
Redox-Regulation in Cancer Stem Cells

Institute of Medical Biochemistry and Molecular Biology, University Medicine Greifswald, D-17475 Greifswald, Germany
*
Author to whom correspondence should be addressed.
Biomedicines 2022, 10(10), 2413; https://doi.org/10.3390/biomedicines10102413
Submission received: 4 July 2022
/
Revised: 19 September 2022
/
Accepted: 21 September 2022
/
Published: 27 September 2022
(This article belongs to the Special Issue Oxidative Stress– and Redox–Based Therapeutic Strategy in Cancers)
{kind=link}
{kind=link}
Abstract
:Cancer stem cells (CSCs) represent a small subset of slowly dividing cells with tumor-initiating ability. They can self-renew and differentiate into all the distinct cell populations within a tumor. CSCs are naturally resistant to chemotherapy or radiotherapy. CSCs, thus, can repopulate a tumor after therapy and are responsible for recurrence of disease. Stemness manifests itself through, among other things, the expression of stem cell markers, the ability to induce sphere formation and tumor growth in vivo, and resistance to chemotherapeutics and irradiation. Stemness is maintained by keeping levels of reactive oxygen species (ROS) low, which is achieved by enhanced activity of antioxidant pathways. Here, cellular sources of ROS, antioxidant pathways employed by CSCs, and underlying mechanisms to overcome resistance are discussed.
1. Introduction
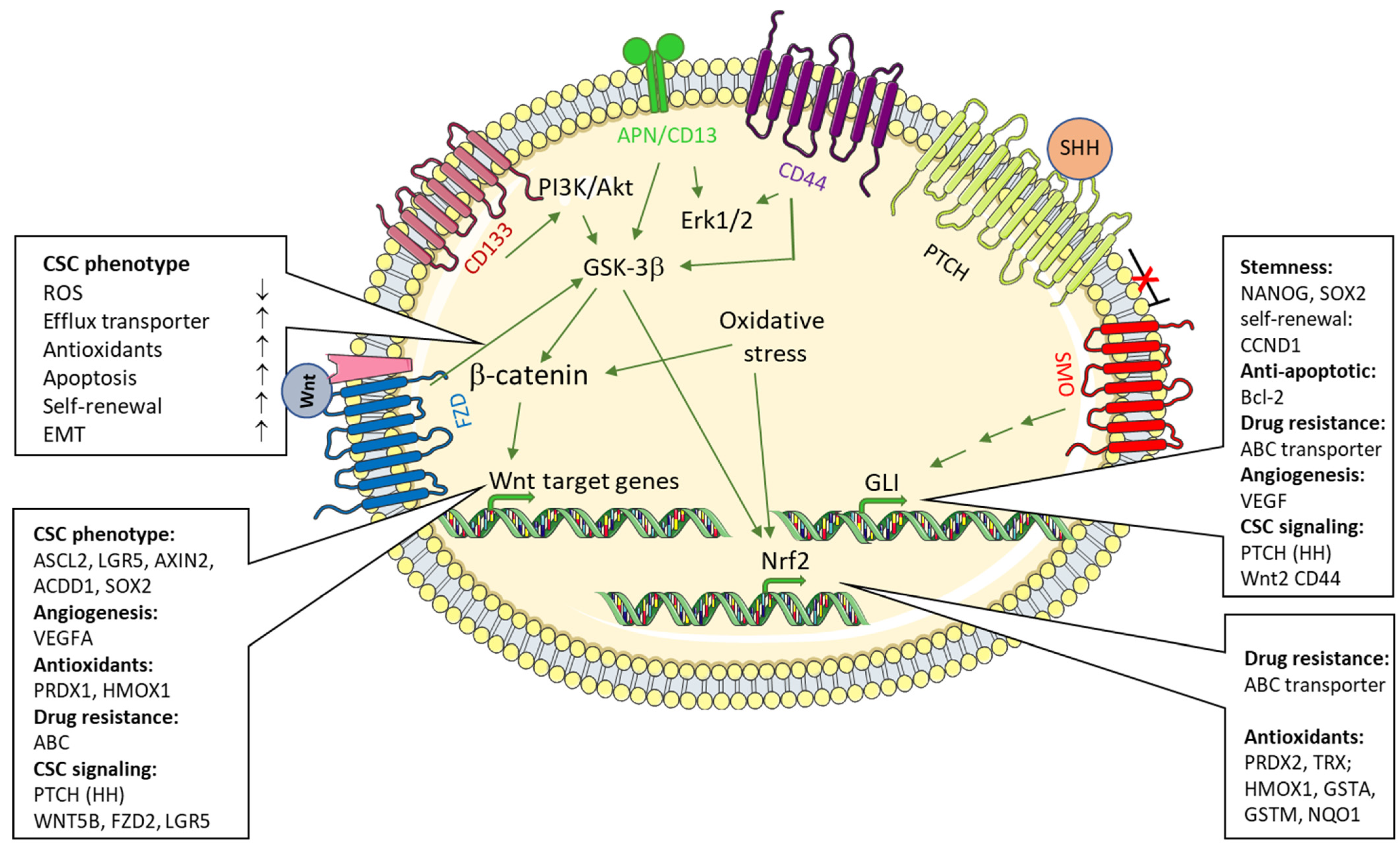
In cancer therapy, a significant limitation of many currently used chemotherapeutic protocols, and thus a longer-term remission, is that so-called tumor stem cells (cancer stem cells, CSC) evade the effect of the therapeutic agents. CSC show a progenitor cell-like phenotype, are found as a regular cellular population in most tumors or tumor cell lines, and, due to their ability to self-renew, contribute to tumor initiation, progression, and spread as well as to its resistance, recurrence, and metastasis after therapy [1,2]. Cancer stem cells are a self-renewing subpopulation of tumor cells that, due to their capacity for multilineage differentiation, give rise to all cell types present in a given tumor sample. CSCs evade chemotherapeutic attacks by expressing highly efficient efflux pumps [3]. The rapid extrusion of Hoechst 33342 is an established FACS-based technique with which this CSC (which can be defined and represented in FACS as a so-called “side population”, SP) can be quantified and enriched by “sorting” [4,5]. The CSC phenotype is characterized by, among other things, the expression of various surface markers. A continuous transition between CSC and mature tumor cells as well as CSC and non-tumor cells contributes to the expansion of the tumor mass, and is also regarded as the basis for the indefinite maintenance of tumor cell lines (7). CSC stemness is controlled by multiple signals from CSCs themselves and from the tumor microenvironment, which includes the so-called CSC niche [2]. Among the pathways that regulate both CSC differentiation and non-CSC dedifferentiation to achieve a progenitor phenotype, (non)canonical Wingless-Int (WNT), transforming growth factor/bone morphogenic protein (TGF/BMP), Sonic Hedgehog (HH), and Notch pathways are well established and most relevant signaling pathways [2] (Figure 1). A significant influence on the oncogenic transformation of CSC or the differentiation of CSC from non-malignant stem cells [6] is also attributed to the coagulation system [7,8] and the presence of reactive oxygen species (ROS)/hypoxia [9]. The contribution of the latter to the CSC phenotype and tumor development, maintenance, and progression is the main subject of this review.
2. “Side Population” Cells in Cancer
According to the CSC hypothesis, tumors typically consist of a heterologous mixture of tumor cells. Only a small proportion of tumor cells is in fact capable of inducing tumor growth when transferred to immunodeficient mice or, e.g., semi-solid media in vitro. This stem cell-like subpopulation is characterized by high proliferative potential, the capacity to self-renew and rebuild progeny [10]. Various cell surface proteins, including CD133 and CD44, have been identified as cancer stem cell markers [11,12]. Markers that would allow for the identification and isolation from tumors of CSCs are often lacking specificity and mostly rather represent markers for stem cells in general, as is true for CD133, CD44, and the aldehyde dehydrogenase, ALDH [10]. In the absence of tumor entity-specific CSC markers, the rapid extrusion of Hoechst 33342 by ABC transporters has become an established FACS-based technique with which these CSCs (which can be defined and represented in FACS as a so-called “side population”, SP) can be quantified and enriched by “sorting”.
As CSCs present themselves as a “side population” in FACS analysis, all the typical characteristics of CSCs of course apply to SP. Only a few studies directly investigated the effects of ROS on SP. Results of recent studies demonstrated, however, that in SP cells the intracellular concentration of ROS is low and expression of hypoxia inducible factor 1 (HIF1A) was found largely restricted to SP cells, where it promoted the expression of the anti-oxidant, heme oxygenase-1 [13].
In contrast, the exposure of SP cells to hydrogen peroxide induced CSC differentiation. Along this line, in bladder cancer cells, administration of dexamethasone to induce cell differentiation decreased the fraction of “side population” cells concomitant with the induction of intracellular ROS and loss of stemness-associated factors, including SNAIL, OCT4, or β-catenin [14].
The presence of this side population, which exhibits high drug efflux capacity, was first confirmed in human tumors by Goodell et al. [3] and has been applied since to many cancers [15,16,17] or cancer cell lines [18,19,20,21]. A special contribution to the optimization and standardization of the FACS protocol for the quantification of human and murine SP cells was made by Anna Golebiewska and her group [4]. The SP phenotype has been suggested to explain both resistance to chemotherapy [22,23,24,25] as well as higher tumorigenicity when xenografted into immunodeficient mice [26,27,28,29,30]. A population of glioma-inducing cells from human glioma tissue has been enriched on the basis of cellular autofluorescence and morphology only, instead of applying surface markers for selection [31].
3. Reactive Oxygen Species and Sources in CSC
Tumors, and CSCs in particular, typically exhibit a very tight regulation of ROS concentrations, which is essential for cell signaling, therapy resistance, and tumor recurrence [9]. Alterations in the cellular redox state can be due to both increased production and reduced removal of ROS [32]. In the various cell compartments, very different ROS species are formed, each of which has a special effect on tumorigenesis, tumor progression, or therapy.
ROS, generally, can be divided into two classes: free radical ROS and nonradical ROS. Whereas the former class includes the members superoxide anion (), nitric oxide (), and hydroxyl ion (), the latter class includes hydrogen peroxide (H2O2) and peroxynitrite (−ONOO).
Superoxide anion and hydrogen peroxide are certainly the best-characterized ROS [33,34]. Mitochondria are considered the major source of ROS [35]. In the mitochondrial respiration chain, 0.1–1.0% of all consumed oxygen by mitochondria can undergo one-electron reductions to yield [36,37]. The majority of originates from complexes I and III of the electron transport chain [37]. The superoxide anion radical (O−2) is subsequently converted into H2O2 by either superoxide dismutase (SOD) [38] or spontaneous dismutation. Due to its relative stability and lack of charge, H2O2 may diffuse across longer distances and, thus, can easily attack mitochondrial DNA or diffuse to the nucleus where chromosomal DNA gets damaged.
Another important cellular source of ROS is the family of NADPH oxidases (NOXs). Here, regulation relies on the signal-induced assembly of the NADPH oxidase. Membrane-bound NADPH subunits p22 and gp91 become assembled with the subunits p40, p47, and p67, which are initially located cytosolically, to provide the active NADPH oxidase. The formation of the active NADPH oxidase results in increased oxygen consumption. It has been suggested that the activation of NOXs in cancer cells contributes to oncogene-mediated cancer cell survival [39].
is a short-lived, ubiquitous signaling molecule implicated in a variety of ways in tumorigenesis and progression [40]. NO is formed by constitutively expressed (endothelial NOS/eNOS/NOS3 or neuronal NO synthase/nNOS/NOS1) or inducible isoforms (inducible NOS/iNOS/NOS2) of NO synthase. An increase in the expression/activity of iNOS and the enhanced ∙NO production that results is associated with various pathophysiological conditions, including inflammation, ischemia/hypoxia, and also cancer. Mechanistically, is formed by the oxidation of L-arginine which yields citrulline in addition to . exerts multiple effects on tumor cells, including both pro- and antitumorigenic effects depending on multiple aspects, which include the actual cell type/tissue, the amounts generated, the oxidative/reductive environment, the tumor environment, and surrounding/infiltrating immune cells and their polarization. The diverse roles of , along with most recent NO-related anticancer therapeutics and applications, have been comprehensively and excellently reviewed recently [40].
That NO is relevant to CSCs at all was first suggested in breast cancer, where NO increased expression of CD44 and of signal transducer and activator of transcription 3 (STAT3) [41]. Accumulating evidence shows that increased NO production by upregulated iNOS or eNOS supports a stem cell-like phenotype in various tumors, including prostate and bladder cancer [42,43], glioma [44,45], and liver and colon cancer [46,47]. Mechanistically, activated Notch signaling [46], increased expression of β-catenin [47], and transcriptional activation or protein stabilization of stem-cell associated transcription factors (SOX2, OCT4) or increased expression of stem cell markers such as CD133 and ALDH1 [45,48,49] contribute in NO-regulation of CSCs [50].
4. Reactive Oxygen Species in Tumorigenesis and Disease Progression
Reactive oxygen species (ROS) are generated under physiological conditions where they function as second messengers in numerous redox-sensitive signal transduction pathways. Oxidative stress has been recognized to play a key role in the pathophysiology of cancer. In tumor cells, maintaining a precise redox balance is a lifelong challenge. Depending on the microenvironment, which is formed by surrounding/infiltrating (immune-)cells, cytokines, and cells of the tumor niche, ROS produce two kinds of effects on tumors. Whereas appropriate concentrations of ROS stimulate tumorigenesis and promote disease progression, higher amounts of ROS cause cell death [51]. Solid tumors and leukemic cells have been demonstrated to produce increased amounts of ROS when compared to normal cells [52,53]. Nevertheless, the elevated ROS levels typically observed in tumors appear to be controlled and limited to a tumor-promoting level, which is achieved by means of the strong induction of an antioxidant equipment. However, “oxidative stress” may occur when the increased production of ROS can no longer be neutralized by anti-oxidative mechanisms. In pathophysiological conditions, chronically elevated amounts of ROS cause molecular damage which impairs cellular structures and functions, known as “oxidative stress” [51]. Historically, the term “oxidative stress” was defined as an imbalance between the generation of ROS and the capacity of the ROS-inactivating defense systems that are intended to detoxify them [54]. Some key findings have changed the understanding in recent years: different oxidants affect distinct subsets of target proteins through modifications that are specific both with respect to the oxidant and the site of modification, most frequently well-defined cysteinyl side chains. The antioxidant redox systems in the different cellular compartments, e.g., glutathione, NADPH, thioredoxin (Trx), and peroxidases such as the peroxiredoxins (Prx), are not in equilibrium but independently maintained at distinct redox potentials. Oxidative stress may thus, more contemporarily, be defined as the chronic dysregulation of redox systems and redox-responsive signal transduction pathways [55,56,57].
Similarly to normal stem cells, CSCs contain lower amounts of intracellular ROS than cells of the tumor bulk [58,59], which is mainly due to elevated expression levels of ROS scavengers [59,60,61]. As shown by Dong et al., the zinc finger transcriptional repressor, Snail, which is implicated in epithelial to mesenchymal transition (EMT), can alter cellular metabolism of breast cancer cells to that of CSCs [62]. The reduced expression of fructose-1,6-bisphosphate promoted glycolysis and formation of NADPH via the pentose-phosphate pathway.
Lower levels of ROS in CSCs are not only important in maintaining a stem cell-like phenotype, but also confer resistance to radiation or chemotherapy which is partly due to less DNA damage occurring upon therapy [63]. It could be confirmed in CD13+ liver CSCs that the pharmacological inhibition of APN/CD13 makes CSC more sensitive to the genotoxic chemotherapeutic 5′-fluorouracil (5-FU) [63,64]. Notably, inhibition of APN/CD13 suppressed the self-renewing and tumor-initiating activity of CD13+ dormant CSC [63].
5. Redox-Signaling in CSCs: Role of NRF2
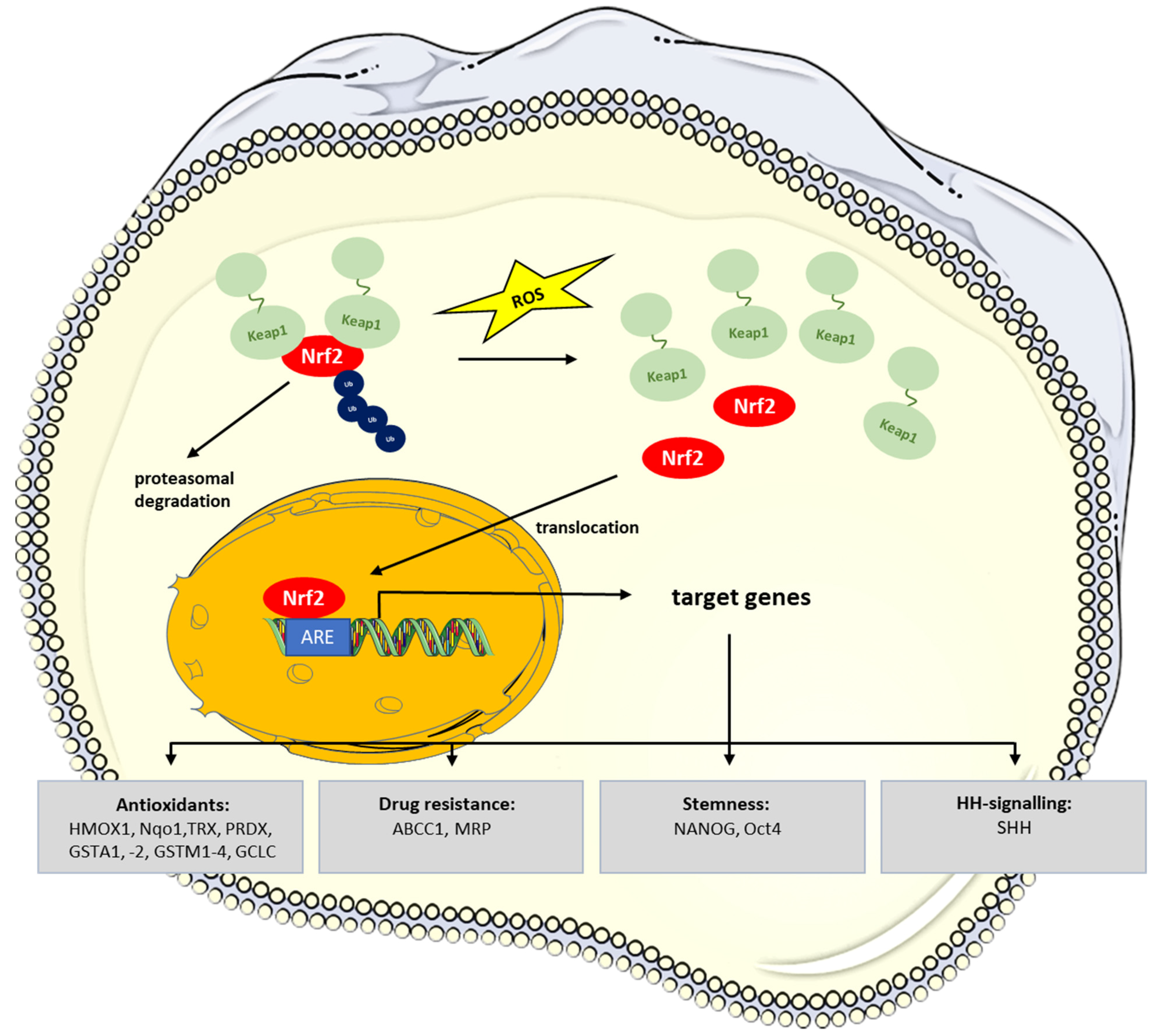
The transcription factor erythroid 2-like 2 (NRF2) is of paramount importance to the defense against oxidative stress by the induction of antioxidant protein, ROS-degrading enzymes, and efflux transporters.
NRF2 plays a special role in CSC maintenance and resistance to cancer therapy protocols. Notably, the low ROS levels which are typically observed in normal stem cells and CSCs and which are required for stem cell maintenance result from Nrf2 signaling [38,65]. Under normal conditions, NRF2 is constantly degraded upon binding to KEAP1 in a ubiquitin-proteasome dependent manner [66,67,68]. When cells encounter oxidative stress, NRF2 is released from Kelch-like ECH associated protein 1 (KEAP1) and translocates into the nucleus, where it activates antioxidant response element (ARE)-containing genes such as the antioxidants heme oxygenase 1 (HMOX1), NAD(P)H quinone oxidase (Nqo1), thioredoxin (TRX), peroxiredoxin 1 (PRDX), glutathione-S-transferases (GSTA1, -2, GSTM1-4), or the efflux pump ATP binding cassette subfamily C member 1 (ABCC1, MRP) [38,69,70] (Figure 2).
Early work identified Nrf2 activators as chemopreventive substances in animal models of chemically induced carcinogenesis and diseases associated with oxidative stress/inflammation [73,74]. Despite being protective for normal cells, NRF2 has stimulating effects on tumor growth/progression and increased resistance to chemotherapy, as has been shown for a variety of human cancers [70,75,76,77]. Persistent activation of Nrf2 signaling has been detected in many different human cancers [70] and is frequently the result of somatic mutations of the NRF2/KEAP1 genes [78]. Hypermethylation of the KEAP1 promoter and resulting downregulation of KEAP1 expression levels represent another relevant mechanism leading to a constitutive NRF2 activation in cancer [79,80,81,82,83].
In CSCs, low ROS levels are a prerequisite for maintaining stemness, which is achieved to a large extent by NRF2 keeping these cells in a stem cell-like and undifferentiated state [84]. Accordingly, downregulation of NRF2 reduced the expression of stem cell markers in various types of cancer cells and provoked cellular differentiation in a glioma model [85,86,87]. Notably, NRF2 has been shown to bind to upstream regions of the genes for stem cell factors, NANOG HOMEOBOX and OCT4 [88].
Breast cancer CSCs show an increased expression of the NRF2 target gene, glutamate-cysteine ligase catalytic subunit (GCLC) [89]. GCLC is a rate-limiting enzyme of glutathione synthesis and, thus, adds to the CSCs’ antioxidative equipment.
Exposure to chronic and low levels of arsenic stress induces transformation of the human bronchial epithelial cells, BEAS-2B, to cancer stem-like cells. This dedifferentiation of BEAS-2B cells into CSC-like cells was accompanied by features of EMT and enhanced chemoresistance [90]. Exposure to arsenic also multiplied the skin carcinogenesis in Tg.AC mice, a mouse strain where the activation of the v-Ha-ras transgene increased the induction of 12-O-tetradecanoyl phorbol-13-acetate (TPA)-induced squamous cell carcinomas, along with increasing the number of CD13-positive CSC [91]. A Nrf2-dependent metabolic shift towards glycolysis very likely contributes to this effect of arsenic [92].
NRF2, via induction of its target genes, also induces anticancer drug resistance in CSCs. Increased expression of NRF2 and ABCB1 (MDR1) was associated with increased resistance to doxorubicin of CD44+CD133+ cells [93]. Likewise, increased expression of Sonic Hedgehog (SHH) and NRF2, correlated with induced CSC characteristics and chemoresistance in head and neck squamous cell carcinoma [94].
Recent work suggests that histamine N-methyltransferase (HNMT), which is regulated by ROS, is co-expressed with human epidermal growth factor receptor (HER2) [95]. The expression of both HER2 and HNMT is increased in breast and lung cancer [95,96]. HNMT directly interacts with HER2 to regulate NRF2 DNA binding activity and target gene expression, which is in part mediated by phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) [95]. In non-small-cell lung cancer (NSCLC) CSCs, expression levels of the HNMT-inhibiting miRs, miR3065/223, are low, leading to elevated expression/activity of NRF2. This contributes to enhanced chemoresistance to cisplatin [95].
NRF2 also plays an important role in macrophage polarization and, thus, macrophages are considered key players in cancer progression [75,97,98,99]. Infiltration of macrophages in and around tumors (tumor-associated macrophages, TAMs) can often be observed, and their presence is associated with poor prognosis in solid tumors [100,101]. It is considered proven that M2 macrophages support angiogenesis and neovascularization, remodeling of stroma and the tumor niche, and dissemination of tumor cells. All these processes contribute to tumor progression. Fast-growing solid tumors typically contain hypoxic areas due to insufficient oxygen supply. These areas are often located in the tumor center or in perinecrotic areas. Mechanistically, under hypoxic conditions, ROS-dependent induction of IL4 has been shown to contribute to the recruitment and M2 polarization of macrophages. TAMs preferentially localize in these hypoxic areas of a tumor [37]. Lactate, produced by tumor cells via anaerobic glycolysis, enhances macrophage ROS production and stimulates Nrf2 activation [102]. Furthermore, M2-derived vascular endothelial growth factor (VEGF) leads to the activation of NRF2 in neighboring tumor cells to support epithelial-to-mesenchymal transition (EMT) [102]. The presence of TAMs reduces the effectiveness of tumor therapy protocols, which applies to both chemotherapy and radiotherapy as well as angiogenesis inhibition [100,103,104]. Activation of NRF2 in cancer cells—via induction of target gene expression—promotes cancer progression, metastasis, and resistance to radio- or chemotherapy [98].
By applying the CSC model, sphere culture of HT-116 colon cancer cells, it could be demonstrated that the expression of the colon cancer CSC marker CD133 correlates with that of Nrf2. Silencing of CD133 reduced the expression of stem cell markers Kruppel-like factor 4 (KLF4) and ABCC2, but decreased the expression of NRF2 [105]. Mechanistically, this effect seems to be mediated via PI3K/GSK-3β. Vice versa, silencing of NRF2 impaired sphere formation and led to a decrease in CSC markers [105]. Furthermore, in the CD133high population isolated from HT-116 cells, it was shown that CSCs exhibit higher expression of NRF2, and this is associated with enhanced CSC-like properties such as the extent of sphere formation, colony formation, and resistance to anticancer therapy. The authors conclude that the CD133/NRF2 axis contributes to the development of a CSC-like phenotype [105]. High expression of the antioxidant gene peroxiredoxin-2 (PRDX2) has been attributed to CD133+CD44+ cells and is associated with colon cancer CSC stemness [106]. Depletion of PRDX2 substantially suppressed maintenance of stemness, together with migration, invasion, and metastasis. Notably, knockdown of PRDX2 increased ROS-levels in CD133+CD44+, making CSCs sensitive to oxidative stress and chemotherapy [106].
An association between CSC markers and Nrf2 has been detected in several studies. The CD44highCD24low CSC population exhibited higher expression of CSC makers and of Nrf2. Nrf2 levels could be further increased by addition to the cells of hyaluronic acid, a ligand for CD44 [107]. Accordingly, high expression levels of CD133 and CD44 go along with increased NRF2 and its target gene, efflux transporters ABCC1 [93]. In ovarian CSCs, Nrf2 levels were found to be associated with levels of ALDH [89].
6. Signaling Pathways Contributing in the Regulation of ROS Levels in CSC
Different signaling pathways support and enable the self-renewal of cancer stem cells. These include the Wingless-Int (Wnt), Notch, and Sonic Hedgehog (HH) pathways [2,108,109]. Notably, all these pathways to some extent contribute to CSC maintenance by regulating CSCs’ redox balance.
6.1. Wnt-Signaling Pathway
Hyperactivation of the Wnt/β-catenin signaling pathway has been identified as one of the most frequent events occurring in CSCs [110]. Activation of the pathway leads to stabilization and nuclear translocation of β-catenin and, eventually, transcriptional upregulation of target genes [111,112]. Notably, the Wnt/β-catenin pathway has been heavily implicated in liver CSCs [113,114]. In hepatic CSCs, glutamine synthase 1 (GLS1) is highly expressed and regulates antioxidant defense function by increasing GSH levels and decreasing reactive oxygen species (ROS) levels, which in turn protects cells from oxidative stress. Silencing GLS1 expression or inhibiting GLS1 activity dysregulated the redox homeostasis of cancer cells [115,116,117]. GLS1 expression is positively associated with the stemness phenotype in hepatocellular carcinoma (HCC), and targeting GLS1 inhibits CSC markers expression and stem-like properties in vitro and in vivo. Mechanistically, glutamine deprivation or GLS1 inhibition leads to an increased accumulation of ROS, which suppresses the translocation of β-catenin from the cytoplasm to the nucleus and consequently decreases the expression of stemness-related genes [118]. Similarly, glutamine has been shown to support the maintenance of stemness by promoting glutathione synthesis. Accordingly, deprivation of glutamine in the culture medium reduced the proportion of side population cells of the non-small lung cancer A549 cell line [119]. Deprivation of glutamine, similar to the addition of H2O2, increased the phosphorylation of β-catenin [111], the central player in the canonical Wnt-pathway [111,112], and, thus, downregulated SOX2 and other Wnt-regulated genes such as baculoviral IAP repeat containing 5 (BIRC5, survivin) and AXIN1 [118]. Inactivation of the adenomatous polyposis coli (APC) gene is frequently observed in colorectal carcinoma. It is considered the initiating event in colorectal tumorigenesis [120]. Inactive APC mutants exhibit constitutive activation of the β-catenin signaling [111]. APC is a negative regulator of the canonical Wnt-pathway, which provides a self-renewal signal for different types of stem cells [121]. Mechanistically, LGR5+/intestinal stem cell expansion after loss of APC was dependent on RAC1 and ROS production, as well as on efficient NF-κB (nuclear factor ‘kappa-light-chain-enhancer’) of activated B-cells (NF-κB)—signaling [122].
6.2. Sonic Hedgehog Signaling
The Sonic Hedgehog signaling pathway is crucially implicated in carcinogenesis and the tumor microenvironment. Aberrant HH signaling activation accelerates tumor growth and, as far as CSCs are involved, contributes to immune tolerance and drug resistance [125,126]. In gastrointestinal CSCs, various factors lead to the activation of HH signaling and, thereby, increase stemness. These factors include chemotherapy, CCN1 and NOTCH1 [127], SKI [128], and vasohibin 2 [129], which upregulate stem cell markers such as CD24, CD44, CD133, SOX2, OCT4, and NANOG as well as efflux transporters ABCC1 and ABCG2, and hypoxia-induced factor 1α (HIF1α) [130]. The latter is a key transcription factor, which is crucial for adapting cells and tissues to diminished oxygen availability [131] by stimulating oxygen supply via increasing angiogenesis and erythropoiesis and limiting oxygen consumption via shifting energy metabolism from oxidative phosphorylation to glycolysis [132]. This metabolic switch is considered to contribute to low ROS levels as they are required for maintaining stemness [133]. Activation of the HH pathway has been shown to stimulate transcription of the glycolytic gate keepers hexokinase 2 (HK2) and pyruvate kinase 2 (PKM2) [134]. Transcriptional activation of these genes is mediated by activation of GLI transcription factors, which are central to the canonical HH pathway.
Positive expression of GLI family zinc finger 1 (GLI1) has been recognized as a reliable indicator of poor prognosis in patients with highly aggressive gastric cancer [135]. The poor prognosis of patients with strong expression of GLI1 very likely is due to reactive oxygen species (ROS) generated by NADPH oxidase 4 (NOX4). Of note, increased production of ROS in hypoxia leads to GLI1 induction and EMT [136,137]. Interestingly, in human colon cancer biopsies, overexpression of the HH pathway components sonic hedgehog (SHH), patched 1 (PTCH), or GLI1, was found to be an indicator of poor prognosis [125]. Likewise, overexpression of the frizzled class receptor smoothend (SMO) has been suggested to be associated with poor prognosis in colorectal cancer [138,139].
Tumor-associated hypoxia contributes both to drug resistance and self-renewal. Inhibition of HH signaling was reported to suppress self-renewal and chemo-resistance of, e.g., pancreatic CSCs [140]. GLI target genes regulate not only proliferation, migration/invasion, and angiogenesis, but also stem cell regeneration (via target genes FST1, BMP4) [141]. EMT has been demonstrated to endow cells with stem cell-like properties. Accordingly, cananginone, a secondary metabolite of certain plants, was shown to inhibit HH signaling, EMT, and the acquisition of stem cell features in parallel. Inhibition of HH, and of GLI1 in particular, mediated this induction of EMT, largely by interfering with transcriptional activation of, e.g., SNAIL and VIM [142]. Likewise, engrailed (EN1), a homeo-box containing transcription factor that regulates embryonic development, has been shown to control tumorigenic capacity and resistance to radiation of glioma by regulating intracellular ROS levels [143]. Again, regulation of HH by altering GLI1 expression level has been identified as the underlying mechanism.
ROS-induced activation of NRF2 controls drug resistance, for example against sorafenib [144]. Mechanistically, the authors could demonstrate that NRF2 physically binds to the promoter of Sonic Hedgehog Signaling Molecule (SHH) [144], and that the administration of recombinant SHH abolished the effects resulting from NRF2 knockdown [144].
Of note, there are other cellular signaling pathways that interact with HH signaling. This holds true, e.g., for the Wnt pathway, transforming growth factor/bone morphogenic protein (TGF/BMP) pathway, and the epidermal growth factor (EGF) receptor pathway [145]. As far as the Wnt pathway is concerned, in the developing nervous system, upstream HH activation is absolutely required for the expression of T cell factors 3/4 (TCF3/4), major transcriptional mediators of the canonical Wnt signaling pathway [145].
6.3. CD13/APN in CSC
A positive correlation between multidrug resistance and CD13 expression has been established [146,147,148]. A set of surface markers representative of liver cancer stem cell (LCSC) subpopulations has been identified in in hepatocellular carcinoma (HCC). These include CD133, CD44, CD13, CD90, CD47, epithelial cell adhesion molecule (EpCAM), an antigen detected by “oval cell” antibody, OV-6, and the side population. CD13+ cells were mainly detected in the G0/G1 phase. They comprised dormant or slow-growing cancer cell populations, which are associated with chemoresistance due to ABC transporter expression and recurrence [149]. Similar to CD44, CD13 in combination with other surface markers, including CD133 or CD90, could effectively initiate tumor formation, as observed by increased HCC tumorigenesis [63]. CD13+CD133+ and CD13+CD90+ cells increase HCC tumor initiation and genotoxic chemoresistance to, e.g., doxorubicin (DXR) and fluorouracil (5′Fu) in parallel [150]. Accordingly, expression of hepatic stem cell markers (CD13, CD90, CD133), the proportion of side population, ability to induce sphere formation, and in vivo tumorigenesis were highest in selected HCC cells resistant to doxorubicin or 5′-fluorouracil [151]. Recent data suggest that CD13+ CSCs depend on aerobic metabolism of tyrosine rather than glucose as an energy source [143]. The acetyl-CoA formed by the metabolism of tyrosine facilitates acetylation/stabilization of transcription factor FOXD3, which in turn contributes to quiescence and chemoresistance [152].
CD13 (APN, EC3.4.11.2) is a Zn2+-dependent ectopeptidase that preferentially cleaves small neutral amino acids from the N-terminus of small peptides. APN/CD13 is associated with diverse physiological functions, including proliferation, differentiation, invasion, migration, and angiogenesis [153]. Increased expression or activity of APN/CD13 has been described for a variety of different tumors, with aggressive and rapidly growing tumors often being strongly CD13-positive (CD13+). For colon cancer, expression of CD13 has been shown to be associated with reduced disease-free and overall survival [154].
APN/CD13 is a neo-angiogenesis marker that is found on neo-angiogenetically active, but not on normal, endothelia [155]. This fact makes APN/CD13 an interesting and specific receptor for so-called “tumor-homing peptides” (NRG peptides), which bind to APN with high affinity, and thus represent a promising target for the inhibition of tumor-induced angiogenesis as well as for the tumor-selective administration of, e.g., cytotoxic substances [156,157,158]. In this way, APN/CD13 can, in principle, also be used for molecular imaging of tumor masses and (neo)angiogenesis in animal models and also in patients [159]. Pharmacological inhibition of the enzyme has been shown to reduce tumor growth and progression in a number of APN/CD13-positive tumors. The inhibition of angiogenesis (or “tube formation” in vitro) is only one obvious aspect, because therapeutic effects of APN inhibitors (ubenimex, actinonin) have also been described for acute or chronic myeloid leukemia (AML, CML) [160,161,162].
CD13 has been identified as a CSC-specific membrane marker (e.g., for hepatocellular carcinoma, cholangiocarcinoma) [163,164]. CD13 is preferentially expressed on cells of the side population (SP), which are characterized by extremely high chemoresistance and tumorigenicity in a transplantation model [165,166]. Functionally, CD13 is known to reduce the extent of DNA damage induced by ROS production after genotoxic stress and thereby protect CSC from apoptosis [165]. In hepatic cancer cells, the TGF-β-induced epithelial-to-mesenchymal transition (EMT) is associated with increased expression of CD13, which prevents a further increase in ROS levels and, thus, supports CSCs’ survival [64]. The pharmacological inhibition of APN/CD13 stimulates CSCs apoptosis [64] and has been shown to increase the antitumor effects of chemotherapeutic agents, e.g., 5-fluorouracil [148,167], which is why APN/CD13 inhibitors are considered cancer chemosensitizers [168]. A sensitization of the CSC to chemotherapeutic agents has also been achieved by the administration of APN/CD13 inhibitors in colorectal and hepatocellular carcinoma [150,169].
CD13 expression was found to be higher in metastatic HCC samples, and was associated with poor prognosis for patients after surgical resection [170]. CD13 stimulated HCC proliferation, invasion, and progression through the cell cycle, and caused resistance to sorafenib. Mechanistically, this was achieved by an activation of nuclear factor kappa B (NF-κB)-signaling, which was initiated by a direct interaction with and stabilization of histone deacetylase 5 (HDAC5), leading to lysin-specific demethylase 1 (LSD1) deacetylation/stabilization [170].
Available data suggest that CD13 induces multi-drug resistance (MDR) in tumor cells [148]. MDR is a phenomenon that makes certain cells resistant to the action of chemotherapy drugs, and is regarded as a characteristic stem cell feature. MDR often is achieved through increased expression of efflux transporters. In CSCs, the induction of ABC transporter expression via CD13 is caused by dysregulated HH signaling [171], where CD13 can act as a pseudoligand of the receptor PATCHED to sensitize the pathway, and to upregulate expression of ABCG2, ABCB1 (P-glycoprotein), ABCC2 (MRP2), and ABCC3 (MRP3) to increase drug resistance [172].
There is a negative correlation between CD13 expression and the cellular level of ROS [173]. In contrast, CD13 is positively correlated with expression of anti-apoptotic BCL2 and BCL-XL [174]. Mechanistically, ROS as second messenger and mitogen-activated protein kinases (MAPK) are involved in regulation of drug resistance. Of note, inhibition of APN/CD13 has been demonstrated to provoke induction/activation of mitogen-activated protein kinases (MAPK) Erk1/2 in the human T cell line KARPAS-299 [175].
7. Conclusions
The CSC phenotype is characterized by a high self-renewing capability and resistance to chemotherapy drugs, among other things. Its maintenance is crucially dependent on ensuring low intracellular ROS levels and induced expression of efflux transporters. Low ROS levels are largely due to the increased expression of anti-oxidative structures/mechanisms. The NRF2 plays an important role in this. Furthermore, metabolic balancing between anaerobic glycolysis and oxidative phosphorylation minimizes ROS production. Signaling pathways and molecules associated with the CSC phenotype, such as the Wnt- and HH pathways, and inhibition of APN/CD13 enzymatic activity regulate the maintenance of the CSC phenotype in several ways. Direct or indirect regulation of the CSC redox balance is crucially implicated in CSC maintenance.
Pharmacological targeting of the signaling pathways involved can represent a promising strategy for overcoming the limitations of current tumor therapy protocols. APN/CD13 facilitates the targeted inhibition of tumor angiogenesis and the sensitization of CSC for radiotherapy and chemotherapy. Likewise, HH (SMO and GLI) inhibitors are promising tools to address stemness-associated features of CSC. The contribution of ROS in these pathways requires further investigation.
Author Contributions
U.L. and C.W. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
U.L. receives funding from the Deutsche Forschungsgemeinschaft (DFG; LE900/13-1, LE90016-1).
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare that they have no competing interest.
References
- Boesch, M.; Zeimet, A.G.; Fiegl, H.; Wolf, B.; Huber, J.; Klocker, H.; Gastl, G.; Sopper, S.; Wolf, D. High prevalence of side population in human cancer cell lines. Oncoscience 2016, 3, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Fan, Z. Cancer stem cells and tumorigenesis. Biophys. Rep. 2018, 4, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct "side population" of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, A.; Brons, N.H.; Bjerkvig, R.; Niclou, S.P. Critical appraisal of the side population assay in stem cell and cancer stem cell research. Cell Stem Cell 2011, 8, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Sales-Pardo, I.; Avendano, A.; Martinez-Munoz, V.; Garcia-Escarp, M.; Celis, R.; Whittle, P.; Barquinero, J.; Domingo, J.C.; Marin, P.; Petriz, J. Flow cytometry of the Side Population: Tips & tricks. Cell Oncol. 2006, 28, 37–53. [Google Scholar]
- Afify, S.M.; Seno, M. Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation. Cancers 2019, 11, 345. [Google Scholar] [CrossRef]
- Garnier, D.; Milsom, C.; Magnus, N.; Meehan, B.; Weitz, J.; Yu, J.; Rak, J. Role of the tissue factor pathway in the biology of tumor initiating cells. Thromb. Res. 2010, 125 (Suppl. S2), S44–S50. [Google Scholar] [CrossRef]
- Unruh, D.; Horbinski, C. Beyond thrombosis: The impact of tissue factor signaling in cancer. J. Hematol. Oncol. 2020, 13, 93. [Google Scholar] [CrossRef]
- Tuy, K.; Rickenbacker, L.; Hjelmeland, A.B. Reactive oxygen species produced by altered tumor metabolism impacts cancer stem cell maintenance. Redox Biol. 2021, 44, 101953. [Google Scholar] [CrossRef]
- Aramini, B.; Masciale, V.; Arienti, C.; Dominici, M.; Stella, F.; Martinelli, G.; Fabbri, F. Cancer Stem Cells (CSCs), Circulating Tumor Cells (CTCs) and Their Interplay with Cancer Associated Fibroblasts (CAFs): A New World of Targets and Treatments. Cancers 2022, 14, 2408. [Google Scholar] [CrossRef]
- Mirantes, C.; Espinosa, I.; Ferrer, I.; Dolcet, X.; Prat, J.; Matias-Guiu, X. Epithelial-to-mesenchymal transition and stem cells in endometrial cancer. Hum. Pathol. 2013, 44, 1973–1981. [Google Scholar] [CrossRef] [PubMed]
- Mizrak, D.; Brittan, M.; Alison, M. CD133: Molecule of the moment. J. Pathol. 2008, 214, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Watanabe, M.; Nakano, K.; Uchimaru, K.; Horie, R. Differentiation of Hodgkin lymphoma cells by reactive oxygen species and regulation by heme oxygenase-1 through HIF-1α. Cancer Sci. 2021, 112, 2542–2555. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Sun, M.; Zhang, X.; Xu, Z.; Miyamoto, H.; Zheng, Y. Activation of Glucocorticoid Receptor Inhibits the Stem-Like Properties of Bladder Cancer via Inactivating the β-Catenin Pathway. Front Oncol. 2020, 10, 1332. [Google Scholar] [CrossRef]
- Foster, B.A.; Gangavarapu, K.J.; Mathew, G.; Azabdaftari, G.; Morrison, C.D.; Miller, A.; Huss, W.J. Human prostate side population cells demonstrate stem cell properties in recombination with urogenital sinus mesenchyme. PLoS ONE 2013, 8, e55062. [Google Scholar] [CrossRef]
- Huang, B.; Huang, Y.J.; Yao, Z.J.; Chen, X.; Guo, S.J.; Mao, X.P.; Wang, D.H.; Chen, J.X.; Qiu, S.P. Cancer stem cell-like side population cells in clear cell renal cell carcinoma cell line 769P. PLoS ONE 2013, 8, e68293. [Google Scholar] [CrossRef]
- Liu, T.; Xu, H.; Huang, M.; Ma, W.; Saxena, D.; Lustig, R.A.; Alonso-Basanta, M.; Zhang, Z.; O’Rourke, D.M.; Zhang, L.; et al. Circulating Glioma Cells Exhibit Stem Cell-like Properties. Cancer Res. 2018, 78, 6632–6642. [Google Scholar] [CrossRef]
- Ho, M.M.; Ng, A.V.; Lam, S.; Hung, J.Y. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007, 67, 4827–4833. [Google Scholar] [CrossRef]
- Platet, N.; Mayol, J.F.; Berger, F.; Herodin, F.; Wion, D. Fluctuation of the SP/non-SP phenotype in the C6 glioma cell line. FEBS Lett. 2007, 581, 1435–1440. [Google Scholar] [CrossRef]
- Xiong, B.; Ma, L.; Hu, X.; Zhang, C.; Cheng, Y. Characterization of side population cells isolated from the colon cancer cell line SW480. Int. J. Oncol. 2014, 45, 1175–1183. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, Y.; Qi, Y.; Liu, D.; Wu, K.; Wen, F.; Zhao, S. Side population cells separated from A549 lung cancer cell line possess cancer stem cell-like properties and inhibition of autophagy potentiates the cytotoxic effect of cisplatin. Oncol. Rep. 2015, 34, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.; Zaiden, N.; Chong, K.H.; See, S.J.; Wong, M.C.; Ang, B.T.; Tang, C. Characterization of a side population of astrocytoma cells in response to temozolomide. J. Neurosurg. 2008, 109, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinulescu, D.M.; Connolly, D.; Foster, R.; Dombkowski, D.; Preffer, F.; Maclaughlin, D.T.; Donahoe, P.K. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 11154–11159. [Google Scholar] [CrossRef]
- Wu, C.; Alman, B.A. Side population cells in human cancers. Cancer Lett. 2008, 268, 1–9. [Google Scholar] [CrossRef]
- Chiba, T.; Kita, K.; Zheng, Y.W.; Yokosuka, O.; Saisho, H.; Iwama, A.; Nakauchi, H.; Taniguchi, H. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology 2006, 44, 240–251. [Google Scholar] [CrossRef]
- Haraguchi, N.; Utsunomiya, T.; Inoue, H.; Tanaka, F.; Mimori, K.; Barnard, G.F.; Mori, M. Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cell. 2006, 24, 506–513. [Google Scholar] [CrossRef]
- Wu, C.; Wei, Q.; Utomo, V.; Nadesan, P.; Whetstone, H.; Kandel, R.; Wunder, J.S.; Alman, B.A. Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer Res. 2007, 67, 8216–8222. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Stingl, J.; Eirew, P.; Ricketson, I.; Shackleton, M.; Vaillant, F.; Choi, D.; Li, H.I.; Eaves, C.J. Purification and unique properties of mammary epithelial stem cells. Nature 2006, 439, 993–997. [Google Scholar] [CrossRef]
- Clement, V.; Marino, D.; Cudalbu, C.; Hamou, M.F.; Mlynarik, V.; de Tribolet, N.; Dietrich, P.Y.; Gruetter, R.; Hegi, M.E.; Radovanovic, I. Marker-independent identification of glioma-initiating cells. Nat. Method. 2010, 7, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor stem cells. Adv. Cancer Res. 2014, 122, 1–67. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.M.; Aykin-Burns, N.; Sim, J.E.; Walsh, S.A.; Higashikubo, R.; Buettner, G.R.; Venkataraman, S.; Mackey, M.A.; Flanagan, S.W.; Oberley, L.W.; et al. Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J. Biol. Chem. 2005, 280, 4254–4263. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A. Mitochondrial production of superoxide radical and hydrogen peroxide. Adv. Exp. Med. Biol. 1977, 78, 67–82. [Google Scholar]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it at-tenuates degenerative disease. Free Radic. Biol. Med. 2015, 88 Pt B, 108–146. [Google Scholar] [CrossRef]
- Weyemi, U.; Redon, C.E.; Parekh, P.R.; Dupuy, C.; Bonner, W.M. NADPH Oxidases NOXs and DUOXs as putative targets for cancer therapy. Anticancer Agent. Med. Chem. 2013, 13, 502–514. [Google Scholar]
- Mintz, J.; Vedenko, A.; Rosete, O.; Shah, K.; Goldstein, G.; Hare, J.M.; Ramasamy, R.; Arora, H. Current Advances of Nitric Oxide in Cancer and Anticancer Therapeutics. Vaccines 2021, 9, 94. [Google Scholar] [CrossRef]
- Switzer, C.H.; Glynn, S.A.; Cheng, R.Y.; Ridnour, L.A.; Green, J.E.; Ambs, S.; Wink, D.A. S-nitrosylation of EGFR and Src activates an oncogenic signaling network in human basal-like breast cancer. Mol. Cancer Res. 2012, 10, 1203–1215. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Gao, S.; Han, D.; Han, W.; Chen, S.; Patalano, S.; Macoska, J.A.; He, H.H.; Cai, C. TMPRSS2-ERG activates NO-cGMP signaling in prostate cancer cells. Oncogene 2019, 38, 4397–4411. [Google Scholar] [CrossRef] [PubMed]
- Belgorosky, D.; Girouard, J.; Langle, Y.V.; Hamelin-Morrissete, J.; Marino, L.; Aguero, E.I.; Malagrino, H.; Reyes-Moreno, C.; Eijan, A.M. Relevance of iNOS expression in tumor growth and maintenance of cancer stem cells in a bladder cancer model. J. Mol. Med. 2020, 98, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, P.; Lombardi, F.; Siragusa, G.; Dehcordi, S.R.; Luzzi, S.; Cimini, A.; Cifone, M.G.; Cinque, B. Involvement of NOS2 Activity on Human Glioma Cell Growth, Clonogenic Potential, and Neurosphere Generation. Int. J. Mol. Sci. 2018, 19, 2801. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, Y.; Tsung, A.; Huang, H.; Du, Q.; Yang, M.; Deng, M.; Xiong, S.; Wang, X.; Zhang, L.; et al. iNOS promotes CD24(+)CD133(+) liver cancer stem cell phenotype through a TACE/ADAM17-dependent Notch signaling pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E10127–E10136. [Google Scholar] [CrossRef]
- Penarando, J.; Lopez-Sanchez, L.M.; Mena, R.; Guil-Luna, S.; Conde, F.; Hernandez, V.; Toledano, M.; Gudino, V.; Raponi, M.; Billard, C.; et al. A role for endothelial nitric oxide synthase in intestinal stem cell proliferation and mesenchymal colorectal cancer. BMC Biol. 2018, 16, 3. [Google Scholar] [CrossRef]
- Maiuthed, A.; Bhummaphan, N.; Luanpitpong, S.; Mutirangura, A.; Aporntewan, C.; Meeprasert, A.; Rungrotmongkol, T.; Rojanasakul, Y.; Chanvorachote, P. Nitric oxide promotes cancer cell dedifferentiation by disrupting an Oct4:caveolin-1 complex: A new regulatory mechanism for cancer stem cell formation. J. Biol. Chem. 2018, 293, 13534–13552. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Chanvorachote, P. Nitric Oxide and Aggressive Behavior of Lung Cancer Cells. Anticancer Res. 2015, 35, 4585–4592. [Google Scholar]
- Gao, W.; Wang, Y.; Yu, S.; Wang, Z.; Ma, T.; Chan, A.M.; Chiu, P.K.; Ng, C.F.; Wu, D.; Chan, F.L. Endothelial nitric oxide synthase (eNOS)-NO signaling axis functions to promote the growth of prostate cancer stem-like cells. Stem Cell Res. Ther. 2022, 13, 188. [Google Scholar] [CrossRef]
- Xing, F.; Hu, Q.; Qin, Y.; Xu, J.; Zhang, B.; Yu, X.; Wang, W. The Relationship of Redox With Hallmarks of Cancer: The Importance of Homeostasis and Context. Front. Oncol. 2022, 12, 862743. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Shen, Q.; Claret, F.X. Novel roles of reactive oxygen species in the pathogenesis of acute myeloid leukemia. J. Leukoc. Biol. 2013, 94, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- Cadenas, E.; Wefers, H.; Müller, A.; Brigelius, R.; Sies, H. Active oxygen metabolites and their action in the hepatocyte. Studies on chemiluminescence responses and alkane production. Agent. Action. 1982, 11, 203–216. [Google Scholar]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2020, 8, 1865–1879. [Google Scholar] [CrossRef]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef]
- Ghezzi, P. Oxidoreduction of protein thiols in redox regulation. Biochem. Soc. Trans. 2005, 33 Pt 6, 1378–1381. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, Y.; Zheng, J.; Pan, J. Reactive oxygen species in cancer stem cells. Antioxid. Redox Signal. 2012, 16, 1215–1228. [Google Scholar] [CrossRef]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Ohkuma, M.; Kim, H.M.; Akita, H.; Takiuchi, D.; Hatano, H.; Nagano, H.; et al. CD13 is a therapeutic target in human liver cancer stem cells. J. Clin. Investig. 2010, 120, 3326–3339. [Google Scholar] [CrossRef]
- Kim, H.M.; Haraguchi, N.; Ishii, H.; Ohkuma, M.; Okano, M.; Mimori, K.; Eguchi, H.; Yamamoto, H.; Nagano, H.; Sekimoto, M.; et al. Increased CD13 expression reduces reactive oxygen species, promoting survival of liver cancer stem cells via an epithelial-mesenchymal transition-like phenomenon. Ann. Surg. Oncol. 2012, 19 (Suppl. S3), S539–S548. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, K.R.; Rachakonda, G.; Freeman, M.L. Cysteine-based regulation of the CUL3 adaptor protein Keap1. Toxicol. Appl. Pharmacol. 2010, 244, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Gene. Cell. 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: An insight into cullin-ring ubiquitin ligases. Antioxid Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Choi, B.H.; Kim, J.M.; Kwak, M.K. The multifaceted role of NRF2 in cancer progression and cancer stem cells maintenance. Arch. Pharm. Res. 2021, 44, 263–280. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Suzuki, M.; Otsuki, A.; Keleku-Lukwete, N.; Yamamoto, M. Overview of redox-regulation by Keap1-Nrf2 system in toxicology and cancer. Curr. Opinion. Toxicol. 2016, 1, 29–36. [Google Scholar] [CrossRef]
- Kwak, M.K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145. [Google Scholar] [CrossRef]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637–651. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Mostafazadeh, M.; Kahroba, H.; Haiaty, S.; TazeKand, A.P.; Samadi, N.; Rahbarghazi, R.; Nouri, M. In vitro exosomal transfer of Nrf2 led to the oxaliplatin resistance in human colorectal cancer LS174T cells. Cell Biochem. Funct. 2022, 40, 391–402. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Kang, Q.; Pan, C.; Zhang, T.; Feng, C.; Chen, L.; Wei, S.; Wang, J. Nrf2 Overexpression Decreases Vincristine Chemotherapy Sensitivity Through the PI3K-AKT Pathway in Adult B-Cell Acute Lymphoblastic Leukemia. Front. Oncol. 2022, 12, 876556. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trend. Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef]
- Guo, D.; Wu, B.; Yan, J.; Li, X.; Sun, H.; Zhou, D. A possible gene silencing mechanism: Hypermethylation of the Keap1 promoter abrogates binding of the transcription factor Sp1 in lung cancer cells. Biochem. Biophys. Res. Commun. 2012, 428, 80–85. [Google Scholar] [CrossRef]
- Hanada, N.; Takahata, T.; Zhou, Q.; Ye, X.; Sun, R.; Itoh, J.; Ishiguro, A.; Kijima, H.; Mimura, J.; Itoh, K.; et al. Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 2012, 12, 66. [Google Scholar] [CrossRef]
- Sparaneo, A.; Fabrizio, F.P.; la Torre, A.; Graziano, P.; Di Maio, M.; Fontana, A.; Bisceglia, M.; Rossi, A.; Pizzolitto, S.; De Maglio, G.; et al. Effects of KEAP1 Silencing on the Regulation of NRF2 Activity in Neuroendocrine Lung Tumors. Int. J. Mol. Sci. 2019, 20, 2531. [Google Scholar] [CrossRef]
- Wang, D.; Ma, Y.; Yang, X.; Xu, X.; Zhao, Y.; Zhu, Z.; Wang, X.; Deng, H.; Li, C.; Gao, F.; et al. Hypermethylation of the Keap1 gene inactivates its function, promotes Nrf2 nuclear accumulation, and is involved in arsenite-induced human keratinocyte transformation. Free Radic. Biol. Med. 2015, 89, 209–219. [Google Scholar] [CrossRef]
- Wang, R.; An, J.; Ji, F.; Jiao, H.; Sun, H.; Zhou, D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem. Biophys. Res. Commun. 2008, 373, 151–154. [Google Scholar] [CrossRef]
- Kahroba, H.; Shirmohamadi, M.; Hejazi, M.S.; Samadi, N. The Role of Nrf2 signaling in cancer stem cells: From stemness and self-renewal to tumorigenesis and chemoresistance. Life Sci. 2019, 239, 116986. [Google Scholar] [CrossRef]
- Gao, L.; Morine, Y.; Yamada, S.; Saito, Y.; Ikemoto, T.; Tokuda, K.; Takasu, C.; Miyazaki, K.; Shimada, M. Nrf2 signaling promotes cancer stemness, migration, and expression of ABC transporter genes in sorafenib-resistant hepatocellular carcinoma cells. PLoS ONE 2021, 16, e0256755. [Google Scholar] [CrossRef]
- Kim, D.; Choi, B.H.; Ryoo, I.G.; Kwak, M.K. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: Inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 2018, 9, 896. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, H.; Fan, Y.; Hu, Y.; Ji, X.; Sun, Q.; Liu, H. Knockdown of nuclear factor erythroid 2-related factor 2 by lentivirus induces differentiation of glioma stem-like cells. Oncol. Rep. 2014, 32, 1170–1178. [Google Scholar] [CrossRef]
- Jang, J.; Wang, Y.; Lalli, M.A.; Guzman, E.; Godshalk, S.E.; Zhou, H.; Kosik, K.S. Primary Cilium-Autophagy-Nrf2 (PAN) Axis Activation Commits Human Embryonic Stem Cells to a Neuroectoderm Fate. Cell 2016, 165, 410–420. [Google Scholar] [CrossRef]
- Kim, D.H.; Jang, J.H.; Kwon, O.S.; Cha, H.J.; Youn, H.J.; Chun, K.S.; Surh, Y.J. Nuclear Factor Erythroid-Derived 2-Like 2-Induced Reductive Stress Favors Self-Renewal of Breast Cancer Stem-Like Cells via the FoxO3a-Bmi-1 Axis. Antioxid. Redox Signal. 2020, 32, 1313–1329. [Google Scholar] [CrossRef]
- Chang, Q.; Bi, Z.; Fu, Y.; Rice, M.K.A.; Zhang, Q.; Wadgaonkar, P.; Almutairy, B.; Zhang, W.; Lu, Y.; Xu, L.; et al. Characterization of Arsenic-Induced Cancer Stem-Like Cells. Method. Mol. Biol. 2020, 2117, 293–303. [Google Scholar]
- Waalkes, M.P.; Liu, J.; Germolec, D.R.; Trempus, C.S.; Cannon, R.E.; Tokar, E.J.; Tennant, R.W.; Ward, J.M.; Diwan, B.A. Arsenic exposure in utero exacerbates skin cancer response in adulthood with contemporaneous distortion of tumor stem cell dynamics. Cancer Res. 2008, 68, 8278–8285. [Google Scholar] [CrossRef]
- Bi, Z.; Zhang, Q.; Fu, Y.; Wadgaonkar, P.; Zhang, W.; Almutairy, B.; Xu, L.; Rice, M.; Qiu, Y.; Thakur, C.; et al. Nrf2 and HIF1alpha converge to arsenic-induced metabolic reprogramming and the formation of the cancer stem-like cells. Theranostics 2020, 10, 4134–4149. [Google Scholar] [CrossRef]
- Goto, S.; Kawabata, T.; Li, T.S. Enhanced Expression of ABCB1 and Nrf2 in CD133-Positive Cancer Stem Cells Associates with Doxorubicin Resistance. Stem Cells Int. 2020, 2020, 8868849. [Google Scholar] [CrossRef]
- Noman, A.S.M.; Parag, R.R.; Rashid, M.I.; Rahman, M.Z.; Chowdhury, A.A.; Sultana, A.; Jerin, C.; Siddiqua, A.; Rahman, L.; Shirin, A.; et al. Widespread expression of Sonic hedgehog (Shh) and Nrf2 in patients treated with cisplatin predicts outcome in resected tumors and are potential therapeutic targets for HPV-negative head and neck cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920911229. [Google Scholar] [CrossRef]
- Kuo, K.T.; Lin, C.H.; Wang, C.H.; Pikatan, N.W.; Yadav, V.K.; Fong, I.H.; Yeh, C.T.; Lee, W.H.; Huang, W.C. HNMT Upregulation Induces Cancer Stem Cell Formation and Confers Protection against Oxidative Stress through Interaction with HER2 in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2022, 23, 1663. [Google Scholar] [CrossRef]
- von Mach-Szczypinski, J.; Stanosz, S.; Sieja, K.; Stanosz, M. Histamine and its metabolizing enzymes in tissues of primary ductal breast cancer. Eur. J. Gynaecol. Oncol. 2009, 30, 509–511. [Google Scholar]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Lendeckel, U.; Venz, S.; Wolke, C. Macrophages: Shapes and functions. ChemTexts 2022, 8, 12. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal. 2018, 16, 54. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef]
- Park, J.; Kim, S.K.; Hallis, S.P.; Choi, B.H.; Kwak, M.K. Role of CD133/NRF2 Axis in the Development of Colon Cancer Stem Cell-Like Properties. Front. Oncol. 2021, 11, 808300. [Google Scholar] [CrossRef]
- Peng, L.; Xiong, Y.; Wang, R.; Xiang, L.; Zhou, H.; Fu, Z. The critical role of peroxiredoxin-2 in colon cancer stem cells. Aging 2021, 13, 11170–11187. [Google Scholar] [CrossRef]
- Ryoo, I.G.; Choi, B.H.; Ku, S.K.; Kwak, M.K. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: Implications for cancer stem cell resistance. Redox Biol. 2018, 17, 246–258. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Wang, R.; Sun, Q.; Wang, P.; Liu, M.; Xiong, S.; Luo, J.; Huang, H.; Du, Q.; Geller, D.A.; Cheng, B. Notch and Wnt/beta-catenin signaling pathway play important roles in activating liver cancer stem cells. Oncotarget 2016, 7, 5754–5768. [Google Scholar] [CrossRef]
- Fodde, R.; Brabletz, T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 2007, 19, 150–158. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Wolke, C.; Antileo, E.; Lendeckel, U. WNT signaling in atrial fibrillation. Exp. Biol. Med. 2021, 246, 1112–1120. [Google Scholar] [CrossRef]
- Yamashita, T.; Budhu, A.; Forgues, M.; Wang, X.W. Activation of hepatic stem cell marker EpCAM by Wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007, 67, 10831–10839. [Google Scholar] [CrossRef]
- Yang, W.; Yan, H.X.; Chen, L.; Liu, Q.; He, Y.Q.; Yu, L.X.; Zhang, S.H.; Huang, D.D.; Tang, L.; Kong, X.N.; et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008, 68, 4287–4295. [Google Scholar] [CrossRef]
- Martin-Rufian, M.; Nascimento-Gomes, R.; Higuero, A.; Crisma, A.R.; Campos-Sandoval, J.A.; Gomez-Garcia, M.C.; Cardona, C.; Cheng, T.; Lobo, C.; Segura, J.A.; et al. Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. J. Mol. Med. 2014, 92, 277–290. [Google Scholar] [CrossRef]
- Ulanet, D.B.; Couto, K.; Jha, A.; Choe, S.; Wang, A.; Woo, H.K.; Steadman, M.; DeLaBarre, B.; Gross, S.; Driggers, E.; et al. Mesenchymal phenotype predisposes lung cancer cells to impaired proliferation and redox stress in response to glutaminase inhibition. PLoS ONE 2014, 9, e115144. [Google Scholar] [CrossRef]
- Xiang, Y.; Stine, Z.E.; Xia, J.; Lu, Y.; O’Connor, R.S.; Altman, B.J.; Hsieh, A.L.; Gouw, A.M.; Thomas, A.G.; Gao, P.; et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Investig. 2015, 125, 2293–2306. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Cao, Y.; Meng, G.; Qian, L.; Xu, T.; Yan, C.; Luo, O.; Wang, S.; Wei, J.; Ding, Y.; et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine 2019, 39, 239–254. [Google Scholar] [CrossRef]
- Liao, J.; Liu, P.P.; Hou, G.; Shao, J.; Yang, J.; Liu, K.; Lu, W.; Wen, S.; Hu, Y.; Huang, P. Regulation of stem-like cancer cells by glutamine through beta-catenin pathway mediated by redox signaling. Mol. Cancer 2017, 16, 51. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Pelicci, P.G.; Dalton, P.; Giorgio, M. The other face of ROS: A driver of stem cell expansion in colorectal cancer. Cell Stem Cell 2013, 12, 635–636. [Google Scholar] [CrossRef]
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.L.; Athineos, D.; et al. ROS production and NF-kappaB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 2013, 12, 761–773. [Google Scholar] [CrossRef]
- Suh, H.N.; Kim, M.J.; Jung, Y.S.; Lien, E.M.; Jun, S.; Park, J.I. Quiescence Exit of Tert(+) Stem Cells by Wnt/beta-Catenin Is Indispensable for Intestinal Regeneration. Cell Rep. 2017, 21, 2571–2584. [Google Scholar] [CrossRef]
- Povinelli, B.J.; Nemeth, M.J. Wnt5a regulates hematopoietic stem cell proliferation and repopulation through the Ryk receptor. Stem Cell. 2014, 32, 105–115. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, J.; Zeng, X.; Nie, M.; Luan, J.; Wang, Y.; Ju, D.; Yin, K. Hedgehog signaling in gastrointestinal carcinogenesis and the gastrointestinal tumor microenvironment. Acta Pharm. Sin. B 2021, 11, 609–620. [Google Scholar] [CrossRef]
- Yoon, C.; Park, D.J.; Schmidt, B.; Thomas, N.J.; Lee, H.J.; Kim, T.S.; Janjigian, Y.Y.; Cohen, D.J.; Yoon, S.S. CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clin. Cancer Res. 2014, 20, 3974–3988. [Google Scholar] [CrossRef]
- Haque, I.; De, A.; Majumder, M.; Mehta, S.; McGregor, D.; Banerjee, S.K.; Van Veldhuizen, P.; Banerjee, S. The matricellular protein CCN1/Cyr61 is a critical regulator of Sonic Hedgehog in pancreatic carcinogenesis. J. Biol. Chem. 2012, 287, 38569–38579. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Chen, X.; Gao, S.; Zhang, C.; Qu, C.; Wang, P.; Liu, L. Ski modulate the characteristics of pancreatic cancer stem cells via regulating sonic hedgehog signaling pathway. Tumor Biol. 2016, 37, 16115–16125. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, X.; Zhao, X.; Qin, L.; Shen, Y.; Dou, H.; Sun, J.; Wang, T.; Yang, D.Q. Vasohibin 2 promotes malignant behaviors of pancreatic cancer cells by inducing epithelial-mesenchymal transition via Hedgehog signaling pathway. Cancer Med. 2018, 7, 5567–5576. [Google Scholar] [CrossRef]
- Liang, Y.; Yang, L.; Xie, J. The Role of the Hedgehog Pathway in Chemoresistance of Gastrointestinal Cancers. Cells 2021, 10, 2030. [Google Scholar] [CrossRef]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Mimeault, M.; Batra, S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J. Cell Mol. Med. 2013, 17, 30–54. [Google Scholar] [CrossRef]
- Di Magno, L.; Manzi, D.; D’Amico, D.; Coni, S.; Macone, A.; Infante, P.; Di Marcotullio, L.; De Smaele, E.; Ferretti, E.; Screpanti, I.; et al. Druggable glycolytic requirement for Hedgehog-dependent neuronal and medul-loblastoma growth. Cell Cycle 2014, 13, 3404–3413. [Google Scholar] [CrossRef]
- Lu, L.; Wu, M.; Zhao, F.; Fu, W.; Li, W.; Li, X.; Liu, T. Prognostic and clinicopathological value of Gli-1 expression in gastric cancer: A meta-analysis. Oncotarget 2016, 7, 69087–69096. [Google Scholar] [CrossRef]
- Tang, C.-T.; Lin, X.-L.; Wu, S.; Liang, Q.; Yang, L.; Gao, Y.-J.; Ge, Z.-Z. NOX4-driven ROS formation regulates proliferation and apoptosis of gastric cancer cells through the GLI1 pathway. Cell. Signal. 2018, 46, 52–63. [Google Scholar] [CrossRef]
- Liu, Z.; Tu, K.; Wang, Y.; Yao, B.; Li, Q.; Wang, L.; Dou, C.; Liu, Q.; Zheng, X. Hypoxia Accelerates Aggressiveness of Hepa-tocellular Carcinoma Cells Involving Oxidative Stress, Epithelial-Mesenchymal Transition and Non-Canonical Hedgehog Signaling. Cell. Physiol. Biochem. 2017, 44, 1856–1868. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Liao, X.; Lochhead, P.; Morikawa, T.; Yamauchi, M.; Nishihara, R.; Inamura, K.; Kim, S.A.; Mima, K.; Sukawa, Y.; et al. SMO expression in colorectal cancer: Associations with clinical, pathological, and molecular features. Ann. Surg. Oncol. 2014, 21, 4164–4173. [Google Scholar] [CrossRef]
- Xu, M.; Li, X.; Liu, T.; Leng, A.; Zhang, G. Prognostic value of hedgehog signaling pathway in patients with colon cancer. Med. Oncol. 2011, 29, 1010–1016. [Google Scholar] [CrossRef]
- Huang, F.-T.; Zhuan-Sun, Y.-X.; Zhuang, Y.-Y.; Wei, S.-L.; Tang, J.; Chen, W.-B.; Zhang, S.-N. Zhang, Inhibition of hedgehog signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. Int. J. Oncol. 2012, 41, 1707–1714. [Google Scholar] [CrossRef]
- Lee, D.-H.; Lee, S.-Y.; Oh, S.C. Hedgehog signaling pathway as a potential target in the treatment of advanced gastric cancer. Tumor Biol. 2017, 39, 1–10. [Google Scholar] [CrossRef]
- Bose, C.; Das, U.; Kuilya, T.K.; Mondal, J.; Bhadra, J.; Banerjee, P.; Goswami, R.K.; Sinha, S. Cananginone Abrogates EMT in Breast Cancer Cells through Hedgehog Signaling. Chem. Biodivers. 2022, 19, e202100823. [Google Scholar] [CrossRef]
- Chang, J.; Guo, C.; Li, J.; Liang, Z.; Wang, Y.; Yu, A.; Liu, R.; Guo, Y.; Chen, J.; Huang, S. EN1 Regulates Cell Growth and Pro-liferation in Human Glioma Cells via Hedgehog Signaling. Int. J. Mol. Sci. 2022, 23, 1123. [Google Scholar] [CrossRef] [PubMed]
- Leung, H.W.; Lau, E.Y.T.; Leung, C.O.N.; Lei, M.M.L.; Mok, E.H.K.; Ma, V.W.S.; Cho, W.C.S.; Ng, I.O.L.; Yun, J.P.; Cai, S.H.; et al. NRF2/SHH signaling cascade promotes tumor-initiating cell lineage and drug resistance in hepa-tocellular carcinoma. Cancer Lett. 2020, 476, 856. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Medina, R.; Le Dreau, G.; Ros, M.; Martí, E. Hedgehog activation is required upstream of Wnt signalling to control neural progenitor proliferation. Development 2009, 136, 3301–3309. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jing, F.J.; Qu, H.J.; Xu, W.; Han, B.; Xing, X.M.; Ji, H.Y.; Jing, F.B. Ubenimex Reverses MDR in Gastric Cancer Cells by Activating Caspase-3-Mediated Apoptosis and Suppressing the Expression of Membrane Transport Proteins. Biomed Res. Int. 2019, 2019, 4390839. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jing, F.J.; Xu, W.; Li, X.; Li, X.; Sun, J.L.; Xing, X.M.; Zhou, C.K.; Jing, F.B. Ubenimex induces autophagy inhibition and EMT suppression to overcome cisplatin resistance in GC cells by perturbing the CD13/EMP3/PI3K/AKT/NF-kappaB axis. Aging 2019, 12, 80–105. [Google Scholar] [CrossRef]
- Guo, Q.; Sui, Z.G.; Xu, W.; Quan, X.H.; Sun, J.L.; Li, X.; Ji, H.Y.; Jing, F.B. Ubenimex suppresses Pim-3 kinase expression by targeting CD13 to reverse MDR in HCC cells. Oncotarget 2017, 8, 72652–72665. [Google Scholar] [CrossRef]
- Liu, Y.C.; Yeh, C.T.; Lin, K.H. Cancer Stem Cell Functions in Hepatocellular Carcinoma and Comprehensive Therapeutic Strategies. Cells 2020, 9, 1331. [Google Scholar] [CrossRef]
- Yamashita, M.; Wada, H.; Eguchi, H.; Ogawa, H.; Yamada, D.; Noda, T.; Asaoka, T.; Kawamoto, K.; Gotoh, K.; Umeshita, K.; et al. A CD13 inhibitor, ubenimex, synergistically enhances the effects of anticancer drugs in hepatocellular carcinoma. Int. J. Oncol. 2016, 49, 89–98. [Google Scholar] [CrossRef]
- Vu, N.B.; Nguyen, T.T.; Tran, L.C.; Do, C.D.; Nguyen, B.H.; Phan, N.K.; Pham, P.V. Doxorubicin and 5-fluorouracil resistant hepatic cancer cells demonstrate stem-like properties. Cytotechnology 2013, 65, 491–503. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, L.; Chen, J.; Li, C.; Sun, H.; Wang, J.; Xiao, H. Activation of Tyrosine Metabolism in CD13+ Cancer Stem Cells Drives Relapse in Hepatocellular Carcinoma. Cancer Res. Treat. 2020, 52, 604–621. [Google Scholar] [CrossRef]
- Lu, C.; Amin, M.A.; Fox, D.A. CD13/Aminopeptidase N Is a Potential Therapeutic Target for Inflammatory Disorders. J. Immunol. 2020, 204, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Hashida, H.; Takabayashi, A.; Kanai, M.; Adachi, M.; Kondo, K.; Kohno, N.; Yamaoka, Y.; Miyake, M. Aminopeptidase N is involved in cell motility and angiogenesis: Its clinical significance in human colon cancer. Gastroenterology 2002, 122, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, S.V.; Lahdenranta, J.; Giordano, R.; Arap, W.; Pasqualini, R.; Shapiro, L.H. CD13/APN is activated by angiogenic signals and is essential for capillary tube formation. Blood 2001, 97, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.X.; Zhang, H.L.; Xu, W.F.; Qu, X.J. 13F-1, a novel 5-fluorouracil prodrug containing an Asn-Gly-Arg (NO2) COOCH3 tripeptide, inhibits human colonic carcinoma growth by targeting Aminopeptidase N (APN/CD13). Eur. J. Pharmacol. 2014, 734, 50–59. [Google Scholar] [CrossRef]
- Pasqualini, R.; Koivunen, E.; Kain, R.; Lahdenranta, J.; Sakamoto, M.; Stryhn, A.; Ashmun, R.A.; Shapiro, L.H.; Arap, W.; Ruoslahti, E. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000, 60, 722–727. [Google Scholar]
- Seidi, K.; Jahanban-Esfahlan, R.; Monhemi, H.; Zare, P.; Minofar, B.; Daei Farshchi Adli, A.; Farajzadeh, D.; Behzadi, R.; Mesgari Abbasi, M.; Neubauer, H.A.; et al. NGR (Asn-Gly-Arg)-targeted delivery of coagulase to tumor vasculature arrests cancer cell growth. Oncogene 2018, 37, 3967–3980. [Google Scholar] [CrossRef]
- Meng, Y.; Zhang, Z.; Liu, K.; Ye, L.; Liang, Y.; Gu, W. Aminopeptidase N (CD13) targeted MR and NIRF dual-modal imaging of ovarian tumor xenograft. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 93, 968–974. [Google Scholar] [CrossRef]
- Kanamori, H.; Takasaki, H.; Takabayashi, M.; Yamaji, S.; Koharazawa, H.; Fujimaki, K.; Taguchi, J.; Ishigatsubo, Y. Long-term cytogenetic remission with ubenimex monotherapy in a case of chronic myeloid leukemia. Anticancer Drug. 2004, 15, 729–731. [Google Scholar] [CrossRef]
- Fujii, H.; Yosizawa, K.; Maruyama, S.; Abe, F. Growth inhibitory effects of ubenimex on leukemic cell lines resistant to chemotherapeutic agents. Jpn. J. Antibiot. 1996, 49, 1109–1115. [Google Scholar]
- Usuka, Y.; Saito, Y. Bestatin treatment of myelodysplastic syndromes and chronic myelogenous leukemia. Biomed. Pharmacother. 1991, 45, 87–93. [Google Scholar] [CrossRef]
- Cardinale, V.; Renzi, A.; Carpino, G.; Torrice, A.; Bragazzi, M.C.; Giuliante, F.; DeRose, A.M.; Fraveto, A.; Onori, P.; Napoletano, C.; et al. Profiles of cancer stem cell subpopulations in cholangiocarcinomas. Am. J. Pathol. 2015, 185, 1724–1739. [Google Scholar] [CrossRef] [PubMed]
- Castelli, G.; Pelosi, E.; Testa, U. Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Cancers 2017, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, N.; Inoue, H.; Tanaka, F.; Mimori, K.; Utsunomiya, T.; Sasaki, A.; Mori, M. Cancer stem cells in human gastrointestinal cancers. Hum. Cell 2006, 19, 24–29. [Google Scholar] [CrossRef]
- Mu, X.; Espanol-Suner, R.; Mederacke, I.; Affo, S.; Manco, R.; Sempoux, C.; Lemaigre, F.P.; Adili, A.; Yuan, D.; Weber, A.; et al. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J. Clin. Investig. 2015, 125, 3891–3903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fang, C.; Qu, M.; Wu, H.; Wang, X.; Zhang, H.; Ma, H.; Zhang, Z.; Huang, Y.; Shi, L.; et al. CD13 Inhibition Enhances Cytotoxic Effect of Chemotherapy Agents. Front. Pharmacol. 2018, 9, 1042. [Google Scholar] [CrossRef]
- Xing, X.; Li, F.; Hu, Y.; Zhang, L.; Hui, Q.; Qin, H.; Jiang, Q.; Jiang, W.; Fang, C.; Zhang, L. Discovery of Novel Tetrahydro-beta-carboline Containing Aminopeptidase N Inhibitors as Cancer Chemosensitizers. Front. Oncol. 2022, 12, 894842. [Google Scholar] [CrossRef]
- Sun, Z.P.; Zhang, J.; Shi, L.H.; Zhang, X.R.; Duan, Y.; Xu, W.F.; Dai, G.; Wang, X.J. Aminopeptidase N inhibitor 4cc synergizes antitumor effects of 5-fluorouracil on human liver cancer cells through ROS-dependent CD13 inhibition. Biomed. Pharmacother. 2015, 76, 65–72. [Google Scholar] [CrossRef]
- Hu, B.; Xu, Y.; Li, Y.C.; Huang, J.F.; Cheng, J.W.; Guo, W.; Yin, Y.; Gao, Y.; Wang, P.X.; Wu, S.Y.; et al. CD13 promotes hepatocellular carcinogenesis and sorafenib resistance by activating HDAC5-LSD1-NF-kappaB oncogenic signaling. Clin. Transl. Med. 2020, 10, e233. [Google Scholar] [CrossRef]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Laukkanen, M.O.; Castellone, M.D. Hijacking the Hedgehog Pathway in Cancer Therapy. Anticancer Agent. Med. Chem. 2016, 16, 309–317. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R., Jr.; Yang, D.H.; Chen, Z.S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updat. 2018, 41, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Kesavardhana, S.; Kanneganti, T.D. Stressed-out ROS take a silent death route. Nat. Immunol. 2018, 19, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Lendeckel, U.; Kahne, T.; Arndt, M.; Frank, K.; Ansorge, S. Inhibition of alanyl aminopeptidase induces MAP-kinase p42/ERK2 in the human T cell line KARPAS-299. Biochem. Biophys. Res. Commun. 1998, 252, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.; Ishii, H.; Haraguchi, N.; Kano, Y.; Fukusumi, T.; Ohta, K.; Ozaki, M.; Sakai, D.; Satoh, T.; Nagano, H.; et al. Genotoxic therapy stimulates error-prone DNA repair in dormant hepatocellular cancer stem cells. Exp. Ther. Med. 2012, 3, 959–962. [Google Scholar] [CrossRef]
- Haraguchi, N.; Ishii, H.; Nagano, H.; Doki, Y.; Mori, M. The future prospects and subject of the liver cancer stem cells study for the clinical application. Gastroenterology 2011, 140, 1355. [Google Scholar] [CrossRef]
Figure 1.
Pathways contributing to the maintenance of the cancer stem cell (CSC) phenotype. Keeping intracellular ROS levels low is required for a stem cell to maintain quiescence and self-renewal properties. This is achieved to a large extent by expression of Nrf2-induced target genes. Thereby, antioxidative defense molecules and detoxifying enzymes including thioredoxin, peroxiredoxin, glutathione-S-transferases, heme oxygenase I are provided to keep the levels of reactive oxygen species derived from, e.g., NADH oxidases (NOX), the respiratory chain, infiltrating/surrounding immune cells, or chemotherapeutic drugs low. Nrf2 and pathways initiated by the CSC surface receptors and markers to minimize ROS production in response to chemotherapeutics like Sonic Hedgehog, Wnt family members, CD13, CD44 or CD133 add to the strong induction of drug efflux transporters. In addition, pathways that contribute to the CSC phenotype act via preventing apoptosis, promoting angiogenesis and expression of stem cell transcription factors such as NANOG homeobox (NANOG HOMEOBOX) and SRY-box transcription factor 2 (SOX2). Note that the canonical β-catenin-dependent Wnt pathway, and GSK-3β in particular, is a downstream mediator shared by many of the CSC markers and CSC pathways.
Figure 1.
Pathways contributing to the maintenance of the cancer stem cell (CSC) phenotype. Keeping intracellular ROS levels low is required for a stem cell to maintain quiescence and self-renewal properties. This is achieved to a large extent by expression of Nrf2-induced target genes. Thereby, antioxidative defense molecules and detoxifying enzymes including thioredoxin, peroxiredoxin, glutathione-S-transferases, heme oxygenase I are provided to keep the levels of reactive oxygen species derived from, e.g., NADH oxidases (NOX), the respiratory chain, infiltrating/surrounding immune cells, or chemotherapeutic drugs low. Nrf2 and pathways initiated by the CSC surface receptors and markers to minimize ROS production in response to chemotherapeutics like Sonic Hedgehog, Wnt family members, CD13, CD44 or CD133 add to the strong induction of drug efflux transporters. In addition, pathways that contribute to the CSC phenotype act via preventing apoptosis, promoting angiogenesis and expression of stem cell transcription factors such as NANOG homeobox (NANOG HOMEOBOX) and SRY-box transcription factor 2 (SOX2). Note that the canonical β-catenin-dependent Wnt pathway, and GSK-3β in particular, is a downstream mediator shared by many of the CSC markers and CSC pathways.

Figure 2.
Nrf2 signaling pathway activation by ROS and induction of target genes. Under physiological conditions, NRF2 bound to KEAP1 is ubiquitylated and degraded in the proteasome. In response to ROS and various other stress factors, KEAP1 is released from NRF2, leading to its stabilization and translocation into the nucleus. Target gene expression is induced upon binding of NRF2 to AREs (antioxidant response element) in the promoters of target genes. Target genes comprise genes coding for the cell’s antioxidative equipment, efflux pumps enabling drug resistance, stemness-regulating transcription factors, as well as SHH (modified from [71,72]).
Figure 2.
Nrf2 signaling pathway activation by ROS and induction of target genes. Under physiological conditions, NRF2 bound to KEAP1 is ubiquitylated and degraded in the proteasome. In response to ROS and various other stress factors, KEAP1 is released from NRF2, leading to its stabilization and translocation into the nucleus. Target gene expression is induced upon binding of NRF2 to AREs (antioxidant response element) in the promoters of target genes. Target genes comprise genes coding for the cell’s antioxidative equipment, efflux pumps enabling drug resistance, stemness-regulating transcription factors, as well as SHH (modified from [71,72]).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lendeckel, U.; Wolke, C. Redox-Regulation in Cancer Stem Cells. Biomedicines 2022, 10, 2413. https://doi.org/10.3390/biomedicines10102413
AMA Style
Lendeckel U, Wolke C. Redox-Regulation in Cancer Stem Cells. Biomedicines. 2022; 10(10):2413. https://doi.org/10.3390/biomedicines10102413
Chicago/Turabian StyleLendeckel, Uwe, and Carmen Wolke. 2022. "Redox-Regulation in Cancer Stem Cells" Biomedicines 10, no. 10: 2413. https://doi.org/10.3390/biomedicines10102413
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.