
Neuron Protection by EDTA May Explain the Successful Outcomes of Toxic Metal Chelation Therapy in Neurodegenerative Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Toxic Metals
3. Toxic Metals as Risk Factors for ND
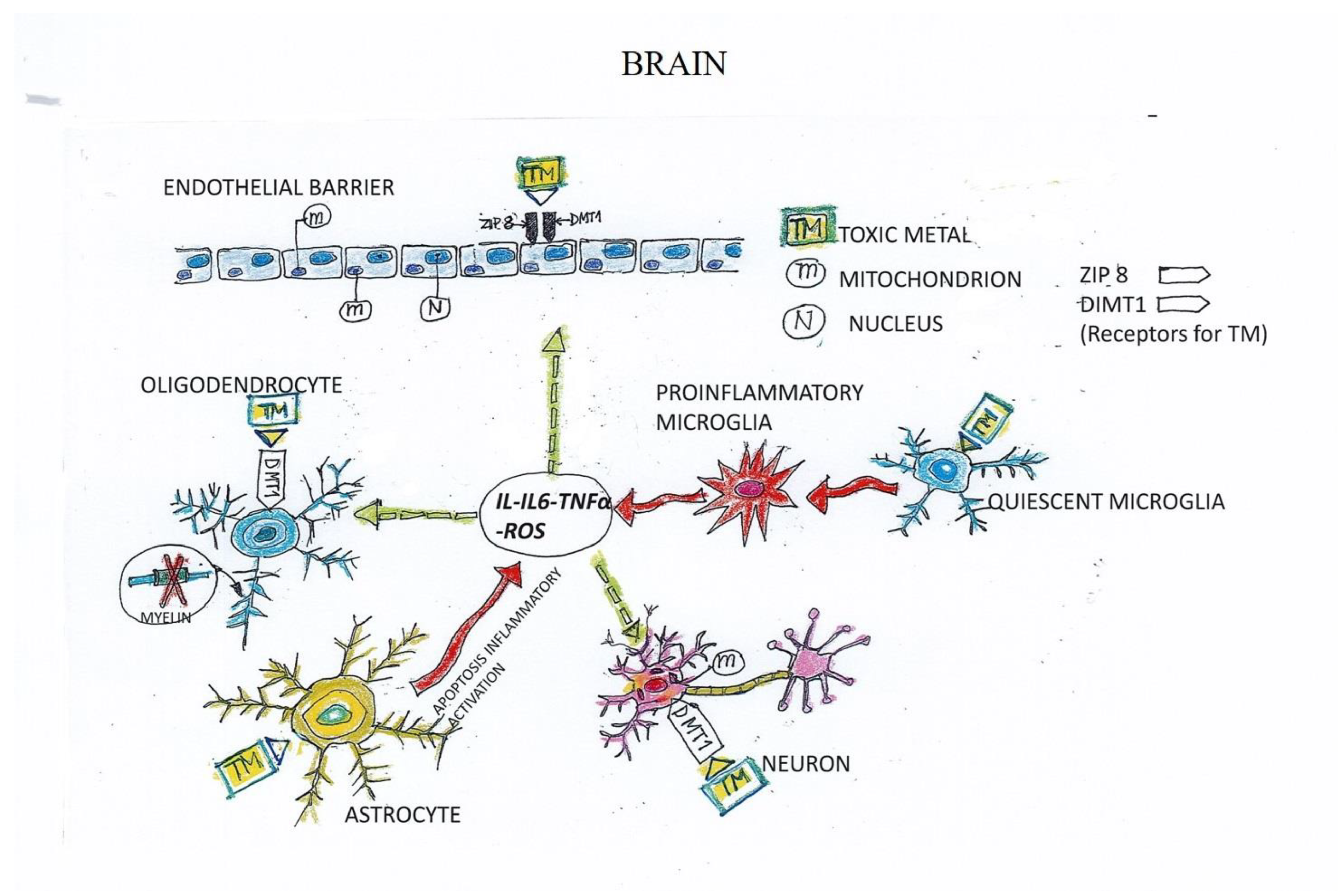
4. Mechanisms of Toxicity Induced by Toxic Metals in ND
5. Mechanism of TM-Induced Neuron and Endothelial Cell (EC) Damage
6. Chelation Therapy with EDTA
7. Case Report
8. Patients
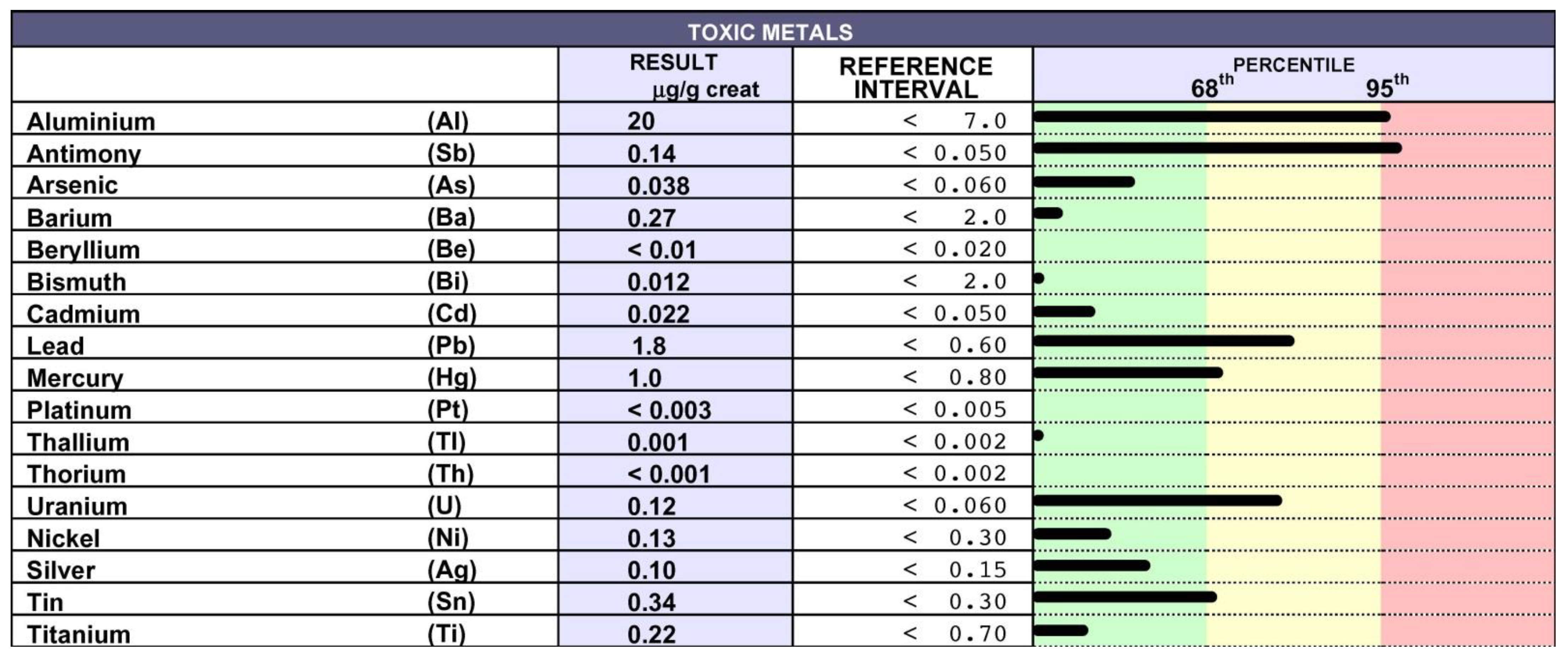
9. Chelation Test
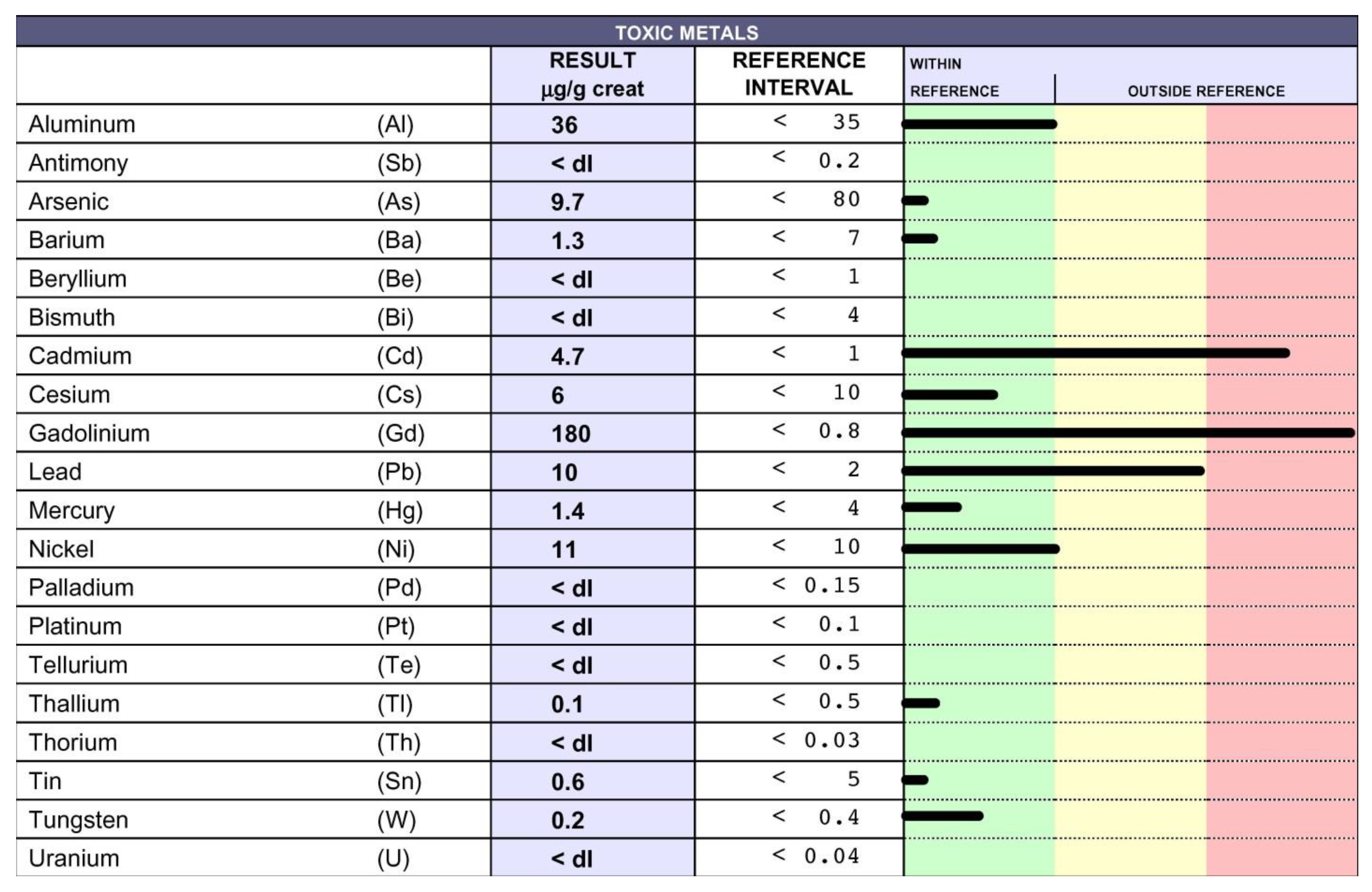
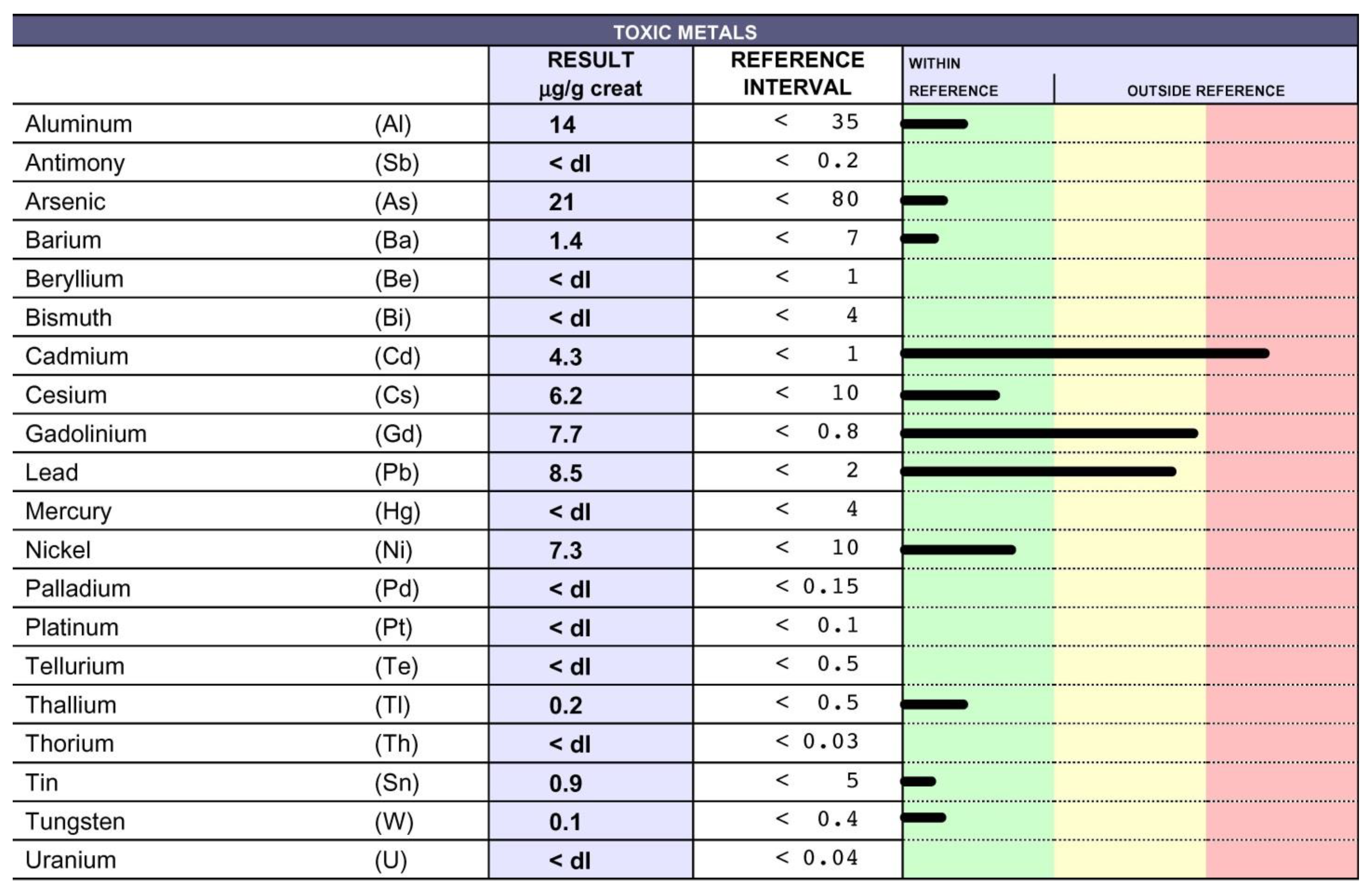
10. Chelation Therapy and Toxic Metal Analysis
11. Clinical Course and Treatments
12. Highlights
- The close association between toxic metals (TMs) and ND definitively points to TMs as relevant etiological agents in ND.
- We support the hypothesis that the endothelium, whose activation is instrumental to the pathogenesis and spread of ND, could also represent a relevant target in these diseases.
- Why do only some patients develop ND following exposure to TMs?The accumulation of TMs in the cells is dependent not only on their high levels, but also on the capacity of the cells to eliminate them. Successful elimination relies on the physiological, individual sources of antioxidants (e.g., enzymes, vitamins, reduced glutathione, metallothioneins), and functional mitochondria. These form part of the individual assets and may explain the aggravating role of age, which is associated with a biological decline in all of these functions. Consistently, we achieved our best outcomes with chelation therapy with young people, who benefit in terms of improved deambulation, disturbance of fine motor skills, paresthesia and ataxia, and quality of life, substantiating the need for early diagnosis and therapeutic intervention.
- Many researchers emphasize the importance of using the appropriate chelating agent for each metal. For instance, the iron (Fe) chelator PBT434 also modulates the uptake of Fe2+ by human brain microvascular endothelial cells [85]. However, most patients are intoxicated by multiple TMs, making it difficult to apply a tailored therapy. Moreover, in our long-lasting experience, we have learned that therapy with the chelator EDTA has widespread effectiveness in promoting the excretion of all TMs.
- EDTA therapy is a non-invasive treatment and is not associated with either early or late side effects
- In our experience, we could not find tight correlation between MRI and the clinical progression of MS [86].
- Finally, we observed the relevance of the early detection of TM poisoning and the ensuing EDTA chelation in achieving successful clinical outcomes.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alessandro, F.; Elena, F.M. EDTA Chelation Therapy for the Treatment of Neurotoxicity. Int. J. Mol. Sci. 2019, 20, 1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulgenzi, A.; Vietti, D.; Ferrero, M.E. EDTA Chelation Therapy in the Treatment of Neurodegenerative Diseases: An Update. Biomedicines 2020, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Fulgenzi, A.; Vietti, D.; Ferrero, M.E. Chronic toxic-metal poisoning and neurodegenerative diseases. Int. J. Curr. Res. 2017, 9, 57899–57999. [Google Scholar]
- Nies, D.H. Microbial heavy-metal resistance. Appl. Microbiol. Biotechnol. 1999, 51, 730–750. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Yedjou, C.G.; Patlolla, A.K.; Sutton, D.J. Heavy metal toxicity and the environment. Mol. Clin. Environ. Toxicol. 2012, 101, 133–164. [Google Scholar] [CrossRef] [Green Version]
- Rahman, Z.; Singh, V.P. The relative impact of toxic heavy metals (THMs) (arsenic (As), cadmium (Cd), chromium (Cr)(VI), mercury (Hg), and lead (Pb)) on the total environment: An overview. Environ. Monit. Assess. 2019, 191, 419. [Google Scholar] [CrossRef]
- Caito, S.; Aschner, M. Neurotoxicity of metals. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 131, pp. 169–189. [Google Scholar] [CrossRef]
- Duffus, J.H. Heavy metals-a meaningless term? Pure Appl. Chem. 2002, 74, 793–807. [Google Scholar] [CrossRef] [Green Version]
- Gunnarsson, L.-G.; Bodin, L. Occupational Exposures and Neurodegenerative Diseases—A Systematic Literature Review and Meta-Analyses. Int. J. Environ. Res. Public Health 2019, 16, 337. [Google Scholar] [CrossRef] [Green Version]
- Abbaszadeh, S.; Tabary, M.; Aryannejad, A.; Abolhasani, R.; Araghi, F.; Khaheshi, I.; Azimi, A. Air pollution and multiple sclerosis: A comprehensive review. Neurol. Sci. 2021, 42, 4063–4072. [Google Scholar] [CrossRef]
- Sarihi, S.; Niknam, M.; Mahjour, S.; Hosseini-Bensenjan, M.; Moazzen, F.; Soltanabadi, S.; Akbari, H. Toxic heavy metal concentrations in multiple sclerosis patients: A systematic review and meta-analysis. EXCLI J. 2021, 20, 1571–1584. [Google Scholar] [CrossRef]
- Qin, X.; Wu, P.; Wen, T.; Jia, R.; Zhang, R.; Jin, J.; Hu, F.; Chen, Q.Y.; Dang, J. Comparative assessment of blood Metal/metalloid levels, clinical heterogeneity, and disease severity in amyotrophic lateral sclerosis patients. NeuroToxicology 2022, 89, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Patti, F.; Fiore, M.; Chisari, C.G.; D’Amico, E.; Fermo, S.L.; Toscano, S.; Copat, C.; Ferrante, M.; Zappia, M. CSF neurotoxic metals/metalloids levels in amyotrophic lateral sclerosis patients: Comparison between bulbar and spinal onset. Environ. Res. 2020, 188, 109820. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-D.; Little, J.; Gomes, J.; Cashman, N.R.; Krewski, D. Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. NeuroToxicology 2017, 61, 101–130. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A. Childhood Lead Exposure and Adult Neurodegenerative Disease. J. Alzheimer’s Dis. 2018, 64, 17–42. [Google Scholar] [CrossRef]
- Kullmann, J.A.P.; Pamphlett, R. A Comparison of Mercury Exposure from Seafood Consumption and Dental Amalgam Fillings in People with and without Amyotrophic Lateral Sclerosis (ALS): An International Online Case-Control Study. Int. J. Environ. Res. Public Health 2018, 15, 2874. [Google Scholar] [CrossRef] [Green Version]
- Tomljenovic, L. Aluminum and Alzheimer’s Disease: After a Century of Controversy, Is there a Plausible Link? J. Alzheimer’s Dis. 2011, 23, 567–598. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Du, Y. Cadmium and Its Neurotoxic Effects. Oxidative Med. Cell. Longev. 2013, 2013, 898034. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Zhang, W.; Liu, X.; Zhang, C.; Wang, P.; Zhao, X. Circulatory Levels of Toxic Metals (Aluminum, Cadmium, Mercury, Lead) in Patients with Alzheimer’s Disease: A Quantitative Meta-Analysis and Systematic Review. J. Alzheimer’s Dis. 2018, 62, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Bakulski, K.M.; Seo, Y.A.; Hickman, R.C.; Brandt, D.; Vadari, H.S.; Hu, H.; Park, S.K. Heavy Metals Exposure and Alzheimer’s Disease and Related Dementias. J. Alzheimer’s Dis. 2020, 76, 1215–1242. [Google Scholar] [CrossRef]
- Colomina, M.T.; Peris-Sampedro, F. Aluminum and Alzheimer’s Disease. Adv. Neurobiol. 2017, 18, 183–197. [Google Scholar] [CrossRef]
- Mold, M.; Linhart, C.; Gómez-Ramírez, J.; Villegas-Lanau, A.; Exley, C. Aluminum and Amyloid-β in Familial Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 73, 1627–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mold, M.J.; O’Farrell, A.; Morris, B.; Exley, C. Aluminum and Tau in Neurofibrillary Tangles in Familial Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2021, 5, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Zhao, L.; Hai, Y.; Fahim, M.; Alwayli, D.; Wang, X.; Li, H. “Metal elements and pesticides as risk factors for Parkinson’s disease—A review”. Toxicol. Rep. 2021, 8, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Karri, V.; Schuhmacher, M.; Kumar, V. A systems toxicology approach to compare the heavy metal mixtures (Pb, As, MeHg) impact in neurodegenerative diseases. Food Chem. Toxicol. 2020, 139, 111257. [Google Scholar] [CrossRef]
- Pamphlett, R.; Jew, S.K. Inorganic mercury within motor neurons does not cause the TDP-43 changes seen in sporadic ALS. Toxicol. Lett. 2011, 201, 58–61. [Google Scholar] [CrossRef]
- Karri, V.; Kumar, V.; Ramos, D.; Oliveira, E.; Schuhmacher, M. Comparative In Vitro Toxicity Evaluation of Heavy Metals (Lead, Cadmium, Arsenic, and Methylmercury) on HT-22 Hippocampal Cell Line. Biol. Trace Element Res. 2017, 184, 226–239. [Google Scholar] [CrossRef] [Green Version]
- Maiuolo, J.; Macrì, R.; Bava, I.; Gliozzi, M.; Musolino, V.; Nucera, S.; Carresi, C.; Scicchitano, M.; Bosco, F.; Scarano, F.; et al. Myelin Disturbances Produced by Sub-Toxic Concentration of Heavy Metals: The Role of Oligodendrocyte Dysfunction. Int. J. Mol. Sci. 2019, 20, 4554. [Google Scholar] [CrossRef] [Green Version]
- Ijomone, O.M.; Ifenatuoha, C.W.; Aluko, O.M.; Ijomone, O.K.; Aschner, M. The aging brain: Impact of heavy metal neurotoxicity. Crit. Rev. Toxicol. 2020, 50, 801–814. [Google Scholar] [CrossRef]
- Garza-Lombó, C.; Posadas, Y.; Quintanar, L.; Gonsebatt, M.E.; Franco, R. Neurotoxicity Linked to Dysfunctional Metal Ion Homeostasis and Xenobiotic Metal Exposure: Redox Signaling and Oxidative Stress. Antioxidants Redox Signal. 2018, 28, 1669–1703. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Finley, E.J.; Chakraborty, S.; Fretham, S.J.B.; Aschner, M. Cellular transport and homeostasis of essential and nonessential metals. Metallomics 2012, 4, 593–605. [Google Scholar] [CrossRef]
- Prakash, C.; Soni, M.; Kumar, V. Mitochondrial oxidative stress and dysfunction in arsenic neurotoxicity: A review. J. Appl. Toxicol. 2015, 36, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qu, W.; Kadiiska, M.B. Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicol. Appl. Pharmacol. 2009, 238, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavesi, T.; Moreira, J.C. Mechanisms and individuality in chromium toxicity in humans. J. Appl. Toxicol. 2020, 40, 1183–1197. [Google Scholar] [CrossRef]
- Shilpa, O.; Anupama, K.P.; Antony, A.; Gurushankara, H.P. Lead (Pb) induced Oxidative Stress as a Mechanism to Cause Neurotoxicity in Drosophila melanogaster. Toxicology 2021, 462, 152959. [Google Scholar] [CrossRef]
- Zimmermann, L.T.; Santos, D.B.; Naime, A.A.; Leal, R.B.; Dórea, J.G.; Barbosa, F., Jr.; Aschner, M.; Rocha, J.B.T.; Farina, M. Comparative study on methyl- and ethylmercury-induced toxicity in C6 glioma cells and the potential role of LAT-1 in mediating mercurial-thiol complexes uptake. NeuroToxicology 2013, 38, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Roth, A.D.; Núñez, M.T. Oligodendrocytes: Functioning in a Delicate Balance between High Metabolic Requirements and Oxidative Damage. Adv. Exp. Med. Biol. 2016, 949, 167–181. [Google Scholar] [CrossRef]
- Karri, V.; Ramos, D.; Martinez, J.B.; Odena, A.; Oliveira, E.; Coort, S.L.; Evelo, C.T.; Mariman, E.C.; Schuhmacher, M.; Kumar, V. Differential protein expression of hippocampal cells associated with heavy metals (Pb, As, and MeHg) neurotoxicity: Deepening into the molecular mechanism of neurodegenerative diseases. J. Proteom. 2018, 187, 106–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Xia, M.; Zorec, R.; Parpura, V.; Verkhratsky, A. Astrocytes in heavy metal neurotoxicity and neurodegeneration. Brain Res. 2021, 1752, 147234. [Google Scholar] [CrossRef]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal Toxicity Links to Alzheimer’s Disease and Neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef]
- Alizadeh-Ghodsi, M.; Zavvari, A.; Ebrahimi-Kalan, A.; Shiri-Shahsavar, M.R.; Yousefi, B. The hypothetical roles of arsenic in multiple sclerosis by induction of inflammation and aggregation of tau protein: A commentary. Nutr. Neurosci. 2016, 21, 92–96. [Google Scholar] [CrossRef]
- Morelli, A.; Ravera, S.; Calzia, D.; Panfoli, I. Impairment of heme synthesis in myelin as potential trigger of multiple sclerosis. Med. Hypotheses 2012, 78, 707–710. [Google Scholar] [CrossRef]
- Ravera, S.; Morelli, A.M.; Panfoli, I. Myelination increases chemical energy support to the axon without modifying the basic physicochemical mechanism of nerve conduction. Neurochem. Int. 2020, 141, 104883. [Google Scholar] [CrossRef]
- Ravera, S.; Bartolucci, M.; Calzia, D.; Morelli, A.M.; Panfoli, I. Efficient extra-mitochondrial aerobic ATP synthesis in neuronal membrane systems. J. Neurosci. Res. 2021, 99, 2250–2260. [Google Scholar] [CrossRef]
- Takahashi, T.; Shimohata, T. Vascular Dysfunction Induced by Mercury Exposure. Int. J. Mol. Sci. 2019, 20, 2435. [Google Scholar] [CrossRef] [Green Version]
- Prozialeck, W.C.; Edwards, J.R.; Nebert, D.W.; Woods, J.M.; Barchowsky, A.; Atchison, W.D. The Vascular System as a Target of Metal Toxicity. Toxicol. Sci. 2007, 102, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Lamas, G.A.; Ujueta, F.; Navas-Acien, A. Lead and Cadmium as Cardiovascular Risk Factors: The Burden of Proof Has Been Met. J. Am. Heart Assoc. 2021, 10, e018692. [Google Scholar] [CrossRef]
- Chowdhury, R.; Ramond, A.; O’Keeffe, L.; Shahzad, S.; Kunutsor, S.; Muka, T.; Gregson, J.; Willeit, P.; Warnakula, S.; Khan, H.; et al. Environmental toxic metal contaminants and risk of cardiovascular disease: Systematic review and meta-analysis. BMJ 2018, 362, k3310. [Google Scholar] [CrossRef] [Green Version]
- Skalny, A.; Kopylov, P.; Paoliello, M.; Chang, J.-S.; Aschner, M.; Bobrovnitsky, I.; Chao, J.; Aaseth, J.; Chebotarev, S.; Tinkov, A. Hair Lead, Aluminum, and Other Toxic Metals in Normal-Weight and Obese Patients with Coronary Heart Disease. Int. J. Environ. Res. Public Health 2021, 18, 8195. [Google Scholar] [CrossRef]
- Hu, X.F.; Lowe, M.; Chan, H.M. Mercury exposure, cardiovascular disease, and mortality: A systematic review and dose-response meta-analysis. Environ. Res. 2020, 193, 110538. [Google Scholar] [CrossRef]
- Clasen, S.C.; Dinh, P.C.; Hou, L.; Fung, C.; Sesso, H.D.; Travis, L.B. Cisplatin, environmental metals, and cardiovascular disease: An urgent need to understand underlying mechanisms. Cardio-Oncology 2021, 7, 34. [Google Scholar] [CrossRef]
- Choi, J.W.; Moon, W.-J. Gadolinium Deposition in the Brain: Current Updates. Korean J. Radiol. 2019, 20, 134–147. [Google Scholar] [CrossRef]
- Talukder, M.; Bi, S.-S.; Jin, H.-T.; Ge, J.; Zhang, C.; Lv, M.-W.; Li, J.-L. Cadmium induced cerebral toxicity via modulating MTF1-MTs regulatory axis. Environ. Pollut. 2021, 285, 117083. [Google Scholar] [CrossRef]
- Skjã¸rringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: Implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Howitt, J.; Putz, U.; Lackovic, J.; Doan, A.; Dorstyn, L.; Cheng, H.; Yang, B.; Chan-Ling, T.; Silke, J.; Kumar, S.; et al. Divalent metal transporter 1 (DMT1) regulation by Ndfip1 prevents metal toxicity in human neurons. Proc. Natl. Acad. Sci. USA 2009, 106, 15489–15494. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Huang, W.; Xu, F.; Cao, Z.; Jia, F.; Li, Y. Iron Dyshomeostasis Participated in Rat Hippocampus Toxicity Caused by Aluminum Chloride. Biol. Trace Element Res. 2019, 197, 580–590. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Chibowska, K.; Korbecki, J.; Gutowska, I.; Metryka, E.; Tarnowski, M.; Goschorska, M.; Barczak, K.; Chlubek, D.; Baranowska-Bosiacka, I. Pre- and Neonatal Exposure to Lead (Pb) Induces Neuroinflammation in the Forebrain Cortex, Hippocampus and Cerebellum of Rat Pups. Int. J. Mol. Sci. 2020, 21, 1083. [Google Scholar] [CrossRef] [Green Version]
- Metryka, E.; Chibowska, K.; Gutowska, I.; Falkowska, A.; Kupnicka, P.; Barczak, K.; Chlubek, D.; Baranowska-Bosiacka, I. Lead (Pb) Exposure Enhances Expression of Factors Associated with Inflammation. Int. J. Mol. Sci. 2018, 19, 1813. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Barber, D.S.; Zhang, P.; Liu, B. Complex II of the Mitochondrial Respiratory Chain Is the Key Mediator of Divalent Manganese-Induced Hydrogen Peroxide Production in Microglia. Toxicol. Sci. 2013, 132, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Yubolphan, R.; Phuagkhaopong, S.; Sangpairoj, K.; Sibmooh, N.; Power, C.; Vivithanaporn, P. Intracellular nickel accumulation induces apoptosis and cell cycle arrest in human astrocytic cells. Metallomics 2021, 13, mfaa006. [Google Scholar] [CrossRef]
- Wu, L.; Li, S.; Pang, S.; Zhang, B.; Wang, J.; He, B.; Lv, L.; Wang, W.; Zhao, N.; Zhang, Y. Effects of lead exposure on the activation of microglia in mice fed with high-fat diets. Environ. Toxicol. 2021, 36, 1923–1931. [Google Scholar] [CrossRef]
- Kirkley, K.S.; Popichak, K.A.; Afzali, M.F.; Legare, M.E.; Tjalkens, R.B. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. Neuroinflammation 2017, 14, 99. [Google Scholar] [CrossRef] [Green Version]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. J Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Mezzaroba, L.; Alfieri, D.F.; Simão, A.N.C.; Reiche, E.M.V. The role of zinc, copper, manganese and iron in neurodegenerative diseases. NeuroToxicology 2019, 74, 230–241. [Google Scholar] [CrossRef]
- Fujie, T.; Ito, K.; Ozaki, Y.; Takahashi, S.; Yamamoto, C.; Kaji, T. Induction of ZIP8, a ZIP transporter, via NF-κB signaling by the activation of IκBα and JNK signaling in cultured vascular endothelial cells exposed to cadmium. Toxicol. Appl. Pharmacol. 2021, 434, 115802. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, H.; Xing, X.; Zhao, Z.; He, J.; Li, J.; Chen, J.; Wang, M.; He, Y. Lead promotes abnormal angiogenesis induced by CCM3 gene defects via mitochondrial pathway. J. Dev. Orig. Health Dis. 2017, 9, 182–190. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2018, 1865, 1802–1809. [Google Scholar] [CrossRef]
- Liu, S.; Tsui, M.T.-K.; Lee, E.; Fowler, J.; Jia, Z. Uptake, efflux, and toxicity of inorganic and methyl mercury in the endothelial cells (EA.hy926). Sci. Rep. 2020, 10, 9023. [Google Scholar] [CrossRef]
- Cao, X.; Wang, S.; Bi, R.; Tian, S.; Huo, Y.; Liu, J. Toxic effects of Cr(VI) on the bovine hemoglobin and human vascular endothelial cells: Molecular interaction and cell damage. Chemosphere 2019, 222, 355–363. [Google Scholar] [CrossRef]
- O’Donoghue, J.L.; Watson, G.E.; Brewer, R.; Zareba, G.; Eto, K.; Takahashi, H.; Marumoto, M.; Love, T.; Harrington, D.; Myers, G.J. Neuropathology associated with exposure to different concentrations and species of mercury: A review of autopsy cases and the literature. NeuroToxicology 2020, 78, 88–98. [Google Scholar] [CrossRef]
- Ferrero, M.E. Rationale for the Successful Management of EDTA Chelation Therapy in Human Burden by Toxic Metals. BioMed Res. Int. 2016, 2016, 8274504. [Google Scholar] [CrossRef]
- Aneni, E.C.; Escolar, E.; Lamas, G.A. Chronic Toxic Metal Exposure and Cardiovascular Disease: Mechanisms of Risk and Emerging Role of Chelation Therapy. Curr. Atheroscler. Rep. 2016, 18, 81. [Google Scholar] [CrossRef]
- Moreno, R.C.; Navas-Acien, A.; Escolar, E.; Nathan, D.M.; Newman, J.; Schmedtje, J.F.; Diaz, D.; Lamas, G.A.; Fonseca, V. Potential Role of Metal Chelation to Prevent the Cardiovascular Complications of Diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 2931–2941. [Google Scholar] [CrossRef]
- Fulgenzi, A.; Zito, F.; Marchelli, D.; Colombo, F.; Ferrero, M.E. New Insights into EDTA In Vitro Effects on Endothelial Cells and on In Vivo Labeled EDTA Biodistribution. J Heavy Met. Toxic. Distrib. 2016, 1, 7. [Google Scholar]
- Foglieni, C.; Fulgenzi, A.; Ticozzi, P.; Pellegatta, F.; Sciorati, C.; Belloni, D.; Ferrero, E.; Ferrero, M.E. Protective effect of EDTA preadministration on renal ischemia. BMC Nephrol. 2006, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef]
- Waubant, E.; Lucas, R.; Mowry, E.; Graves, J.; Olsson, T.; Alfredsson, L.; Langer-Gould, A. Environmental and genetic risk factors for MS: An integrated review. Ann. Clin. Transl. Neurol. 2019, 6, 1905–1922. [Google Scholar] [CrossRef] [Green Version]
- Kierdorf, K.; Wang, Y.; Neumann, H. Immune-Mediated CNS Damage. Results Probl. Cell Differ. 2009, 51, 173–196. [Google Scholar] [CrossRef]
- Balasa, R.; Barcutean, L.; Balasa, A.; Motataianu, A.; Roman-Filip, C.; Manu, D. The action of TH17 cells on blood brain barrier in multiple sclerosis and experimental autoimmune encephalomyelitis. Hum. Immunol. 2020, 81, 237–243. [Google Scholar] [CrossRef]
- Lebrun, C.; Rocher, F. Cancer Risk in Patients with Multiple Sclerosis: Potential Impact of Disease-Modifying Drugs. CNS Drugs 2018, 32, 939–949. [Google Scholar] [CrossRef]
- Nedredal, G.I.; Picon, R.V.; Chedid, M.F.; Foss, A. Immunosuppression in Liver Transplantation: State of the Art and Future Perspectives. Curr. Pharm. Des. 2020, 26, 3389–3401. [Google Scholar] [CrossRef] [PubMed]
- Basta, F.; Fasola, F.; Triantafyllias, K.; Schwarting, A. Systemic Lupus Erythematosus (SLE) Therapy: The Old and the New. Rheumatol. Ther. 2020, 7, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Fulgenzi, A.; Ferrero, M.E. Which Strategies can be Adopted against Heavy Metal Intoxication? J. Heavy Met. Toxic. Dis. 2016, 1, 1–2. [Google Scholar] [CrossRef]
- Bailey, D.K.; Clark, W.; Kosman, D.J. The iron chelator, PBT434, modulates transcellular iron trafficking in brain microvascular endothelial cells. PLoS ONE 2021, 16, e0254794. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, L.; Allen, C.M.; Williams, T.; Tallantyre, E.; Evangelou, N.; Chataway, J.; Ford, H.L. The contemporary role of MRI in the monitoring and management of people with multiple sclerosis in the UK. Mult. Scler. Relat. Disord. 2021, 55, 103190. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrero, M.E. Neuron Protection by EDTA May Explain the Successful Outcomes of Toxic Metal Chelation Therapy in Neurodegenerative Diseases. Biomedicines 2022, 10, 2476. https://doi.org/10.3390/biomedicines10102476
Ferrero ME. Neuron Protection by EDTA May Explain the Successful Outcomes of Toxic Metal Chelation Therapy in Neurodegenerative Diseases. Biomedicines. 2022; 10(10):2476. https://doi.org/10.3390/biomedicines10102476
Chicago/Turabian StyleFerrero, Maria Elena. 2022. "Neuron Protection by EDTA May Explain the Successful Outcomes of Toxic Metal Chelation Therapy in Neurodegenerative Diseases" Biomedicines 10, no. 10: 2476. https://doi.org/10.3390/biomedicines10102476