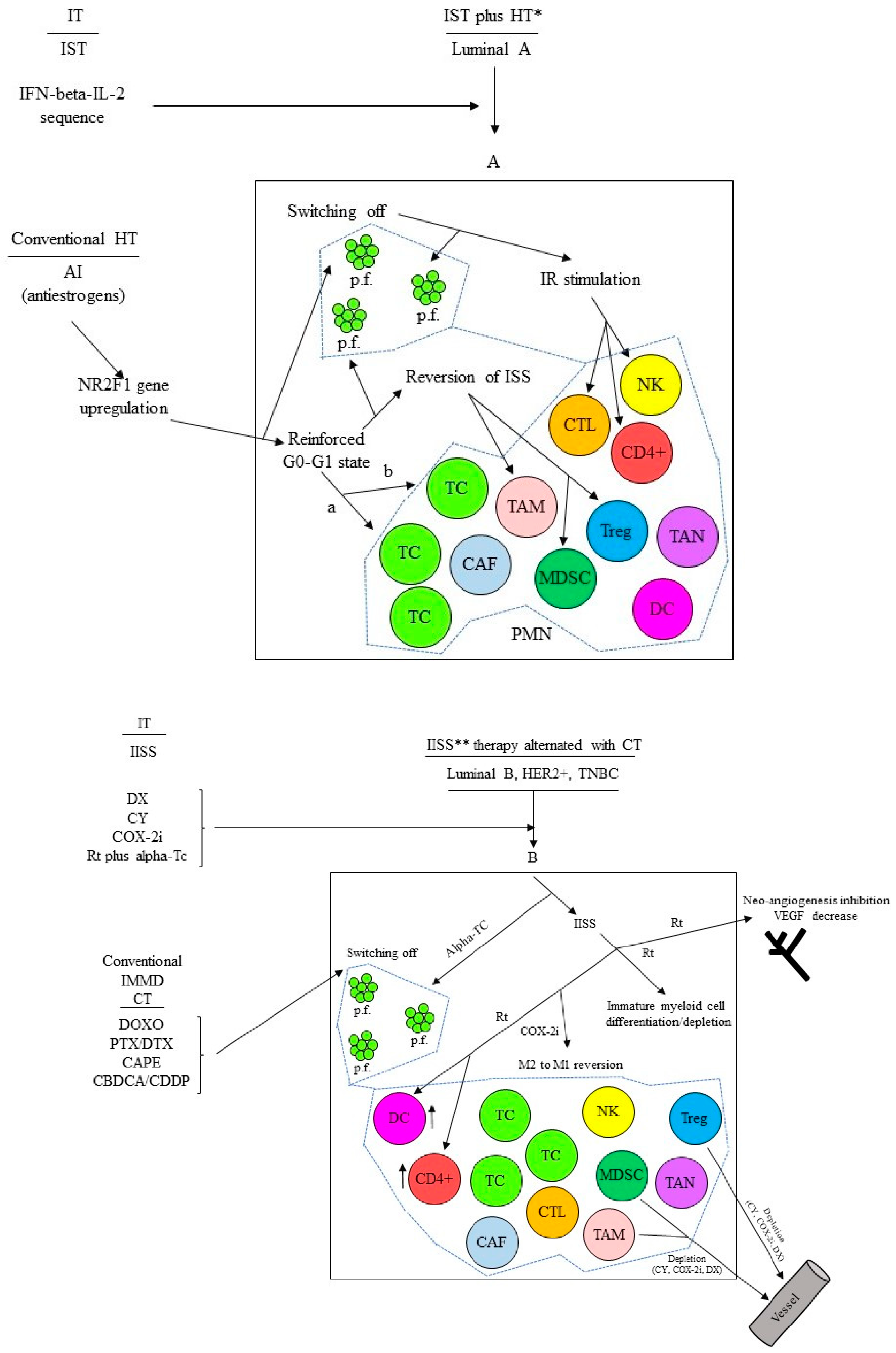
1.6. Locally Advanced Breast Cancer Patients: A New Paradigm for Additional Adjuvant Therapy
1.6.1. The Conventional Adjuvant CT and the Formation of the Pre-Metastatic Niches (PMNs)
1.6.2. The Specific Biology of Disseminated Cancer Cells (DCCs)
2. Discussion and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Globocan. Available online: https://gco.iarc.fr (accessed on 26 July 2022).
- Emens, L.A. Breast Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 4, 511–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriques, B.; Mendes, F.; Martins, D. Immunotherapy in Breast Cancer: When, How, and What Challenges? Biomedicines 2021, 9, 1687. [Google Scholar] [CrossRef]
- Senkus, E.; Cardoso, F.; Pagani, O. Time for more optimism in metastatic breast cancer? Cancer Treat. Rev. 2014, 40, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [Green Version]
- Tong, C.W.S.; Wu, M.; Cho, W.C.S.; To, K.K.W. Recent Advances in the Treatment of Breast Cancer. Front. Oncol. 2018, 8, 227. [Google Scholar] [CrossRef] [Green Version]
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110. [Google Scholar] [CrossRef]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast cancer: Biology, biomarkers, and treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef]
- Arab, A.; Yazdian-Robati, R.; Behravan, J. HER2-Positive Breast Cancer Immunotherapy: A Focus on Vaccine Development. Arch. Immunol. Ther. Exp. 2020, 68, 2. [Google Scholar] [CrossRef]
- Sivaganesh, V.; Promi, N.; Maher, S.; Peethambaran, B. Emerging Immunotherapies against Novel Molecular Targets in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 2433. [Google Scholar] [CrossRef]
- Tavares, D.F.; Ribeiro, V.C.; Andrade, M.; Cardoso-Júnior, L.M.; Teixeira, T.R.G.; Varrone, G.R.; Britto, R.L. Immunotherapy using PD-1/PDL-1 inhibitors in metastatic triple-negative breast cancer: A systematic review. Oncol. Rev. 2021, 15, 497. [Google Scholar] [CrossRef]
- Barroso-Sousa, R.; Keenan, T.E.; Li, T.; Tayob, N.; Trippa, L.; Pastorello, R.G.; Richardson Iii, E.T.; Dillon, D.; Amoozgar, Z.; Overmoyer, B.; et al. Nivolumab in combination with cabozantinib for metastatic triple-negative breast cancer: A phase II and biomarker study. NPJ Breast Cancer 2021, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Sternschuss, M.; Yerushalmi, R.; Saleh, R.R.; Amir, E.; Goldvaser, H. Efficacy and safety of neoadjuvant immune checkpoint inhibitors in early-stage triple-negative breast cancer: A systematic review and meta-analysis. J. Cancer Res. Clin. Oncol. 2021, 147, 3369–3379. [Google Scholar] [CrossRef] [PubMed]
- Galván Morales, M.A.; Barrera Rodríguez, R.; Santiago Cruz, J.R.; Teran, L.M. Overview of New Treatments with Immunotherapy for Breast Cancer and a Proposal of a Combination Therapy. Molecules 2020, 25, 5686. [Google Scholar] [CrossRef] [PubMed]
- Luen, S.J.; Savas, P.; Fox, S.B.; Salgado, R.; Loi, S. Tumour-infiltrating lymphocytes and the emerging role of immunotherapy in breast cancer. Pathology 2017, 49, 141–155. [Google Scholar] [CrossRef]
- Kim, H.M.; Lee, J.; Koo, J.S. Clinicopathological and prognostic significance of programmed death ligand-1 expression in breast cancer: A meta-analysis. BMC Cancer 2017, 17, 690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salemme, V.; Centonze, G.; Cavallo, F.; Defilippi, P.; Conti, L. The Crosstalk Between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front. Oncol. 2021, 11, 610303. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019, 10, 2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planes-Laine, G.; Rochigneux, P.; Bertucci, F.; Chrétien, A.S.; Viens, P.; Sabatier, R.; Gonçalves, A. PD-1/PD-L1 Targeting in Breast Cancer: The First Clinical Evidences Are Emerging. A Literature Review. Cancers 2019, 11, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, T.; Yao, S.; Zhu, G.; Flies, A.S.; Flies, S.J.; Chen, L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 2008, 111, 3635–3643. [Google Scholar] [CrossRef]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef]
- Bell, R.B.; Feng, Z.; Bifulco, C.B.; Leidner, R.; Weinberg, A.; Fox, B.A. Immunotherapy. In Oral, Head and Neck Oncology and Reconstructive Surgery; Bell, R.B., Fernandes, R.P., Andersen, P.E., Eds.; Elsevier: New York, NY, USA, 2018; pp. 314–340. [Google Scholar]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Fellner, C. Ipilimumab (yervoy) prolongs survival in advanced melanoma: Serious side effects and a hefty price tag may limit its use. Pharm. Ther. 2012, 37, 503–530. [Google Scholar]
- Yang, Q.; Cao, W.; Wang, Z.; Zhang, B.; Liu, J. Regulation of cancer immune escape: The roles of miRNAs in immune checkpoint proteins. Cancer Lett. 2018, 431, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef]
- Nallasamy, P.; Chava, S.; Verma, S.S.; Mishra, S.; Gorantla, S.; Coulter, D.W.; Byrareddy, S.N.; Batra, S.K.; Gupta, S.C.; Challagundla, K.B. PD-L1, inflammation, non-coding RNAs, and neuroblastoma: Immuno-oncology perspective. Semin. Cancer Biol. 2018, 52 Pt 2, 53–65. [Google Scholar] [CrossRef]
- Vennapusa, B.; Baker, B.; Kowanetz, M.; Boone, J.; Menzl, I.; Bruey, J.M.; Fine, G.; Mariathasan, S.; McCaffery, I.; Mocci, S.; et al. Development of a PD-L1 Complementary Diagnostic Immunohistochemistry Assay (SP142) for Atezolizumab. Appl. Immunohistochem. Mol. Morphol. 2019, 27, 92–100. [Google Scholar] [CrossRef]
- García-Aranda, M.; Redondo, M. Immunotherapy: A Challenge of Breast Cancer Treatment. Cancers 2019, 11, 1822. [Google Scholar] [CrossRef] [Green Version]
- Dimitrova, N.; Saz Parkinson, Z.; Bramesfeld, A.; Uluturk Tekin, A.; Bocchi, G.; Pylkkanen, L.; Lopez Alcalde, J.; Neamtiu, L.; Ambrosio, M.; Deandrea, S.; et al. European Guidelines for Breast Cancer Screening and Diagnosis–the European Breast Guidelines; EUR 28360 EN, JRC104007; Publications Office of the European Union: Luxembourg, 2016. [Google Scholar]
- Nicolini, A.; Ferrari, P.; Duffy, M.J. Prognostic and predictive biomarkers in breast cancer: Past, present and future. Semin. Cancer Biol. 2018, 52 Pt 1, 56–73. [Google Scholar] [CrossRef]
- Erber, R.; Hartmann, A. Understanding PD-L1 Testing in Breast Cancer: A Practical Approach. Breast Care 2020, 15, 481–490. [Google Scholar] [CrossRef]
- Noske, A.; Wagner, D.C.; Schwamborn, K.; Foersch, S.; Steiger, K.; Kiechle, M.; Karapetyan, S.; Oettler, D.; Hapfelmeier, A.; Roth, W.; et al. Comparison study of different programmed death-ligand 1(PD-L1) assays, readers and scoring methods in triple-negative breast cancer (TNBC). Ann. Oncol. 2021, 32 (Suppl. 2), S26. [Google Scholar] [CrossRef]
- Vigliar, E.; Malapelle, U.; Bono, F.; Fusco, N.; Cortinovis, D.; Valtorta, E.; Spyridon, A.; Bimbatti, M.; Zocchi, M.; Piva, C.; et al. The Reproducibility of the Immunohistochemical PD-L1 Testing in Non-Small-Cell Lung Cancer: A Multicentric Italian Experience. Biomed. Res. Int. 2019, 2019, 6832909. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Abreu, D.R.; Felip, E.; Carcereny, E.; Gottfried, M.; Wehler, T.; Ahn, M.J.; Dolled-Filhart, M.; Zhang, J.; Shentu, Y.; et al. Prevalence of PD-L1 expression in patients with non-small cell lung cancer screened for enrollment in KEYNOTE-001, -010, and -024. Ann. Oncol. 2016, 27 (Suppl. S6), vi363. [Google Scholar] [CrossRef]
- Karlsson, E.; Appelgren, J.; Solterbeck, A.; Bergenheim, M.; Alvariza, V.; Bergh, J. Breast cancer during follow-up and progression—A population based cohort on new cancers and changed biology. Eur. J. Cancer 2014, 50, 2916–2924. [Google Scholar] [CrossRef]
- Lindström, L.S.; Karlsson, E.; Wilking, U.M.; Johansson, U.; Hartman, J.; Lidbrink, E.K.; Hatschek, T.; Skoog, L.; Bergh, J. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J. Clin. Oncol. 2012, 30, 2601–2608. [Google Scholar] [CrossRef]
- Schrijver, W.A.M.E.; Suijkerbuijk, K.P.M.; van Gils, C.H.; van der Wall, E.; Moelans, C.B.; van Diest, P.J. Receptor Conversion in Distant Breast Cancer Metastases: A Systematic Review and Meta-analysis. J. Natl. Cancer Inst. 2018, 110, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Amir, E.; Miller, N.; Geddie, W.; Freedman, O.; Kassam, F.; Simmons, C.; Oldfield, M.; Dranitsaris, G.; Tomlinson, G.; Laupacis, A.; et al. Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer. J. Clin. Oncol. 2012, 30, 587–592. [Google Scholar] [CrossRef]
- Foukakis, T.; Astrom, G.; Lindström, L.; Hatschek, T.; Bergh, J. When to order a biopsy to characterise a metastatic relapse in breast cancer. Ann. Oncol. 2012, 23 (Suppl. 10), 349–353. [Google Scholar] [CrossRef]
- Buisseret, L.; Garaud, S.; de Wind, A.; Van den Eynden, G.; Boisson, A.; Solinas, C.; Gu-Trantien, C.; Naveaux, C.; Lodewyckx, J.N.; Duvillier, H.; et al. Tumor-infiltrating lymphocyte composition, organization and PD-1/ PD-L1 expression are linked in breast cancer. Oncoimmunology 2016, 6, e1257452. [Google Scholar] [CrossRef]
- Núñez Abad, M.; Calabuig-Fariñas, S.; Lobo de Mena, M.; Torres-Martínez, S.; García González, C.; García García, J.Á.; Iranzo González-Cruz, V.; Camps Herrero, C. Programmed Death-Ligand 1 (PD-L1) as Immunotherapy Biomarker in Breast Cancer. Cancers 2022, 14, 307. [Google Scholar] [CrossRef]
- Boman, C.; Zerdes, I.; Mårtensson, K.; Bergh, J.; Foukakis, T.; Valachis, A.; Matikas, A. Discordance of PD-L1 status between primary and metastatic breast cancer: A systematic review and meta-analysis. Cancer Treat. Rev. 2021, 99, 102257. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Molinero, L.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Diéras, V.; Iwata, H.; Barrios, C.H.; Nechaeva, M.; Nguyen-Duc, A.; et al. Atezolizumab and nab-Paclitaxel in Advanced Triple-Negative Breast Cancer: Biomarker Evaluation of the IMpassion130 Study. J. Natl. Cancer Inst. 2021, 113, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.S.P.; Haberberger, J.; Severson, E.; Duncan, D.L.; Hemmerich, A.; Edgerly, C.; Ferguson, N.L.; Williams, E.; Elvin, J.; Vergilio, J.A.; et al. A pan-cancer analysis of PD-L1 immunohistochemistry and gene amplification, tumor mutation burden and microsatellite instability in 48,782 cases. Mod. Pathol. 2021, 34, 252–263. [Google Scholar] [CrossRef]
- Paver, E.C.; Cooper, W.A.; Colebatch, A.J.; Ferguson, P.M.; Hill, S.K.; Lum, T.; Shin, J.S.; O’Toole, S.; Anderson, L.; Scolyer, R.A.; et al. Programmed death ligand-1 (PD-L1) as a predictive marker for immunotherapy in solid tumours: A guide to immunohistochemistry implementation and interpretation. Pathology 2021, 53, 141–156. [Google Scholar] [CrossRef]
- Jeong, S.; Lee, N.; Park, M.J.; Jeon, K.; Song, W. Currently Used Laboratory Methodologies for Assays Detecting PD-1, PD-L1, PD-L2 and Soluble PD-L1 in Patients with Metastatic Breast Cancer. Cancers 2021, 13, 5225. [Google Scholar] [CrossRef]
- Parra, E.R.; Villalobos, P.; Mino, B.; Rodriguez-Canales, J. Comparison of Different Antibody Clones for Immunohistochemistry Detection of Programmed Cell Death Ligand 1 (PD-L1) on Non-Small Cell Lung Carcinoma. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 83–93. [Google Scholar] [CrossRef]
- Beaver, J.A.; Tzou, A.; Blumenthal, G.M.; McKee, A.E.; Kim, G.; Pazdur, R.; Philip, R. An FDA Perspective on the Regulatory Implications of Complex Signatures to Predict Response to Targeted Therapies. Clin. Cancer Res. 2017, 23, 1368–1372. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Wright, G.S.; et al. IMpassion130 Trial Investigators Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Rugo, H.S.; Loi, S.; Adams, S.; Schmid, P.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Winer, E.P.; Kockx, M.M.; et al. PD-L1 Immunohistochemistry Assay Comparison in Atezolizumab Plus nab-Paclitaxel-Treated Advanced Triple-Negative Breast Cancer. J. Natl. Cancer Inst. 2021, 113, 1733–1743. [Google Scholar] [CrossRef]
- Ma, J.; Li, J.; Qian, M.; Han, W.; Tian, M.; Li, Z.; Wan, G.Z.; He, S.; Wu, K. PD-L1 expression and the prognostic significance in gastric cancer: A retrospective comparison of three PD-L1 antibody clones (SP142, 28–8 and E1L3N). Diagn. Pathol. 2018, 13, 91. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef]
- Eckstein, M.; Cimadamore, A.; Hartmann, A.; Lopez-Beltran, A.; Cheng, L.; Scarpelli, M.; Montironi, R.; Gevaert, T. PD-L1 assessment in urothelial carcinoma: A practical approach. Ann. Transl. Med. 2019, 7, 690. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, P.; Leal, L.F.; Duval da Silva, V.; da Silva, E.C.A.; Cordeiro de Lima, V.C.; Reis, R.M. PD-L1 expression by Tumor Proportion Score (TPS) and Combined Positive Score (CPS) are similar in non-small cell lung cancer (NSCLC). J. Clin. Pathol. 2021, 74, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Delord, J.P.; Im, S.A.; Ott, P.A.; Piha-Paul, S.A.; Bedard, P.L.; Sachdev, J.; Le Tourneau, C.; van Brummelen, E.; Varga, A.; et al. Safety and Antitumor Activity of Pembrolizumab in Patients with Estrogen Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 2804–2811. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.; Loi, S.; Toppmeyer, D.; Cescon, D.W.; De Laurentiis, M.; Nanda, R.; Winer, E.P.; Mukai, H.; Tamura, K.; Armstrong, A.; et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: Cohort B of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Winer, E.P.; Lipatov, O.; Im, S.A.; Goncalves, A.; Muñoz-Couselo, E.; Lee, K.S.; Schmid, P.; Tamura, K.; Testa, L.; Witzel, I.; et al. KEYNOTE-119 investigators. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): A randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 499–511. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. KEYNOTE-355 Investigators. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. KEYNOTE-522 Investigators. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Adel, N.G. Current treatment landscape and emerging therapies for metastatic triple-negative breast cancer. Am. J. Manag. Care 2021, 27 (Suppl. 5), S87–S96. [Google Scholar] [PubMed]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G.; et al. International Breast Cancer Study Group and the Breast International Group. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): A single-arm, multicentre, phase 1b-2 trial. Lancet Oncol. 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Ahmed, K.A.; Kim, Y.; Arrington, J.A.; Kim, S.; DeJesus, M.; Soyano, A.E.; Armaghani, A.J.; Costa, R.; Khong, H.T.; Loftus, L.S.; et al. Nivolumab and Stereotactic Radiosurgery for Patients With Breast Cancer Brain Metastases: A Nonrandomized, Open-Label Phase 1b Study. Adv. Radiat. Oncol. 2021, 6, 100798. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Emens, L.A.; Cruz, C.; Eder, J.P.; Braiteh, F.; Chung, C.; Tolaney, S.M.; Kuter, I.; Nanda, R.; Cassier, P.A.; Delord, J.P.; et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol. 2019, 5, 74–82. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. IMpassion130 Investigators. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar]
- Tokumaru, Y.; Joyce, D.; Takabe, K. Current status and limitations of immunotherapy for breast cancer. Surgery 2020, 167, 628–630. [Google Scholar] [CrossRef]
- Franzoi, M.A.; Romano, E.; Piccart, M. Immunotherapy for early breast cancer: Too soon, too superficial, or just right? Ann. Oncol. 2021, 32, 323–336. [Google Scholar] [CrossRef]
- Emens, L.A.; Adams, S.; Barrios, C.H.; Diéras, V.; Iwata, H.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Winer, E.P.; Patel, S.; et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. 2021, 32, 983–993. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): A randomised, double-blind, phase 3 trial. Lancet 2020, 396, 1090–1100. [Google Scholar] [CrossRef]
- Miles, D.; Gligorov, J.; André, F.; Cameron, D.; Schneeweiss, A.; Barrios, C.; Xu, B.; Wardley, A.; Kaen, D.; Andrade, L.; et al. IMpassion131 investigators. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann. Oncol. 2021, 32, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, A.S.; Nichols, E.; Cimino-Mathews, A.; Peer, C.; Cao, L.; Lee, M.J.; Kohn, E.C.; Annunziata, C.M.; Lipkowitz, S.; Trepel, J.B.; et al. A phase I study of the PD-L1 inhibitor, durvalumab, in combination with a PARP inhibitor, olaparib, and a VEGFR1-3 inhibitor, cediranib, in recurrent women’s cancers with biomarker analyses. J. Immunother. Cancer 2019, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.V.; Blohmer, J.U.; Grischke, E.M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: Clinical results and biomarker analysis of GeparNuevo study. Ann. Oncol. 2019, 30, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Bachelot, T.; Filleron, T.; Bieche, I.; Arnedos, M.; Campone, M.; Dalenc, F.; Coussy, F.; Sablin, M.P.; Debled, M.; Lefeuvre-Plesse, C.; et al. Durvalumab compared to maintenance chemotherapy in metastatic breast cancer: The randomized phase II SAFIR02-BREAST IMMUNO trial. Nat. Med. 2021, 27, 250–255. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Zhang, L.; Wang, Z.; Kong, X.; Zhai, J.; Fang, Y.; Wang, J. Efficacy and Safety of Anti-PD-1/ PD-L1 Monotherapy for Metastatic Breast Cancer: Clinical Evidence. Front. Pharmacol. 2021, 12, 653521. [Google Scholar] [CrossRef]
- Jiang, D.M.; Fyles, A.; Nguyen, L.T.; Neel, B.G.; Sacher, A.; Rottapel, R.; Wang, B.X.; Ohashi, P.S.; Sridhar, S.S. Phase I study of local radiation and tremelimumab in patients with inoperable locally recurrent or metastatic breast cancer. Oncotarget 2019, 10, 2947–2958. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.A.; Andresen, N.K.; Russnes, H.G.; Fretland, S.Ø.; Falk, R.S.; Lingjærde, O.C.; Naume, B. ICON: A randomized phase IIb study evaluating immunogenic chemotherapy combined with ipilimumab and nivolumab in patients with metastatic hormone receptor positive breast cancer. J. Transl. Med. 2020, 18, 269. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Mo, H.; Hu, X.; Gao, R.; Zhao, Y.; Liu, B.; Niu, L.; Sun, X.; Yu, X.; et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 2021, 39, 1578–1593.e8. [Google Scholar] [CrossRef]
- Di Spazio, L.; Rivano, M.; Cancanelli, L.; Chiumente, M.; Mengato, D.; Messori, A. The Degree of Programmed Death-Ligand 1 (PD-L1) Positivity as a Determinant of Outcomes in Metastatic Triple-Negative Breast Cancer Treated With First-Line Immune Checkpoint Inhibitors. Cureus 2022, 14, e21065. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.E.; Tolaney, S.M. Role of Immunotherapy in Triple-Negative Breast Cancer. J. Natl. Compr. Cancer Netw. 2020, 18, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilzecker, B.; Buoninfante, O.A.; Jacobs, H. DNA damage tolerance in stem cells, ageing, mutagenesis, disease and cancer therapy. Nucleic Acids Res. 2019, 47, 7163–7181. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Stewart, R.A.; Pilié, P.G.M.; Yap, T.A. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res. 2018, 78, 6717–6725. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. ENGOT-OV16/NOVA Investigators. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef]
- Min, A.; Kim, K.; Jeong, K.; Choi, S.; Kim, S.; Suh, K.J.; Lee, K.H.; Kim, S.; Im, S.A. Homologous repair deficiency score for identifying breast cancers with defective DNA damage response. Sci. Rep. 2020, 10, 12506. [Google Scholar] [CrossRef]
- Timperi, E.; Vissio, E.; Marchiò, C.; Romano, E. The Immune Landscape in Women Cancers. Cancer Treat. Res. 2020, 180, 215–249. [Google Scholar]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Morganella, S.; Purdie, C.A.; Jang, S.J.; Borgen, E.; Russnes, H.; Glodzik, D.; Zou, X.; Viari, A.; Richardson, A.L.; et al. Whole-Genome Sequencing Reveals Breast Cancers with Mismatch Repair Deficiency. Cancer Res. 2017, 77, 4755–4762. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, J.; Chen, J.; Zhou, Q. The developing landscape of combinatorial therapies of immune checkpoint blockade with DNA damage repair inhibitors for the treatment of breast and ovarian cancers. J. Hematol. Oncol. 2021, 14, 206. [Google Scholar] [CrossRef]
- Shevtsov, M.; Sato, H.; Multhoff, G.; Shibata, A. Novel Approaches to Improve the Efficacy of Immuno-Radiotherapy. Front. Oncol. 2019, 9, 156. [Google Scholar] [CrossRef] [Green Version]
- Lai, Q.; Wang, H.; Li, A.; Xu, Y.; Tang, L.; Chen, Q.; Zhang, C.; Gao, Y.; Song, J.; Du, Z. Decitibine improve the efficiency of anti-PD-1 therapy via activating the response to IFN/PD-L1 signal of lung cancer cells. Oncogene 2018, 37, 2302–2312. [Google Scholar] [CrossRef]
- Cao, D.; Zhao, J.; Nguyan, L.N.; Nguyen, L.; Khanal, S.; Dang, X.; Schank, M.; Chand Thakuri, B.K.; Wu, X.Y.; Morrison, Z.D.; et al. Disruption of Telomere Integrity and DNA Repair Machineries by KML001 Induces T Cell Senescence, Apoptosis, and Cellular Dysfunctions. Front. Immunol. 2019, 10, 1152. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Yi, M.; Qin, S.; Chu, Q.; Luo, S.; Wu, K. Prospects for combining immune checkpoint blockade with PARP inhibition. J. Hematol. Oncol. 2019, 12, 98. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.R.; Kofman, E.; Mo, S.S.; Elmarakeby, H.; Van Allen, E. Genomics of response to immune checkpoint therapies for cancer: Implications for precision medicine. Genome Med. 2018, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Cimino-Mathews, A.; Peer, C.J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Cao, L.; Harrell, M.I.; Swisher, E.M.; Houston, N.; et al. Safety and Clinical Activity of the Programmed Death-Ligand 1 Inhibitor Durvalumab in Combination With Poly (ADP-Ribose) Polymerase Inhibitor Olaparib or Vascular Endothelial Growth Factor Receptor 1-3 Inhibitor Cediranib in Women’s Cancers: A Dose-Escalation, Phase I Study. J. Clin. Oncol. 2017, 35, 2193–2202. [Google Scholar] [PubMed]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Rugo, H.S.; Llombart-Cussac, A.; Andre, F.; Robson, M.E.; Saji, S.; Harbeck, N.; Schmid, P.; Cescon, D.W.; Ahn, J.S.; Nanda, R.; et al. KEYLYNK-009: A phase II/III, open-label, randomized study of pembrolizumab (pembro) plus olaparib vs. pembro plus chemotherapy after induction with first-line pembro plus chemotherapy in patients with locally recurrent inoperable or metastatic triple-negative breast cancer (TNBC). J. Clin. Oncol. 2020, 38 (Suppl. 15), TPS596. [Google Scholar]
- Press, M.F.; Pike, M.C.; Chazin, V.R.; Hung, G.; Udove, J.A.; Markowicz, M.; Danyluk, J.; Godolphin, W.; Sliwkowski, M.; Akita, R. Her-2/neu expression in node-negative breast cancer: Direct tissue quantitation by computerized image analysis and association of overexpression with increased risk of recurrent disease. Cancer Res. 1993, 53, 4960–4970. [Google Scholar]
- Musolino, A.; Naldi, N.; Dieci, M.V.; Zanoni, D.; Rimanti, A.; Boggiani, D.; Sgargi, P.; Generali, D.G.; Piacentini, F.; Ambroggi, M.; et al. Immunoglobulin G fragment C receptor polymorphisms and efficacy of preoperative chemotherapy plus trastuzumab and lapatinib in HER2-positive breast cancer. Pharm. J. 2016, 16, 472–477. [Google Scholar] [CrossRef]
- Albanell, J.; Baselga, J. Trastuzumab, a humanized anti-HER2 monoclonal antibody, for the treatment of breast cancer. Drugs Today 1999, 35, 9319–9346. [Google Scholar]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Tóth, G.; Szöőr, Á.; Simon, L.; Yarden, Y.; Szöllősi, J.; Vereb, G. The combination of trastuzumab and pertuzumab administered at approved doses may delay development of trastuzumab resistance by additively enhancing antibody-dependent cell-mediated cytotoxicity. MAbs 2016, 8, 1361–1370. [Google Scholar] [CrossRef] [Green Version]
- Arteaga, C.L.; Winnier, A.R.; Poirier, M.C.; Lopez-Larraza, D.M.; Shawver, L.K.; Hurd, S.D.; Stewart, S.J. p185c-erbB-2 signal enhances cisplatin-induced cytotoxicity in human breast carcinoma cells: Association between an oncogenic receptor tyrosine kinase and drug-induced DNA repair. Cancer Res. 1994, 54, 3758–3765. [Google Scholar] [PubMed]
- Pietras, R.J.; Pegram, M.D.; Finn, R.S.; Maneval, D.A.; Slamon, D.J. Remission of human breast cancer xenografts on therapy with humanized monoclonal antibody to HER-2 receptor and DNA-reactive drugs. Oncogene 1998, 17, 2235–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasniqi, E.; Barchiesi, G.; Pizzuti, L.; Mazzotta, M.; Venuti, A.; Maugeri-Saccà, M.; Sanguineti, G.; Massimiani, G.; Sergi, D.; Carpano, S.; et al. Immunotherapy in HER2-positive breast cancer: State of the art and future perspectives. J. Hematol. Oncol. 2019, 12, 111. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, N.; Crown, J.; Collins, D.M. Immune checkpoint inhibitors: Key trials and an emerging role in breast cancer. Semin. Cancer Biol. 2022, 79, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Gelmon, K.A.; Verma, S.; Wardley, A.; Conte, P.; Miles, D.; Bianchi, G.; Cortes, J.; McNally, V.A.; Ros, S.G.A.; et al. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J. Clin. Oncol. 2010, 28, 1138–1144. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Cortés, J.; Kim, S.B.; Im, S.A.; Hegg, R.; Im, Y.H.; Roman, L.; Pedrini, J.L.; Pienkowski, T.; Knott, A.; et al. CLEOPATRA Study Group. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med. 2012, 366, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.M.; Kim, S.B.; Cortés, J.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Knott, A.; et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): Overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013, 14, 461–471. [Google Scholar] [CrossRef]
- Swain, S.M.; Baselga, J.; Kim, S.B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Heeson, S.; et al. CLEOPATRA Study Group. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Hurvitz, S.A.; Martin, M.; Symmans, W.F.; Jung, K.H.; Huang, C.S.; Thompson, A.M.; Harbeck, N.; Valero, V.; Stroyakovskiy, D.; Wildiers, H.; et al. Neoadjuvant trastuzumab, pertuzumab, and chemotherapy versus trastuzumab emtansine plus pertuzumab in patients with HER2-positive breast cancer (KRISTINE): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2018, 19, 115–126. [Google Scholar] [CrossRef]
- van Ramshorst, M.S.; van der Voort, A.; van Werkhoven, E.D.; Mandjes, I.A.; Kemper, I.; Dezentjé, V.O.; Oving, I.M.; Honkoop, A.H.; Tick, L.W.; van de Wouw, A.J.; et al. Dutch Breast Cancer Research Group (BOOG). Neoadjuvant chemotherapy with or without anthracyclines in the presence of dual HER2 blockade for HER2-positive breast cancer (TRAIN-2): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1630–1640. [Google Scholar] [CrossRef]
- Swain, S.M.; Ewer, M.S.; Viale, G.; Delaloge, S.; Ferrero, J.M.; Verrill, M.; Colomer, R.; Vieira, C.; Werner, T.L.; Douthwaite, H.; et al. BERENICE Study Group. Pertuzumab, trastuzumab, and standard anthracycline- and taxane-based chemotherapy for the neoadjuvant treatment of patients with HER2-positive localized breast cancer (BERENICE): A phase II, open-label, multicenter, multinational cardiac safety study. Ann. Oncol. 2018, 29, 646–653. [Google Scholar] [PubMed]
- Schneeweiss, A.; Chia, S.; Hickish, T.; Harvey, V.; Eniu, A.; Hegg, R.; Tausch, C.; Seo, J.H.; Tsai, Y.F.; Ratnayake, J.; et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: A randomized phase II cardiac safety study (TRYPHAENA). Ann. Oncol. 2013, 24, 2278–2284. [Google Scholar] [CrossRef] [PubMed]
- Burguin, A.; Diorio, C.; Durocher, F. Breast Cancer Treatments: Updates and New Challenges. J. Pers. Med. 2021, 11, 808. [Google Scholar] [CrossRef]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the treatment of breast cancer. Annu. Rev. Med. 2015, 66, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Morganti, S.; Uliano, J.; Giugliano, F.; Crimini, E.; Curigliano, G. Margetuximab for the treatment of HER2-positive metastatic breast cancer. Expert Opin. Biol. Ther. 2021, 21, 127–133. [Google Scholar] [CrossRef]
- Montemurro, F.; Delaloge, S.; Barrios, C.H.; Wuerstlein, R.; Anton, A.; Brain, E.; Hatschek, T.; Kelly, C.M.; Peña-Murillo, C.; Yilmaz, M.; et al. Trastuzumab emtansine (T-DM1) in patients with HER2-positive metastatic breast cancer and brain metastases: Exploratory final analysis of cohort 1 from KAMILLA, a single-arm phase IIIb clinical trial. Ann. Oncol. 2020, 31, 1350–1358. [Google Scholar] [CrossRef]
- Krop, I.E.; Kim, S.B.; Martin, A.G.; LoRusso, P.M.; Ferrero, J.M.; Badovinac-Crnjevic, T.; Hoersch, S.; Smitt, M.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): Final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol. 2017, 18, 743–754. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. DESTINY-Breast01 Investigators. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef]
- Howard, F.M.; Villamar, D.; He, G.; Pearson, A.T.; Nanda, R. The emerging role of immune checkpoint inhibitors for the treatment of breast cancer. Expert Opin. Investig. Drugs 2022, 31, 531–548. [Google Scholar] [CrossRef]
- Emens, L.A.; Esteva, F.J.; Beresford, M.; Saura, C.; De Laurentiis, M.; Kim, S.B.; Im, S.A.; Wang, Y.; Salgado, R.; Mani, A.; et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): A phase 2, multicentre, randomised, double-blind trial. Lancet Oncol. 2020, 21, 1283–1295. [Google Scholar] [CrossRef]
- Walks, A.G.; Keenan, T.; Li, T.; Tayob, N.; Wulf, G.M.; Richardson, E.T.; Mittendorf, E.A.; Overmoyer, B.; Krop, I.E.; Winer, E.P.; et al. A phase Ib study of pembrolizumab (pembro) plus trastuzumab emtansine (T-DM1) for metastatic HER2+ breast cancer (MBC). J. Clin. Oncol. 2020, 38 (Suppl. 15), 1046. [Google Scholar] [CrossRef]
- Huober, J.; Barrios, C.H.; Niikura, N.; Jarząb, M.; Chang, Y.C.; Huggins-Puhalla, S.L.; Pedrini, J.; Zhukova, L.; Graupner, V.; Eiger, D.; et al. Atezolizumab With Neoadjuvant Anti-Human Epidermal Growth Factor Receptor 2 Therapy and Chemotherapy in Human Epidermal Growth Factor Receptor 2-Positive Early Breast Cancer: Primary Results of the Randomized Phase III IMpassion050 Trial. J. Clin. Oncol. 2022, 40, 2946–2956. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Muul, L.M.; Solomon, D.; Rosenberg, S.A. Expansion of human tumor infiltrating lymphocytes for use in immunotherapy trials. J. Immunol. Methods 1987, 102, 127–141. [Google Scholar] [CrossRef]
- Retecki, K.; Seweryn, M.; Graczyk-Jarzynka, A.; Bajor, M. The Immune Landscape of Breast Cancer: Strategies for Overcoming Immunotherapy Resistance. Cancers 2021, 13, 6012. [Google Scholar] [CrossRef]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Capietto, A.H.; Martinet, L.; Fournié, J.J. Stimulated γδ T cells increase the in vivo efficacy of trastuzumab in HER-2+ breast cancer. J. Immunol. 2011, 187, 1031–1038. [Google Scholar] [CrossRef]
- Wu, Y.; Kyle-Cezar, F.; Woolf, R.T.; Naceur-Lombardelli, C.; Owen, J.; Biswas, D.; Lorenc, A.; Vantourout, P.; Gazinska, P.; Grigoriadis, A.; et al. An innate-like Vδ1+ γδ T cell compartment in the human breast is associated with remission in triple-negative breast cancer. Sci. Transl. Med. 2019, 11, eaax9364. [Google Scholar] [CrossRef]
- Rugo, H.S.; Im, S.A.; Cardoso, F.; Cortés, J.; Curigliano, G.; Musolino, A.; Pegram, M.D.; Wright, G.S.; Saura, C.; Escrivá-de-Romaní, S.; et al. SOPHIA Study Group. Efficacy of Margetuximab vs. Trastuzumab in Patients with Pretreated ERBB2-Positive Advanced Breast Cancer: A Phase 3 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 573–584. [Google Scholar] [CrossRef]
- Nicolini, A.; Carpi, A. Beta-interferon and interleukin-2 prolong more than three times the survival of 26 consecutive endocrine dependent breast cancer patients with distant metastases: An exploratory trial. Biomed. Pharmacother. 2005, 59, 253–263. [Google Scholar] [CrossRef]
- Nicolini, A.; Carpi, A.; Rossi, G. An immunotherapy schedule in endocrine-dependent metastatic breast cancer: Correlation between clinical course and immunologic parameters. J. Immunother. 2005, 28, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Rossi, G.; Ferrari, P.; Morganti, R.; Carpi, A. Final results of a 2:1 control-case observational study using interferon beta and interleukin-2, in addition to first-line hormone therapy, in estrogen receptor-positive, endocrine-responsive metastatic breast cancer patients. J. Cancer Metastasis Treat. 2022, 8, 13. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarmarkovich, M.; Farrel, A.; Sison, A., 3rd; di Marco, M.; Raman, P.; Parris, J.L.; Monos, D.; Lee, H.; Stevanovic, S.; Maris, J.M. Immunogenicity and Immune Silence in Human Cancer. Front. Immunol. 2020, 11, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.D.; Agostinis, P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol. Rev. 2017, 280, 126–148. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.J.; Lau, P.K.H.; Unsworth, A.S.; Loi, S.; Darcy, P.K.; Kershaw, M.H.; Slaney, C.Y. Tissue-Dependent Tumor Microenvironments and Their Impact on Immunotherapy Responses. Front. Immunol. 2018, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Dias, A.S.; Almeida, C.R.; Helguero, L.A.; Duarte, I.F. Metabolic crosstalk in the breast cancer microenvironment. Eur. J. Cancer 2019, 121, 154–171. [Google Scholar] [CrossRef]
- Deepak, K.G.K.; Vempati, R.; Nagaraju, G.P.; Dasari, V.R.; Nagini, S.; Rao, D.N.; Malla, R.R. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol. Res. 2020, 153, 104683. [Google Scholar] [CrossRef]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef]
- Mittal, S.; Brown, N.J.; Holen, I. The breast tumor microenvironment: Role in cancer development, progression and response to therapy. Expert Rev. Mol. Diagn. 2018, 18, 227–243. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Yanaba, K.; Tedder, T.F. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: Therapeutic B cell depletion enhances B16 melanoma growth in mice. J. Immunol. 2010, 184, 4006–4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Shaul, M.E.; Fridlender, Z.G. Neutrophils as active regulators of the immune system in the tumor microenvironment. J. Leukoc. Biol. 2017, 102, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, A.; Ferrari, P.; Rossi, G.; Carpi, A. Tumour growth and immune evasion as targets for a new strategy in advanced cancer. Endocr. Relat. Cancer 2018, 25, R577–R604. [Google Scholar] [CrossRef] [Green Version]
- Lapeyre-Prost, A.; Terme, M.; Pernot, S.; Pointet, A.L.; Voron, T.; Tartour, E.; Taieb, J. Immunomodulatory Activity of VEGF in Cancer. Int. Rev. Cell. Mol. Biol. 2017, 330, 295–342. [Google Scholar]
- Olkhanud, P.B.; Damdinsuren, B.; Bodogai, M.; Gress, R.E.; Sen, R.; Wejksza, K.; Malchinkhuu, E.; Wersto, R.P.; Biragyn, A. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4+ T cells to T-regulatory cells. Cancer Res. 2011, 71, 3505–3515. [Google Scholar] [CrossRef] [Green Version]
- Peng, G.L.; Li, L.; Guo, Y.W.; Yu, P.; Yin, X.J.; Wang, S.; Liu, C.P. CD8+ cytotoxic and FoxP3+ regulatory T lymphocytes serve as prognostic factors in breast cancer. Am. J. Transl. Res. 2019, 11, 5039–5053. [Google Scholar] [PubMed]
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T.P. The Role of Macrophages in Cancer Development and Therapy. Cancers 2021, 13, 1946. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Sznol, M.; Chen, L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin. Cancer Res. 2013, 19, 1021–1034. [Google Scholar] [CrossRef] [Green Version]
- Pinton, L.; Solito, S.; Damuzzo, V.; Francescato, S.; Pozzuoli, A.; Berizzi, A.; Mocellin, S.; Rossi, C.R.; Bronte, V.; Mandruzzato, S. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget 2016, 7, 1168–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Wang, Y.; Yan, F.; Zhang, P.; Li, H.; Zhao, H.; Yan, C.; Yan, F.; Ren, X. Noncanonical NF-κB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J. Immunol. 2014, 193, 2574–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, K.; Sakurai, M.; Yamamoto, Y.; Suzuki, E.; Tsuda, M.; Kataoka, T.R.; Hirata, M.; Nishie, M.; Nojiri, T.; Kumazoe, M.; et al. Alteration of specific cytokine expression patterns in patients with breast cancer. Sci. Rep. 2019, 9, 2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraiva, D.P.; Correia, B.F.; Salvador, R.; de Sousa, N.; Jacinto, A.; Braga, S.; Cabral, M.G. Circulating low density neutrophils of breast cancer patients are associated with their worse prognosis due to the impairment of T cell responses. Oncotarget 2021, 12, 2388–2403. [Google Scholar] [CrossRef]
- Rotondo, R.; Bertolotto, M.; Barisione, G.; Astigiano, S.; Mandruzzato, S.; Ottonello, L.; Dallegri, F.; Bronte, V.; Ferrini, S.; Barbieri, O. Exocytosis of azurophil and arginase 1-containing granules by activated polymorphonuclear neutrophils is required to inhibit T lymphocyte proliferation. J. Leukoc. Biol. 2011, 89, 721–727. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, A.; Brooks, M.W.; Houshyar, S.; Reinhardt, F.; Ardolino, M.; Fessler, E.; Chen, M.B.; Krall, J.A.; DeCock, J.; Zervantonakis, I.K.; et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discov. 2016, 6, 630–649. [Google Scholar] [CrossRef]
- Ethier, J.L.; Desautels, D.; Templeton, A.; Shah, P.S.; Amir, E. Prognostic role of neutrophil-to-lymphocyte ratio in breast cancer: A systematic review and meta-analysis. Breast Cancer Res. 2017, 19, 2. [Google Scholar] [CrossRef] [Green Version]
- Bun, A.; Fujimoto, Y.; Higuchi, T.; Sata, A.; Fukui, R.; Ozawa, H.; Miyagawa, Y.; Imamura, M.; Watanabe, T.; Miyoshi, Y. Prognostic Significance of Neutrophil-to-lymphocyte Ratio in Luminal Breast Cancers with Low Levels of Tumour-infiltrating Lymphocytes. Anti Cancer Res. 2020, 40, 2871–2880. [Google Scholar] [CrossRef]
- Bruno, A.; Bassani, B.; D’Urso, D.G.; Pitaku, I.; Cassinotti, E.; Pelosi, G.; Boni, L.; Dominioni, L.; Noonan, D.M.; Mortara, L.; et al. Angiogenin and the MMP9-TIMP2 axis are up-regulated in proangiogenic, decidual NK-like cells from patients with colorectal cancer. FASEB J. 2018, 32, 5365–5377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, I.; Amsalem, H.; Nissan, A.; Darash-Yahana, M.; Peretz, T.; Mandelboim, O.; Rachmilewitz, J. Characterization of tumor infiltrating natural killer cell subset. Oncotarget 2015, 6, 13835–13843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segovia-Mendoza, M.; Morales-Montor, J. Immune Tumor Microenvironment in Breast Cancer and the Participation of Estrogen and Its Receptors in Cancer Physiopathology. Front. Immunol. 2019, 10, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fainaru, O.; Almog, N.; Yung, C.W.; Nakai, K.; Montoya-Zavala, M.; Abdollahi, A.; D’Amato, R.; Ingber, D.E. Tumor growth and angiogenesis are dependent on the presence of immature dendritic cells. FASEB J. 2010, 24, 1411–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S.; et al. Impaired IFN-α production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Tu, Y.; Xu, Y.; Guo, Y.; Yao, F.; Zhang, X. Endoplasmic reticulum stress-induced exosomal miR-27a-3p promotes immune escape in breast cancer via regulating PD-L1 expression in macrophages. J. Cell. Mol. Med. 2020, 24, 9560–9573. [Google Scholar] [CrossRef]
- Sun, Y.; Guo, J.; Yu, L.; Guo, T.; Wang, J.; Wang, X.; Chen, Y. PD-L1+ exosomes from bone marrow-derived cells of tumor-bearing mice inhibit antitumor immunity. Cell. Mol. Immunol. 2021, 18, 2402–2409. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.W.; Chan, L.C.; Wei, Y.; Hsu, J.M.; Xia, W.; Cha, J.H.; Hou, J.; Hsu, J.L.; Sun, L.; et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018, 28, 862–864. [Google Scholar] [CrossRef] [Green Version]
- Morrissey, S.M.; Zhang, F.; Ding, C.; Montoya-Durango, D.E.; Hu, X.; Yang, C.; Wang, Z.; Yuan, F.; Fox, M.; Zhang, H.G.; et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. 2021, 33, 2040–2058.e10. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Frasor, J.; Stossi, F.; Danes, J.M.; Komm, B.; Lyttle, C.R.; Katzenellenbogen, B.S. Selective estrogen receptor modulators: Discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res. 2004, 64, 1522–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasor, J.; Chang, E.C.; Komm, B.; Lin, C.Y.; Vega, V.B.; Liu, E.T.; Miller, L.D.; Smeds, J.; Bergh, J.; Katzenellenbogen, B.S. Gene expression preferentially regulated by tamoxifen in breast cancer cells and correlations with clinical outcome. Cancer Res. 2006, 66, 7334–7340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welte, T.; Zhang, X.H.; Rosen, J.M. Repurposing Antiestrogens for Tumor Immunotherapy. Cancer Discov. 2017, 7, 17–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, A.; Rossi, G.; Ferrari, P.; Morganti, R.; Carpi, A. A new immunotherapy schedule in addition to first-line hormone therapy for metastatic breast cancer patients in a state of clinical benefit during hormone therapy. J. Mol. Med. 2020, 98, 375–382. [Google Scholar] [CrossRef]
- Nicolini, A.; Rossi, G.; Ferrari, P.; Carpi, A. Clinical and laboratory patterns during immune stimulation in hormone responsive metastatic breast cancer. Biomed. Pharmacother. 2014, 68, 171–178. [Google Scholar] [CrossRef]
- Nicolini, A.; Rossi, G.; Ferrari, P.; Carpi, A. Minimal residual disease in advanced or metastatic solid cancers: The G0-G1 state and immunotherapy are key to unwinding cancer complexity. Semin. Cancer Biol. 2022, 79, 68–82. [Google Scholar] [CrossRef]
- Nicolini, A.; Ferrari, P.; Morganti, R.; Carpi, A. Treatment of Metastatic or High-Risk Solid Cancer Patients by Targeting the Immune System and/or Tumor Burden: Six Cases Reports. Int. J. Mol. Sci. 2019, 20, 5986. [Google Scholar] [CrossRef]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef]
- Buonomo, O.C.; Caredda, E.; Portarena, I.; Vanni, G.; Orlandi, A.; Bagni, C.; Petrella, G.; Palombi, L.; Orsaria, P. New insights into the metastatic behavior after breast cancer surgery, according to well-established clinicopathological variables and molecular subtypes. PLoS ONE 2017, 12, e0184680. [Google Scholar] [CrossRef] [Green Version]
- Female Breast Cancer Subtypes-Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/breast-subtypes.html (accessed on 1 September 2022).
- Ott, P.A.; Bang, Y.J.; Piha-Paul, S.A.; Razak, A.; Bennouna, J.; Soria, J.C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated With Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhang, J.; Xu, L.; Yang, H.; Liang, N.; Zhang, L.; Zhang, F.; Zhang, X. Safety and Efficacy of the Rechallenge of Immune Checkpoint Inhibitors After Immune-Related Adverse Events in Patients With Cancer: A Systemic Review and Meta-Analysis. Front. Immunol. 2021, 12, 730320. [Google Scholar] [CrossRef] [PubMed]
- von Minckwitz, G.; Procter, M.; de Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. APHINITY Steering Committee and Investigators. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N. Engl. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef]
- Madariaga, A.; Bowering, V.; Ahrari, S.; Oza, A.M.; Lheureux, S. Manage wisely: Poly (ADP-ribose) polymerase inhibitor (PARPi) treatment and adverse events. Int. J. Gynecol. Cancer 2020, 30, 903–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldie, J.H.; Coldman, A.J. A mathematic model for relating the drug sensitivity of tumors to their spontaneous mutation rate. Cancer Treat. Rep. 1979, 63, 1727–1733. [Google Scholar] [PubMed]
- Goldie, J.H.; Coldman, A.J. Quantitative model for multiple levels of drug resistance in clinical tumors. Cancer Treat. Rep. 1983, 67, 923–931. [Google Scholar]
- Irurzun-Arana, I.; McDonald, T.O.; Trocóniz, I.F.; Michor, F. Pharmacokinetic Profiles Determine Optimal Combination Treatment Schedules in Computational Models of Drug Resistance. Cancer Res. 2020, 80, 3372–3382. [Google Scholar] [CrossRef]
- Kıbış, E.Y.; Büyüktahtakın, I.E. Optimizing multi-modal cancer treatment under 3D spatio-temporal tumor growth. Math. Biosci. 2019, 307, 53–69. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Fertig, E.J.; Jin, K.; Sukumar, S.; Pandey, N.B.; Popel, A.S. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat. Commun. 2014, 5, 4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, R.; Paredes, J.; Ribeiro, A.S. Impact of breast cancer cells’ secretome on the brain metastatic niche remodeling. Semin. Cancer Biol. 2020, 60, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Che, S.P.Y.; Park, J.Y.; Stokol, T. Tissue Factor-Expressing Tumor-Derived Extracellular Vesicles Activate Quiescent Endothelial Cells via Protease-Activated Receptor-1. Front. Oncol. 2017, 7, 261. [Google Scholar] [CrossRef] [Green Version]
- Ham, S.; Lima, L.G.; Chai, E.P.Z.; Muller, A.; Lobb, R.J.; Krumeich, S.; Wen, S.W.; Wiegmans, A.P.; Möller, A. Breast Cancer-Derived Exosomes Alter Macrophage Polarization via gp130/STAT3 Signaling. Front. Immunol. 2018, 9, 871. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wang, L.; Wang, T.; Li, Y.; Xun, Q.; Zhang, R.; Liu, L.; Li, L.; Wang, W.; Tian, Y.; et al. Tumor cell-secreted exosomal miR-22-3p inhibits transgelin and induces vascular abnormalization to promote tumor budding. Mol. Ther. 2021, 29, 2151–2166. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef] [Green Version]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef]
- Nogués, L.; Benito-Martin, A.; Hergueta-Redondo, M.; Peinado, H. The influence of tumour-derived extracellular vesicles on local and distal metastatic dissemination. Mol. Asp. Med. 2018, 60, 15–26. [Google Scholar] [CrossRef]
- Olejarz, W.; Kubiak-Tomaszewska, G.; Chrzanowska, A.; Lorenc, T. Exosomes in Angiogenesis and Anti-angiogenic Therapy in Cancers. Int. J. Mol. Sci. 2020, 21, 5840. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroni, S.; Romero-Cordoba, S.; Plantamura, I.; Dugo, M.; D’Ippolito, E.; Cataldo, A.; Cosentino, G.; Angeloni, V.; Rossini, A.; Daidone, M.G.; et al. Exosome-mediated delivery of miR-9 induces cancer-associated fibroblast-like properties in human breast fibroblasts. Cell Death Dis. 2016, 7, e2312. [Google Scholar] [CrossRef] [Green Version]
- Scognamiglio, I.; Cocca, L.; Puoti, I.; Palma, F.; Ingenito, F.; Quintavalle, C.; Affinito, A.; Roscigno, G.; Nuzzo, S.; Chianese, R.V.; et al. Exosomal microRNAs synergistically trigger stromal fibroblasts in breast cancer. Mol. Ther. Nucleic Acids 2022, 28, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Conte, M.; Rossi, G.; Ferrari, P.; Carpi, A.; Miccoli, P. A new pharmacological approach to gastrointestinal cancer at high risk of relapse based on maintenance of the cytostatic effect. Tumour. Biol. 2010, 31, 523–532. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]


| Trial | Phase | Drug | Setting | Treatment | Pts (n) | Outcome | Ref. |
|---|---|---|---|---|---|---|---|
| Anti HER2 mAbs | |||||||
| Cleopatra NCT00567190 | III | Pertuzumab (PERT)- Trastuzumab (TRST) | HER2+, l. a. or mBC | TRST + DCX + PB vs. TRST + PERT + DCX | 406 vs. 402 | mOS 40.8 vs. 56.5 mo. mPFS 12.4 vs. 18.7 mo. | [121] |
| Berenice NCT02132949e | II | PERT-TRST | HER2+, localized BC, neoadjuvant | DOXO + CY followed by PTX + TRST + PERT vs. FEC followed by DCX + TRST + PERT | 109 vs. 198 | pCR 61.8% vs. 60.7% 6.5% vs. 2% pts with at least one LVEF decline | [124] |
| Antibody–drug conjugates (ADCs) | |||||||
| Kamilla NCT01702571 | IIIb | Ado-trastuzumab emtansine (T-DM1) | Pre-treated, l.a. or mBC with brain metastases (BM) | T-DM1 | 398 | ORR 21.4%, CBR 42.9% (in 126 pts with measurable BM) PFS 55 mo., OS 18.9 mo. | [129] |
| Th3resa NCT01419197 | III | T-DM1 vs. physician choice | Pre-treated, HER2+ aBC | T-DM1 vs. physician choice | 404 vs. 198 | mOS 22.7 vs. 15.8 mo. Serious AEs 25% vs. 22% | [130] |
| Destiny-Breast 01 | II | TRST-Deruxtecan | Pre-treated, HER2+ mBC | TRST-Deruxtecan, 3 different doses | 184 | mRD 14.8 mo., mPFS 16.4 mo. with the recommended dose Grade 3–4 AEs ranging from 7.6 to 20.7% | [131] |
| IMMU-132-01 | I/II | Sacituzumab-govitecan hziy (Trop2 + SN38) | Pre-treated mBC | Sacituzumab-govitecan hziy | 108 | ORR 34.3%, MRD 9.1 mo., CBR 45.4%, mPFS 5.5 mo., mOS 13 mo. | [132] |
| Anti HER2 mAbs or ADCs plus ICIs | |||||||
| PANACEA | Ib/II | Pembrolizumab (PE)-TRST | HER2+ BC progressing on prior TRST | TRST + PE | 58 | ORR 15%, DCR 24% in PDL1 + vs. no ORR in PDL− pts | [64] |
| KATE2 | II | Atezolizumab (ATZ)-T-DM1 | Pre-treated, HER2+ aBC | ATZ + T-DM1 vs. PB + T-DM1 | 133 vs. 69 | mPFS 8.5 vs. 6.8 mo. in PDL1+ | [134] |
| Other investigational immunotherapies | |||||||
| SOPHIA NCT02492711 | III | Margetuximab (MAR) (chimeric antigen receptor) | Pre-treated, HER2+ mBC | MAR + CT vs. TRST + CT | 266 vs. 270 | ORR 22% vs. 16% mPFS 5.7 mo. vs. 4.4 mo. mOS 21.6 mo. vs. 19.8 mo. | [142] |
| 2:1 control-case observational study | II | Sequential IFN-beta-IL-2 plus SERMs | Forst line ER+, HER2− mBC | Sequential IFN-beta-IL-2 plus SERMs vs. AI | 42 vs. 95 | mPFS 33 mo vs. 18 mo. mOS 81 mo. vs. 62 mo. | [145] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolini, A.; Ferrari, P.; Carpi, A. Immune Checkpoint Inhibitors and Other Immune Therapies in Breast Cancer: A New Paradigm for Prolonged Adjuvant Immunotherapy. Biomedicines 2022, 10, 2511. https://doi.org/10.3390/biomedicines10102511
Nicolini A, Ferrari P, Carpi A. Immune Checkpoint Inhibitors and Other Immune Therapies in Breast Cancer: A New Paradigm for Prolonged Adjuvant Immunotherapy. Biomedicines. 2022; 10(10):2511. https://doi.org/10.3390/biomedicines10102511
Chicago/Turabian StyleNicolini, Andrea, Paola Ferrari, and Angelo Carpi. 2022. "Immune Checkpoint Inhibitors and Other Immune Therapies in Breast Cancer: A New Paradigm for Prolonged Adjuvant Immunotherapy" Biomedicines 10, no. 10: 2511. https://doi.org/10.3390/biomedicines10102511