
Inducible T-Cell Costimulator Ligand Plays a Dual Role in Melanoma Metastasis upon Binding to Osteopontin or Inducible T-Cell Costimulator

 ,
,  ,
,  , ,
, ,  , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. ICOSL Cloning
2.3. Cell Transfection and Cell Silencing
2.4. Invasion Assays
2.5. Anchorage-Dependent Growth Assay
2.6. In Vivo Experiments
2.7. Proximity Ligation Assays (PLA)
2.8. Statistics
3. Results
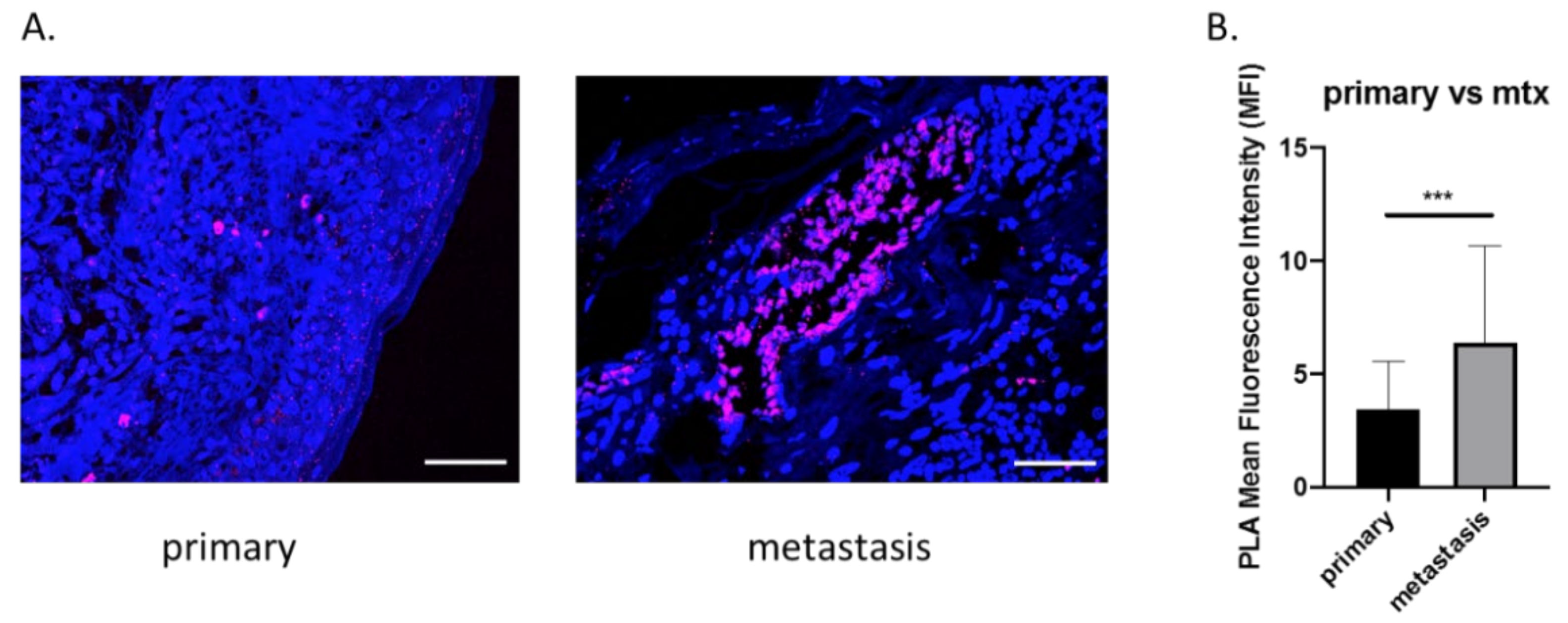
3.1. OPN and ICOSL Highly Interact in Human Melanoma Metastases
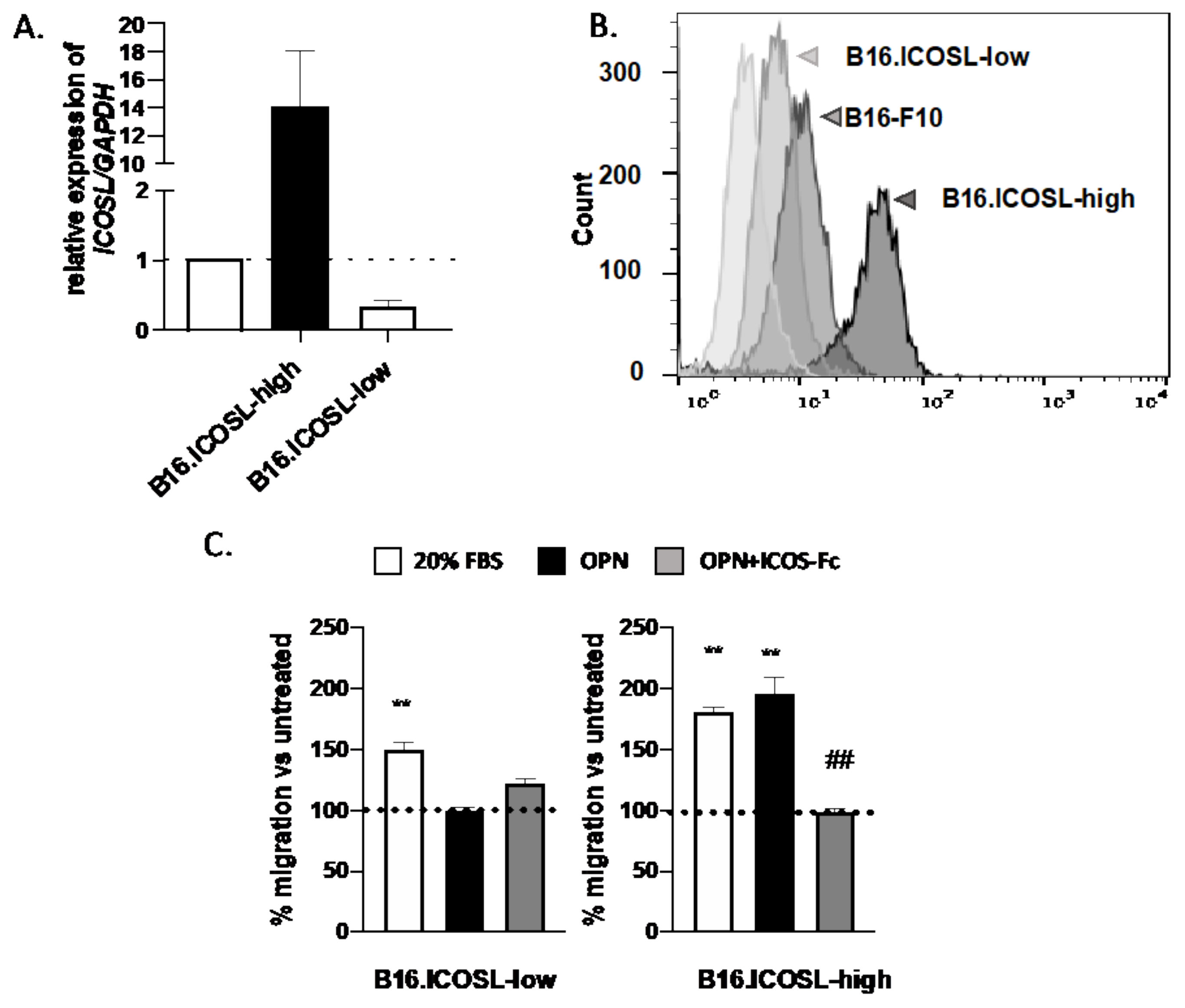
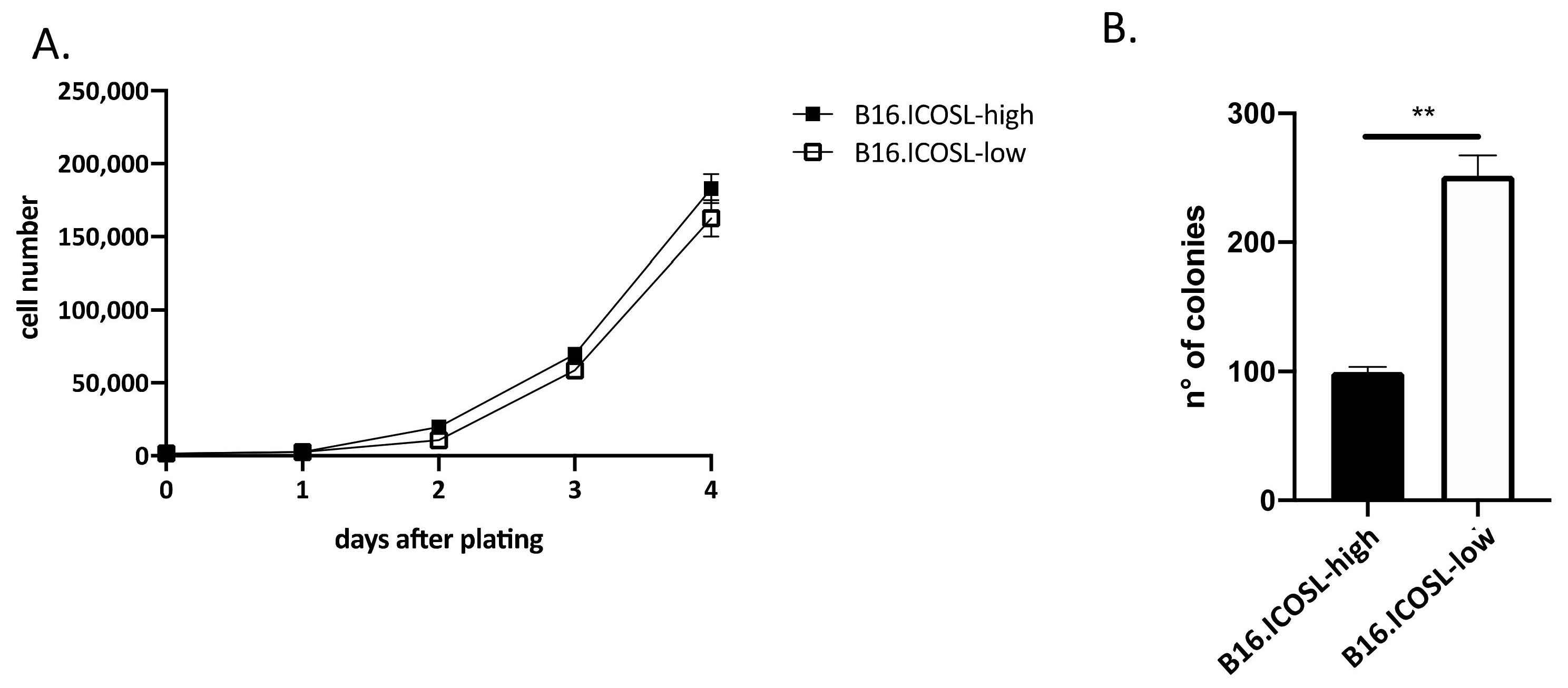
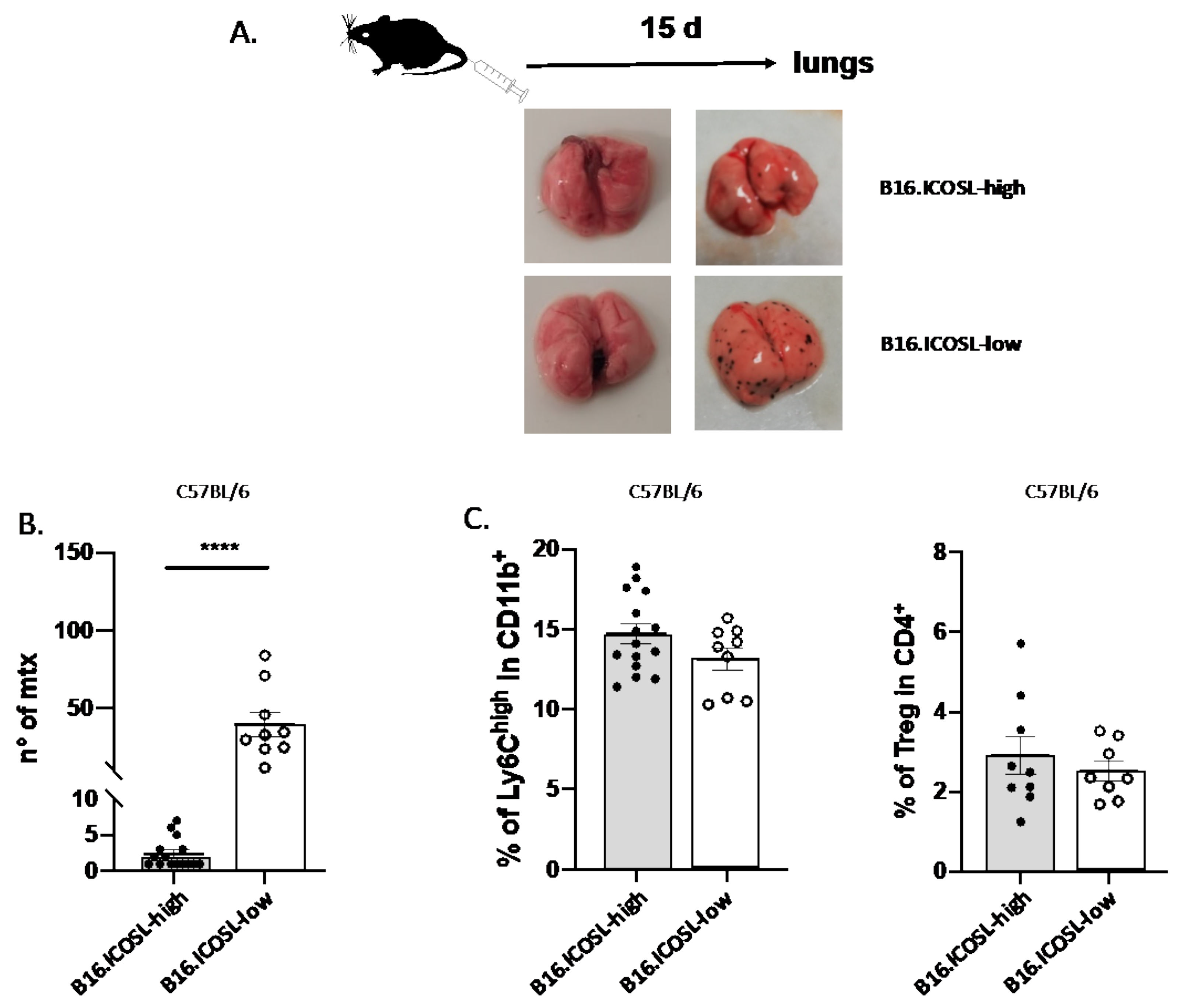
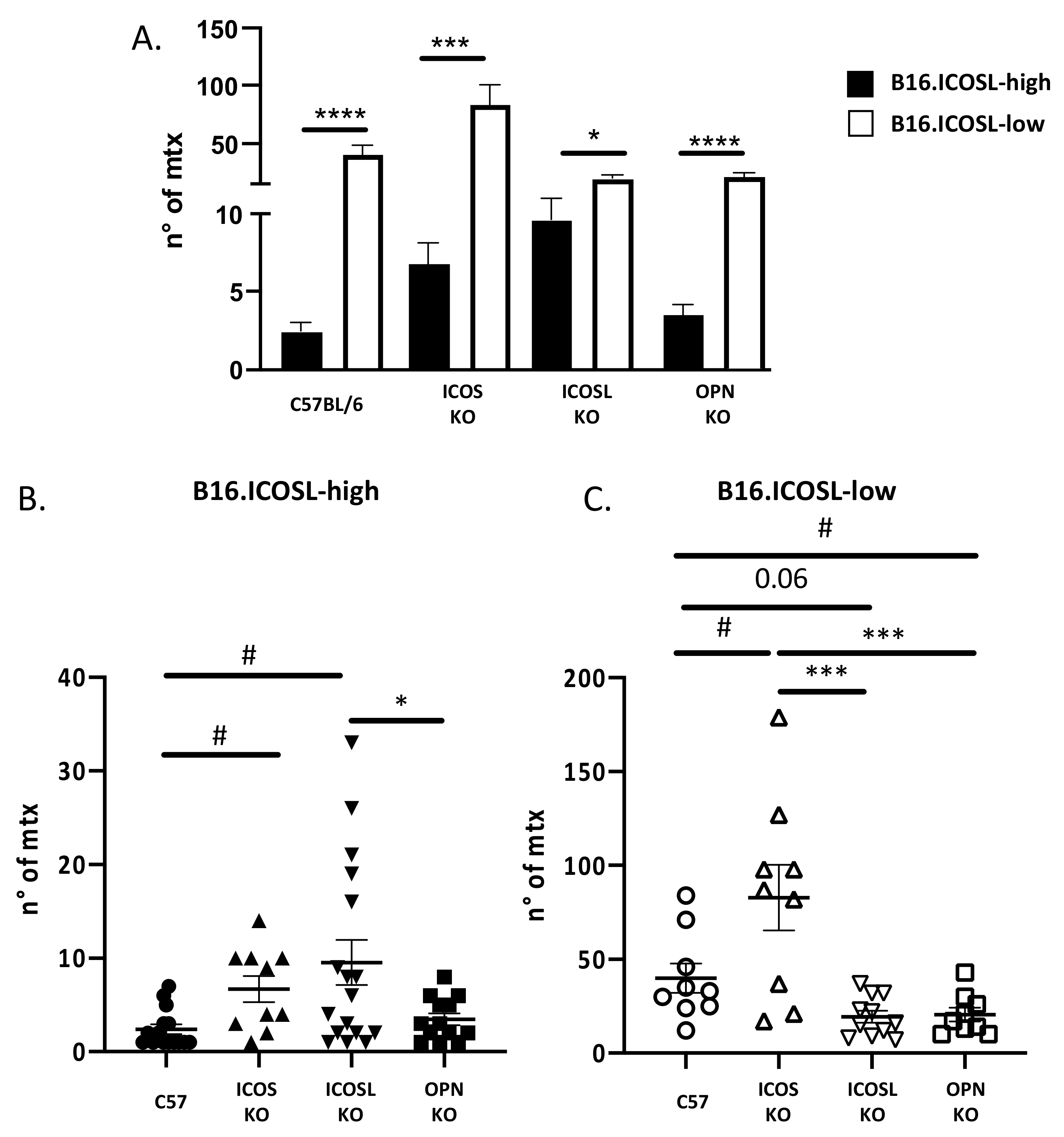
3.2. ICOSL Expression Modulates In Vitro Growth and In Vivo Metastasis of B16-F10 Melanoma Cells
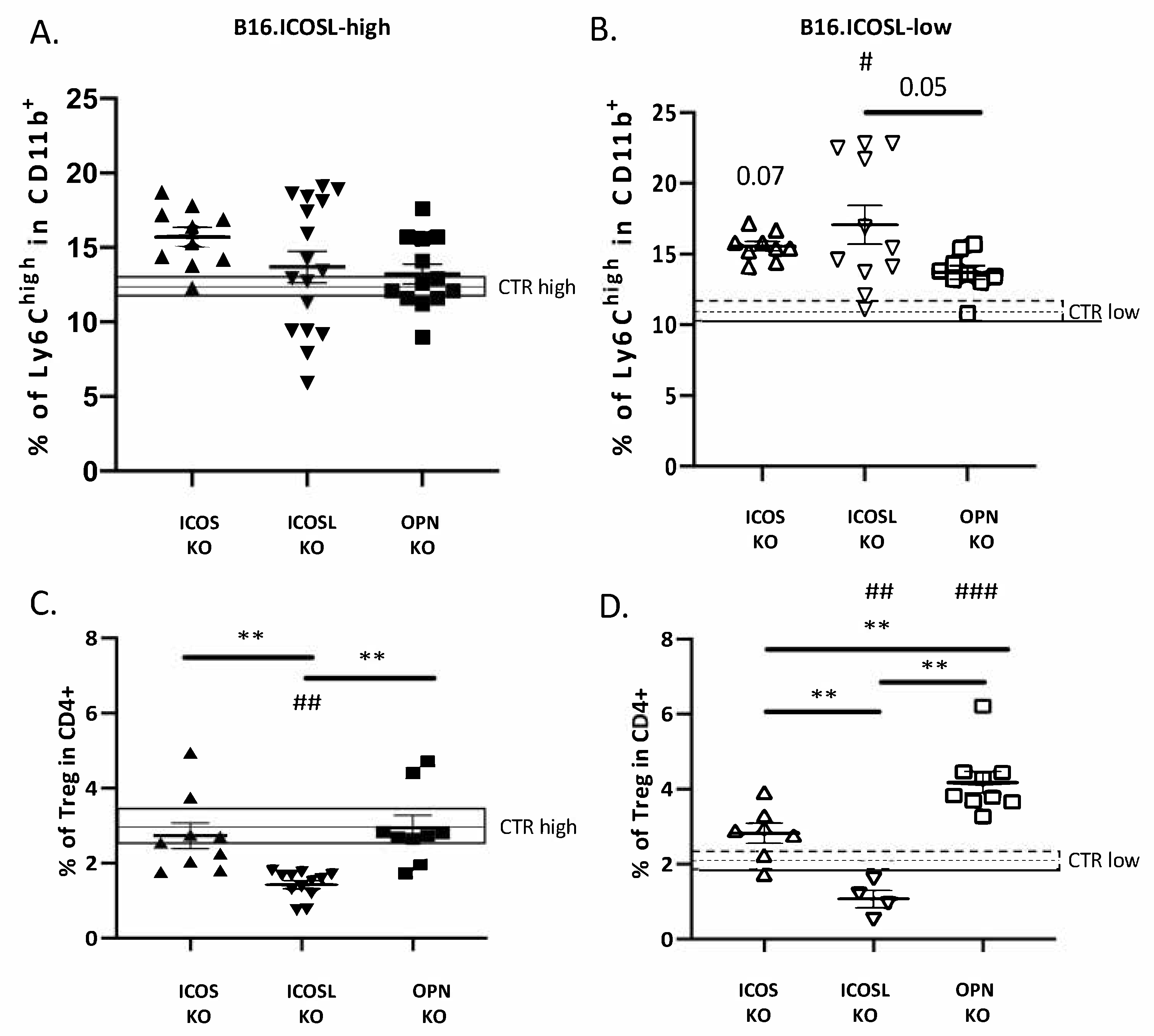
3.3. ICOSL Mediates Different Metastatic Effects When Binding to OPN or to ICOS
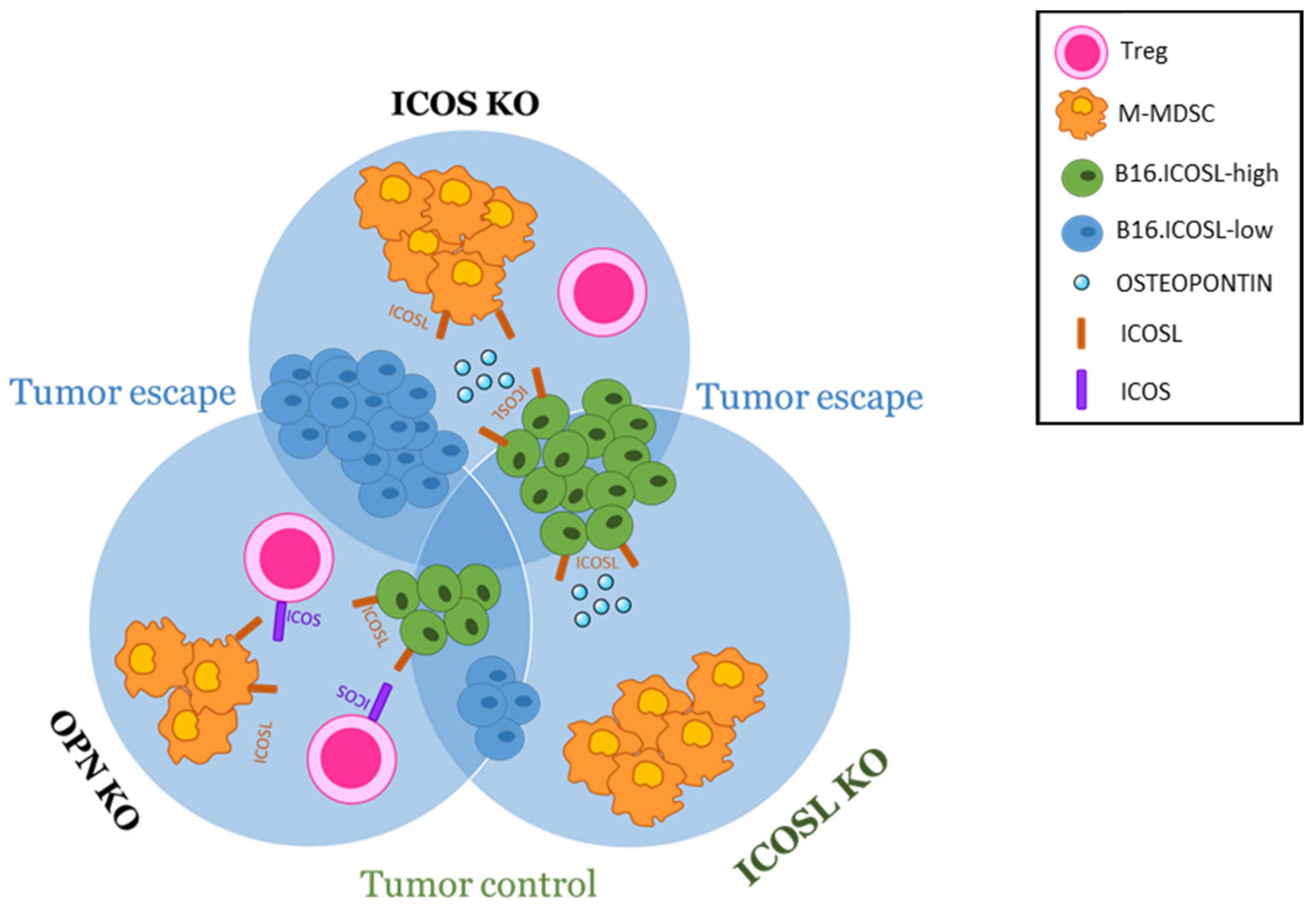
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Liu, Y.; Sheikh, M.S. Melanoma: Molecular Pathogenesis and Therapeutic Management. Mol. Cell Pharmacol. 2014, 6, 228. [Google Scholar]
- Antohe, M.; Nedelcu, R.I.; Nichita, L.; Popp, C.G.; Cioplea, M.; Brinzea, A.; Hodorogea, A.; Calinescu, A.; Balaban, M.; Ion, D.A.; et al. Tumor infiltrating lymphocytes: The regulator of melanoma evolution. Oncol. Lett. 2019, 17, 4155–4161. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Y.; Liu, S.; Wang, H.; Zhu, J.; Ou, L.; Xu, X. Targeting regulatory T cells for immunotherapy in melanoma. Mol. Biomed. 2021, 2, 11. [Google Scholar] [CrossRef]
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10, 2453. [Google Scholar] [CrossRef] [PubMed]
- De Cicco, P.; Ercolano, G.; Ianaro, A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Front. Immunol. 2020, 11, 1680. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Haist, M.; Stege, H.; Grabbe, S.; Bros, M. The Functional Crosstalk between Myeloid-Derived Suppressor Cells and Regulatory T Cells within the Immunosuppressive Tumor Microenvironment. Cancers 2021, 13, 210. [Google Scholar] [CrossRef] [PubMed]
- Castello, L.M.; Raineri, D.; Salmi, L.; Clemente, N.; Vaschetto, R.; Quaglia, M.; Garzaro, M.; Gentilli, S.; Navalesi, P.; Cantaluppi, V.; et al. Osteopontin at the Crossroads of Inflammation and Tumor Progression. Mediat. Inflamm 2017, 2017, 4049098. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Soikkeli, J.; Jahkola, T.; Virolainen, S.; Saksela, O.; Hölttä, E. Osteopontin promotes the invasive growth of melanoma cells by activating integrin αvβ3 and down-regulating tetraspanin CD9. Am. J. Pathol. 2014, 184, 842–858. [Google Scholar] [CrossRef] [Green Version]
- Kiss, T.; Ecsedi, S.; Vizkeleti, L.; Koroknai, V.; Emri, G.; Kovács, N.; Adany, R.; Balazs, M. The role of osteopontin expression in melanoma progression. Tumour Biol. 2015, 36, 7841–7847. [Google Scholar] [CrossRef] [Green Version]
- Klement, J.D.; Paschall, A.V.; Redd, P.S.; Ibrahim, M.L.; Lu, C.; Yang, D.; Celis, E.; Abrams, S.I.; Ozato, K.; Liu, K. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Invest. 2018, 128, 5549–5560. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Tripodo, C.; Sandri, S.; Torselli, I.; Vitali, C.; Ratti, C.; Botti, L.; Burocchi, A.; Porcasi, R.; Tomirotti, A.; et al. Osteopontin shapes immunosuppression in the metastatic niche. Cancer Res. 2014, 74, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Overwijk, W.W.; Restifo, N.P. B16 as a mouse model for human melanoma. Curr. Protoc Immunol. 2001, 39, 20–21. [Google Scholar] [CrossRef]
- Kumar, S.; Sharma, P.; Kumar, D.; Chakraborty, G.; Gorain, M.; Kundu, G.C. Functional characterization of stromal osteopontin in melanoma progression and metastasis. PLoS ONE 2013, 8, e69116. [Google Scholar] [CrossRef] [Green Version]
- Giopanou, I.; Lilis, I.; Papaleonidopoulos, V.; Agalioti, T.; Kanellakis, N.I.; Spiropoulou, N.; Spella, M.; Stathopoulos, G.T. Tumor-derived osteopontin isoforms cooperate with TRP53 and CCL2 to promote lung metastasis. Oncoimmunology 2017, 6, e1256528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raineri, D.; Dianzani, C.; Cappellano, G.; Maione, F.; Baldanzi, G.; Iacobucci, I.; Clemente, N.; Baldone, G.; Boggio, E.; Gigliotti, C.L.; et al. Osteopontin binds ICOSL promoting tumor metastasis. Commun. Biol. 2020, 3, 615. [Google Scholar] [CrossRef] [PubMed]
- Clemente, N.; Boggio, E.; Gigliotti, L.C.; Raineri, D.; Ferrara, B.; Miglio, G.; Argenziano, M.; Chiocchetti, A.; Cappellano, G.; Trotta, F.; et al. Immunotherapy of experimental melanoma with ICOS-Fc loaded in biocompatible and biodegradable nanoparticles. J. Control Release 2020, 320, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Dianzani, C.; Minelli, R.; Gigliotti, C.L.; Occhipinti, S.; Giovarelli, M.; Conti, L.; Boggio, E.; Shivakumar, Y.; Baldanzi, G.; Malacarne, V.; et al. B7h triggering inhibits the migration of tumor cell lines. J. Immunol. 2014, 192, 4921–4931. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Zhu, Y.; Zhu, G.; Augustine, M.; Zheng, L.; Goode, D.J.; Broadwater, M.; Ruff, W.; Flies, S.; Xu, H.; et al. B7-h2 is a costimulatory ligand for CD28 in human. Immunity 2011, 34, 729–740. [Google Scholar] [CrossRef] [Green Version]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999, 397, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.L.; Blair, P.J.; Musser, J.T.; Abe, R.; Tezuka, K.; Tsuji, T.; June, C.H. ICOS costimulation requires IL-2 and can be prevented by CTLA-4 engagement. J. Immunol. 2001, 166, 4943–4948. [Google Scholar] [CrossRef]
- Warnatz, K.; Bossaller, L.; Salzer, U.; Skrabl-Baumgartner, A.; Schwinger, W.; van der Burg, M.; van Dongen, J.J.; Orlowska-Volk, M.; Knoth, R.; Durandy, A.; et al. Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency. Blood 2006, 107, 3045–3052. [Google Scholar] [CrossRef] [Green Version]
- Dianzani, C.; Minelli, R.; Mesturini, R.; Chiocchetti, A.; Barrera, G.; Boscolo, S.; Sarasso, C.; Gigliotti, C.L.; Sblattero, D.; Yagi, J.; et al. B7h triggering inhibits umbilical vascular endothelial cell adhesiveness to tumor cell lines and polymorphonuclear cells. J. Immunol. 2010, 185, 3970–3979. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.I. Regulation of immune and autoimmune responses by ICOS-B7h interaction. Clin. Immunol. 2005, 115, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Occhipinti, S.; Dianzani, C.; Chiocchetti, A.; Boggio, E.; Clemente, N.; Gigliotti, C.L.; Soluri, M.F.; Minelli, R.; Fantozzi, R.; Yagi, J.; et al. Triggering of B7h by the ICOS modulates maturation and migration of monocyte-derived dendritic cells. J. Immunol. 2013, 190, 1125–1134. [Google Scholar] [CrossRef] [Green Version]
- Busse, M.; Krech, M.; Meyer-Bahlburg, A.; Hennig, C.; Hansen, G. ICOS mediates the generation and function of CD4+CD25+Foxp3+ regulatory T cells conveying respiratory tolerance. J. Immunol. 2012, 189, 1975–1982. [Google Scholar] [CrossRef] [Green Version]
- Vocanson, M.; Rozieres, A.; Hennino, A.; Poyet, G.; Gaillard, V.; Renaudineau, S.; Achachi, A.; Benetiere, J.; Kaiserlian, D.; Dubois, B.; et al. Inducible costimulator (ICOS) is a marker for highly suppressive antigen-specific T cells sharing features of TH17/TH1 and regulatory T cells. J. Allergy Clin. Immunol. 2010, 126, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Chan, P.L.; Liu, Y.; Qin, G.; Xiang, Z.; Lam, K.T.; Lewis, D.B.; Lau, Y.L.; Tu, W. ICOS regulates the generation and function of human CD4+ Treg in a CTLA-4 dependent manner. PLoS ONE 2013, 8, e82203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.D.; Tilli, T.M.; Wanderley, J.L.; Palumbo, A., Jr.; Mattos, R.M.; Ferreira, A.C.; Klumb, C.E.; Nasciutti, L.E.; Gimba, E.R. Osteopontin splice variants expression is involved on docetaxel resistance in PC3 prostate cancer cells. Tumour Biol. 2016, 37, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.C.; Zhang, X.F.; Peng, J.; Li, X.F.; Wang, A.L.; Bie, Y.Q.; Shi, L.H.; Lin, M.B.; Zhang, X.F. Survival Mechanisms and Influence Factors of Circulating Tumor Cells. Biomed. Res. Int. 2018, 2018, 6304701. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, Q.; Alam, A.; Cui, J.; Suen, K.C.; Soo, A.P.; Eguchi, S.; Gu, J.; Ma, D. The role of osteopontin in the progression of solid organ tumour. Cell Death Dis. 2018, 9, 356. [Google Scholar] [CrossRef]
- Zhou, Y.; Dai, D.L.; Martinka, M.; Su, M.; Zhang, Y.; Campos, E.I.; Dorocicz, I.; Tang, L.; Huntsman, D.; Nelson, C.; et al. Osteopontin expression correlates with melanoma invasion. J. Invest. Dermatol. 2005, 124, 1044–1052. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Anani, W.; Shurin, M.R. Targeting Myeloid-Derived Suppressor Cells in Cancer. Adv. Exp. Med. Biol 2017, 1036, 105–128. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S. Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol. 2019, 49, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Fu, T.; He, Q.; Sharma, P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 2011, 71, 5445–5454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klement, J.D.; Poschel, D.B.; Lu, C.; Merting, A.D.; Yang, D.; Redd, P.S.; Liu, K. Osteopontin Blockade Immunotherapy Increases Cytotoxic T Lymphocyte Lytic Activity and Suppresses Colon Tumor Progression. Cancers 2021, 13, 1006. [Google Scholar] [CrossRef] [PubMed]
- Moorman, H.R.; Poschel, D.; Klement, J.D.; Lu, C.; Redd, P.S.; Liu, K. Osteopontin: A Key Regulator of Tumor Progression and Immunomodulation. Cancers 2020, 12, 3379. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, E.C.; Mahmoud, S.M.; Bennewith, K.L. Emerging roles of regulatory T cells in tumour progression and metastasis. Cancer Metastasis Rev. 2014, 33, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Li, C.; Wang, J.; Xue, L. Myeloid-derived suppressor cells: Roles in the tumor microenvironment and tumor radiotherapy. Int. J. Cancer 2019, 144, 933–946. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raineri, D.; Cappellano, G.; Vilardo, B.; Maione, F.; Clemente, N.; Canciani, E.; Boggio, E.; Gigliotti, C.L.; Monge, C.; Dianzani, C.; et al. Inducible T-Cell Costimulator Ligand Plays a Dual Role in Melanoma Metastasis upon Binding to Osteopontin or Inducible T-Cell Costimulator. Biomedicines 2022, 10, 51. https://doi.org/10.3390/biomedicines10010051
Raineri D, Cappellano G, Vilardo B, Maione F, Clemente N, Canciani E, Boggio E, Gigliotti CL, Monge C, Dianzani C, et al. Inducible T-Cell Costimulator Ligand Plays a Dual Role in Melanoma Metastasis upon Binding to Osteopontin or Inducible T-Cell Costimulator. Biomedicines. 2022; 10(1):51. https://doi.org/10.3390/biomedicines10010051
Chicago/Turabian StyleRaineri, Davide, Giuseppe Cappellano, Beatrice Vilardo, Federica Maione, Nausicaa Clemente, Elena Canciani, Elena Boggio, Casimiro Luca Gigliotti, Chiara Monge, Chiara Dianzani, and et al. 2022. "Inducible T-Cell Costimulator Ligand Plays a Dual Role in Melanoma Metastasis upon Binding to Osteopontin or Inducible T-Cell Costimulator" Biomedicines 10, no. 1: 51. https://doi.org/10.3390/biomedicines10010051