
DNA Microarray-Based Global Gene Expression Profiling in Human Amniotic Epithelial Cells Predicts the Potential of Microalgae-Derived Squalene for the Nervous System and Metabolic Health

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Squalene-Rich Ethanol Extract of Aurantiochytrium 18W-13a (EEA-SQ)
2.2. Human Amniotic Epithelial Cells (hAECs) Extraction, Culture, and 3D Spheroid Formation
2.3. hAEC Treatment
2.4. RNA Extraction and Quantification
2.5. Microarray Gene Expression
2.6. Microarray Data Processing and Analysis
2.7. Ethics Approval
3. Results
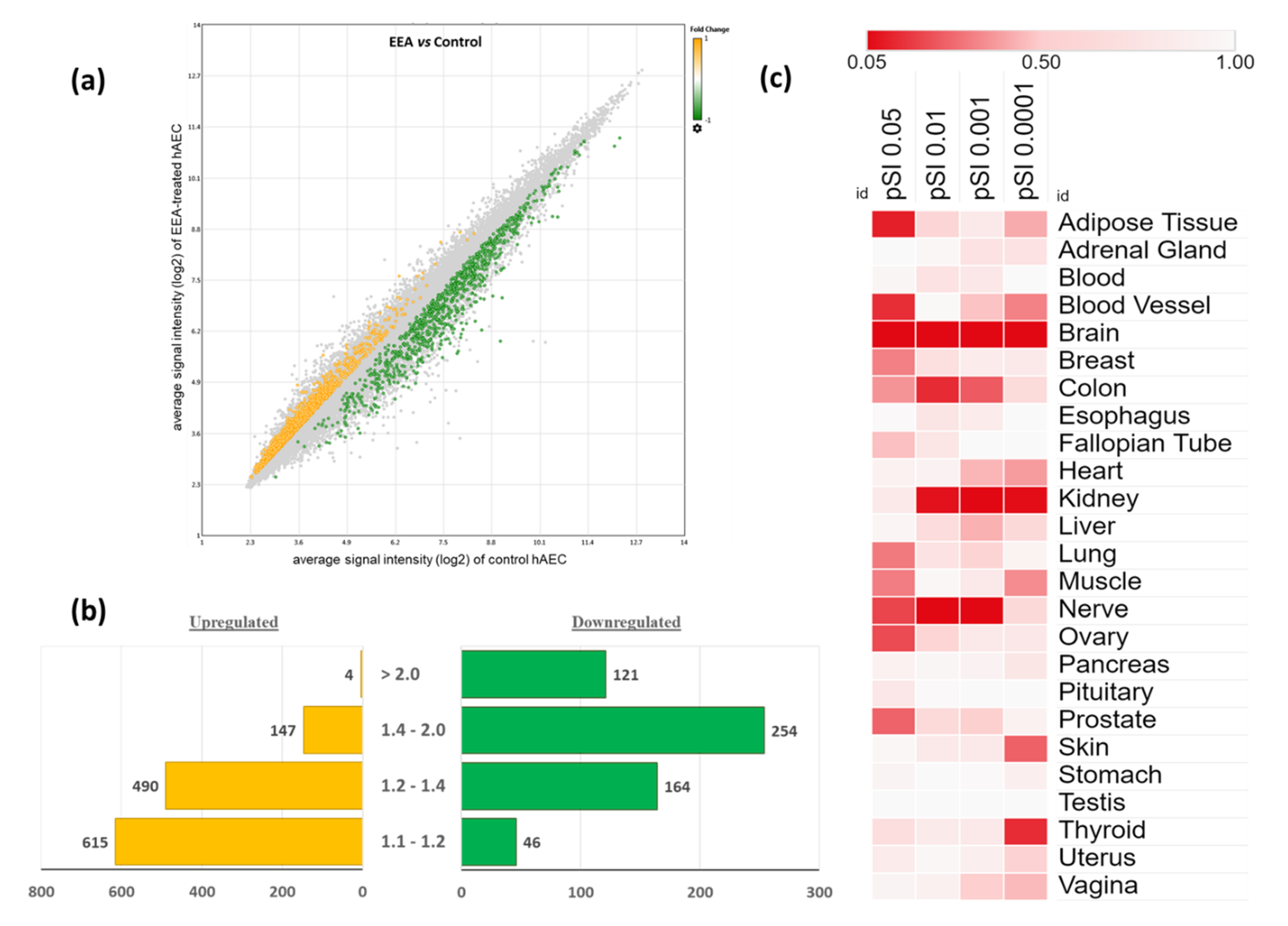
3.1. Characteristics of Gene Expression Profiling in EEA-SQ-Treated hAECs
3.2. Tissue-Specific Enrichment Analysis of the Differentially Expressed Genes (DEGs)
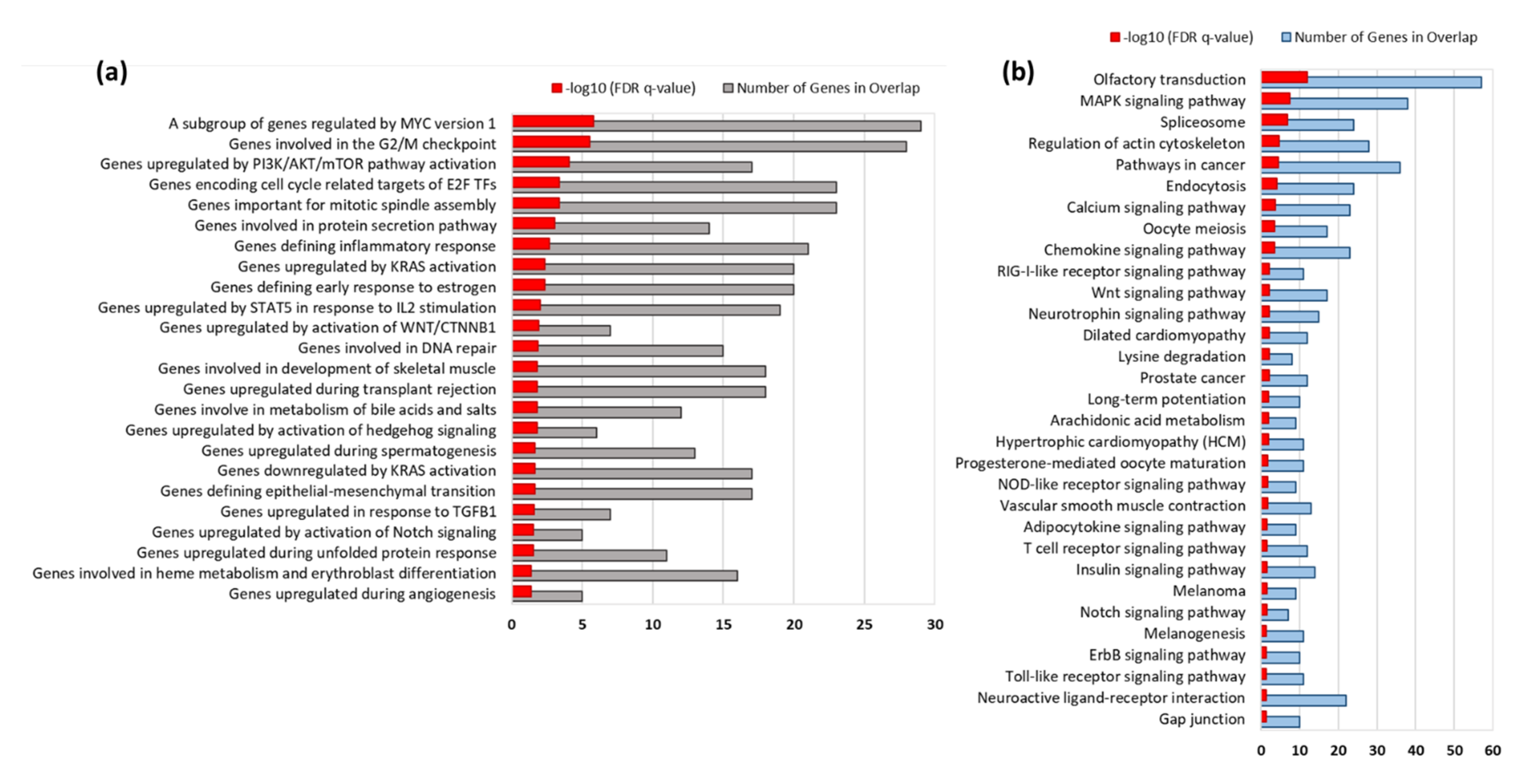
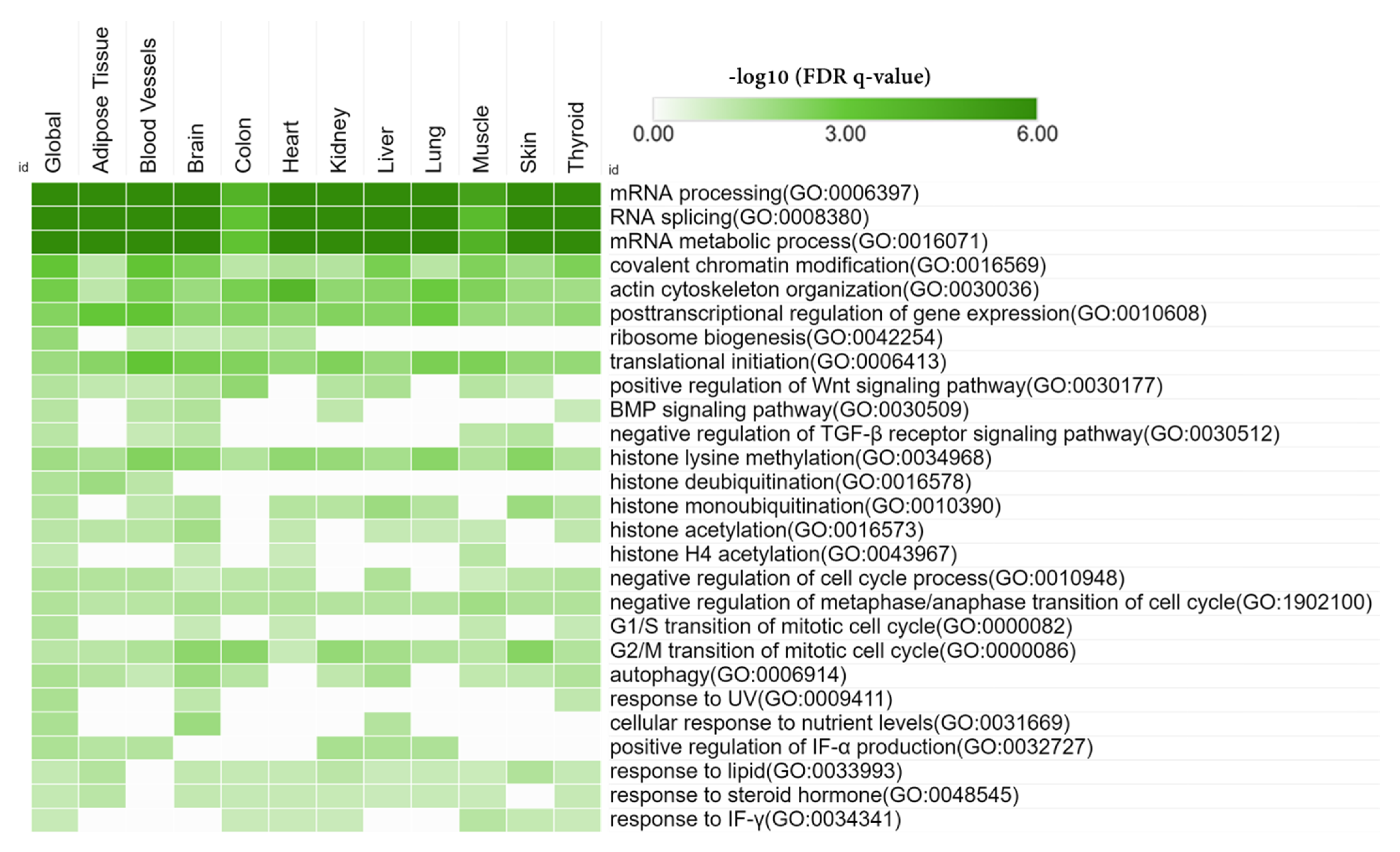
3.3. Gene Set Enrichment Analysis
3.4. KEGG Pathways Enriched by EEA-SQ
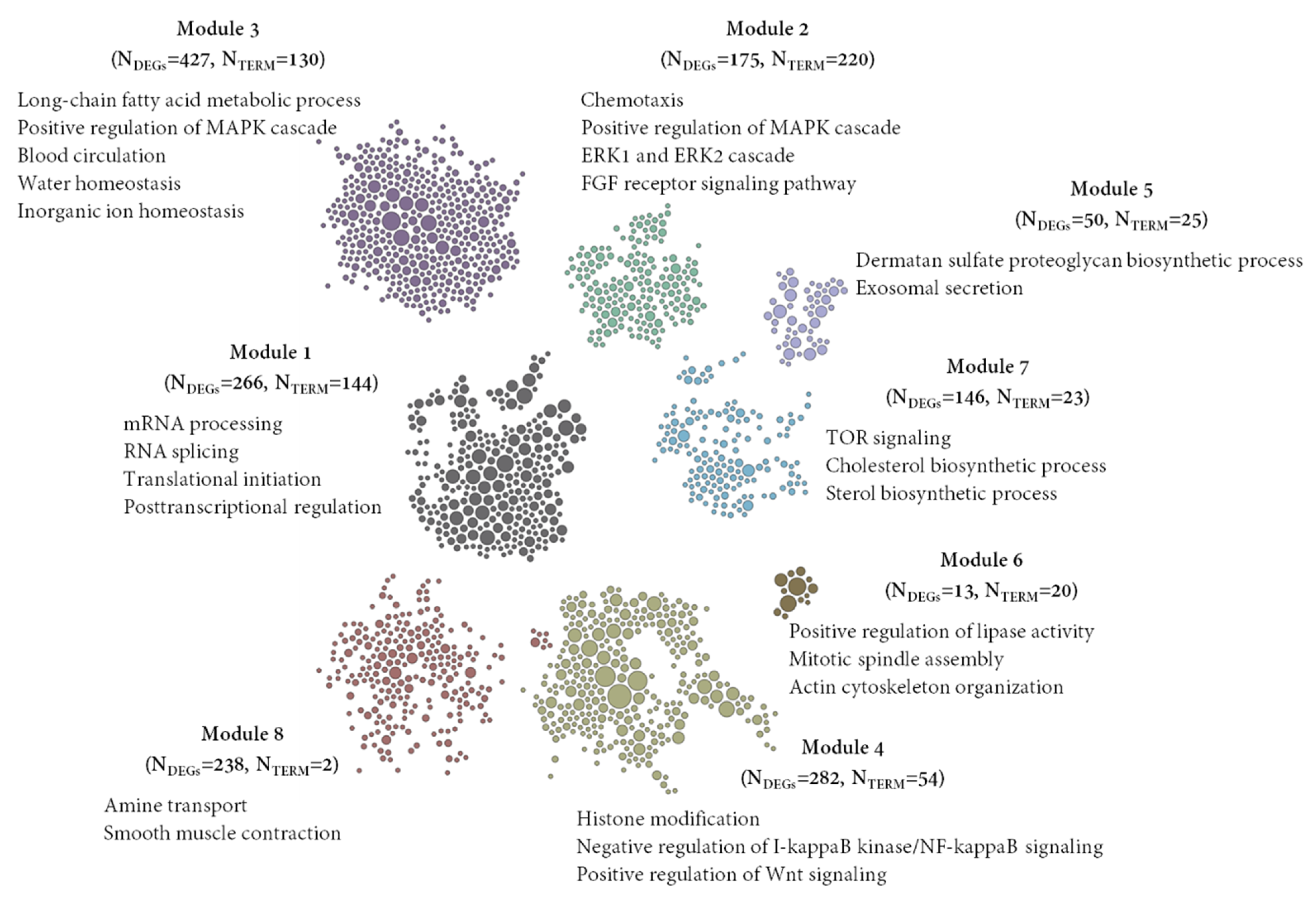
3.5. Detection of Gene Clusters and Functional Module
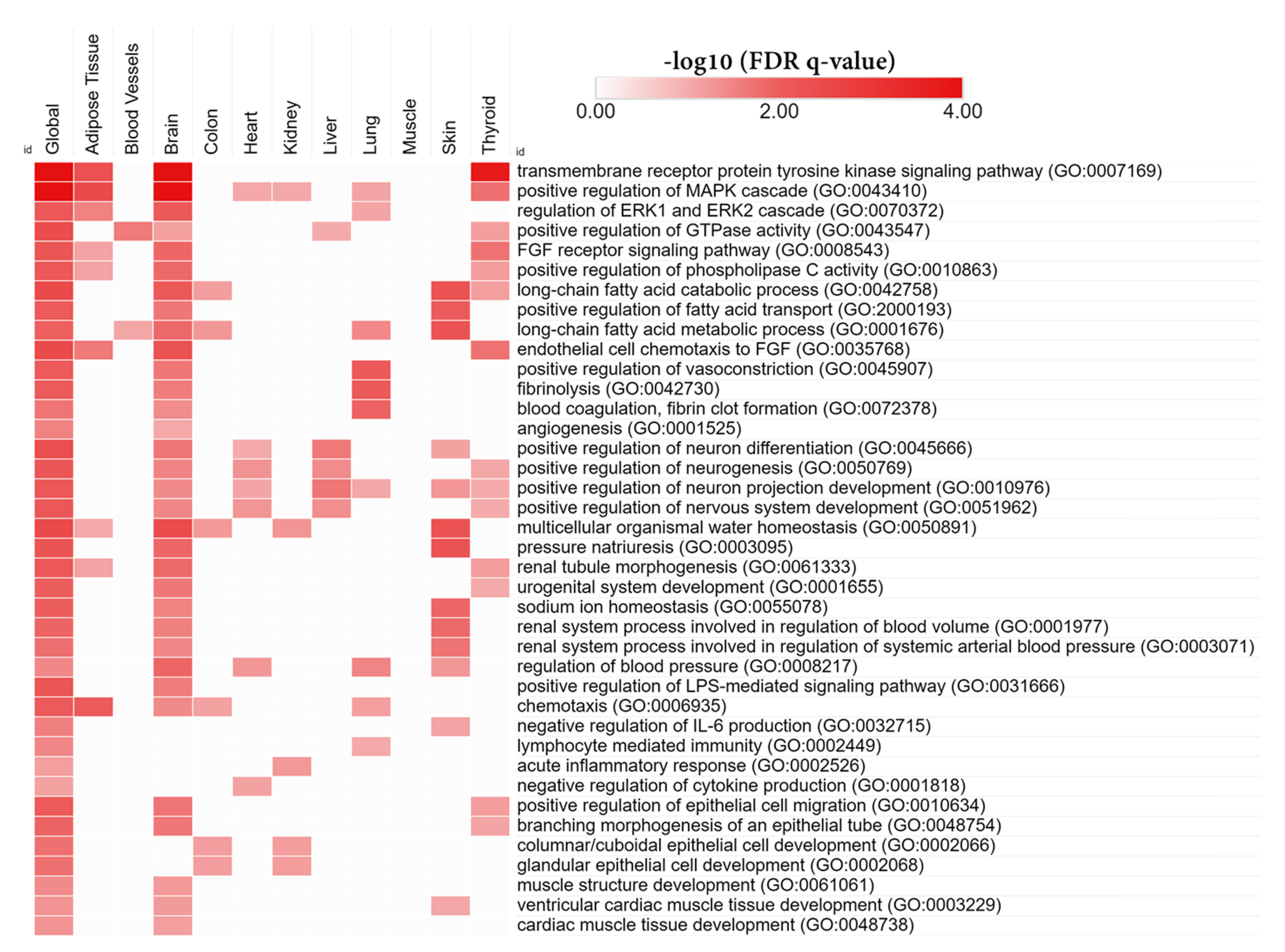
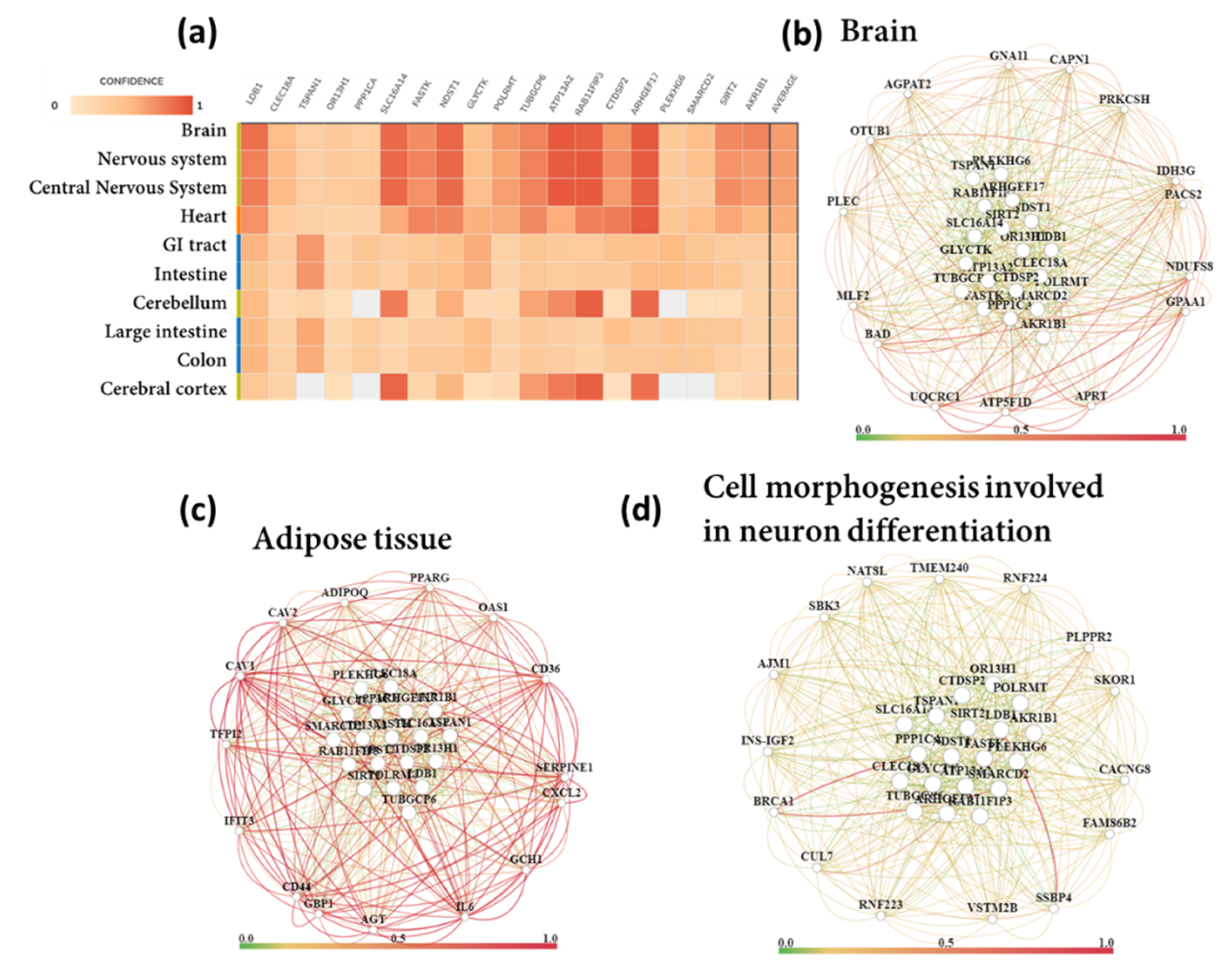
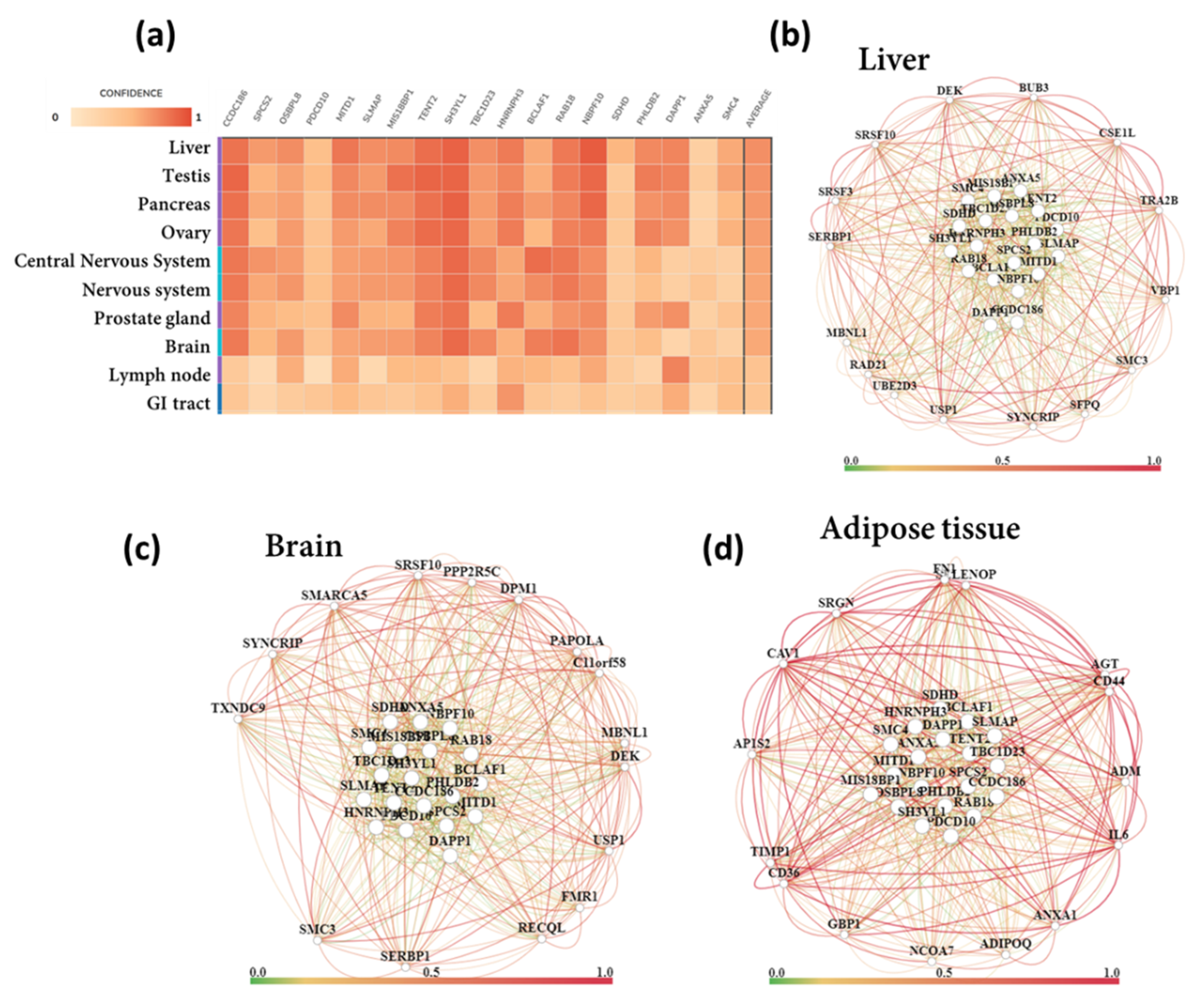
3.6. Tissue-Specific Biological Functions
3.7. Functional Gene Networks
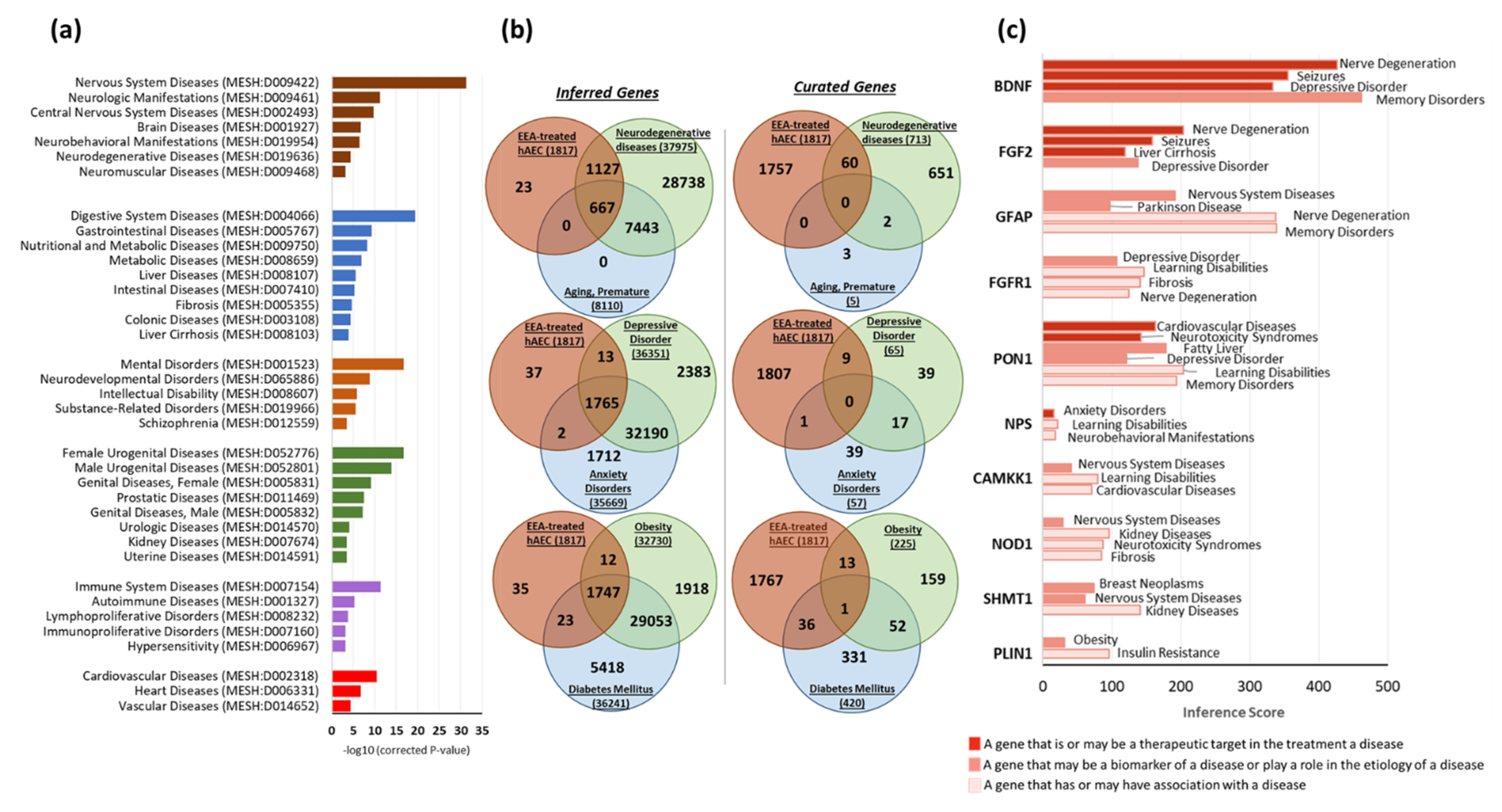
3.8. Gene–Disease Associations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grskovic, M.; Javaherian, A.; Strulovici, B.; Daley, G.Q. Induced pluripotent stem cells—opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 2011, 10, 915–929. [Google Scholar] [CrossRef]
- Laustriat, D.; Gide, J.; Peschanski, M. Human pluripotent stem cells in drug discovery and predictive toxicology. Biochem. Soc. Trans. 2010, 38, 1051–1057. [Google Scholar] [CrossRef] [Green Version]
- McNeish, J. Embryonic stem cells in drug discovery. Nat. Rev. Drug Discov. 2004, 3, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.L.; Haston, K.M. Stem cell biology and drug discovery. BMC Biol. 2011, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.G.; Mallon, B.S.; McKay, R.D.G.; Robey, P.G. Human pluripotent stem cell culture: Considerations for maintenance, expansion, and therapeutics. Cell Stem Cell 2014, 14, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Miki, T. Stem cell characteristics and the therapeutic potential of amniotic epithelial cells. Am. J. Reprod. Immunol. 2018, 80, e13003. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Lehmann, T.; Cai, H.; Stolz, D.B.; Strom, S.C. Stem cell characteristics of amniotic epithelial cells. Stem Cells 2005, 23, 1549–1559. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.; Rosli, S.; Acharya, R.; Mathias, L.; Lim, R.; Wallace, E.; Jenkin, G. Amnion epithelial cell isolation and characterization for clinical use. Curr. Protoc. Stem Cell Biol. 2010, 13, 1E.6.1–1E.6.25. [Google Scholar] [CrossRef] [PubMed]
- Parolini, O.; Alviano, F.; Bagnara, G.P.; Bilic, G.; Bühring, H.J.; Evangelista, M.; Hennerbichler, S.; Liu, B.; Magatti, M.; Mao, N. Concise Review: Isolation and Characterization of Cells from Human Term Placenta: Outcome of the First International Workshop on Placenta Derived Stem Cells. Stem Cells 2008, 26, 300–311. [Google Scholar] [CrossRef] [Green Version]
- Gottipamula, S.; Sridhar, K.N. Large-scale Isolation, Expansion and Characterization of Human Amniotic Epithelial Cells. Int. J. Stem Cells 2018, 11, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Serra, M.; Marongiu, M.; Contini, A.; Miki, T.; Cadoni, E.; Laconi, E.; Marongiu, F. Evidence of amniotic epithelial cell differentiation toward hepatic sinusoidal endothelial cells. Cell Transplant. 2018, 27, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toda, A.; Okabe, M.; Yoshida, T.; Nikaido, T. The potential of amniotic membrane/amnion-derived cells for regeneration of various tissues. J. Pharmacol. Sci. 2007, 105, 215–228. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Fang, T.; Lang, H.; Chen, M.; Shi, P.; Pang, X.; Qi, G. Comparison of the proliferation, migration and angiogenic properties of human amniotic epithelial and mesenchymal stem cells and their effects on endothelial cells. Int. J. Mol. Med. 2017, 39, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.-J.; Yuan, W.-X.; Liu, J.; Li, J.-Y.; Tan, B.; Qiu, C.; Zhu, X.-L.; Qiu, C.; Lai, D.-M.; Guo, L.-H.; et al. Biological characterization of human amniotic epithelial cells in a serum-free system and their safety evaluation. Acta Pharmacol. Sin. 2018, 39, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Ferdousi, F.; Sasaki, K.; Ohkohchi, N.; Zheng, Y.-W.; Isoda, H. Exploring the Potential Role of Rosmarinic Acid in Neuronal Differentiation of Human Amnion Epithelial Cells by Microarray Gene Expression Profiling. Front. Neurosci. 2019, 13, 779. [Google Scholar] [CrossRef] [Green Version]
- Furuya, K.; Zheng, Y.-W.; Sako, D.; Iwasaki, K.; Zheng, D.-X.; Ge, J.-Y.; Liu, L.-P.; Furuta, T.; Akimoto, K.; Yagi, H. Enhanced hepatic differentiation in the subpopulation of human amniotic stem cells under 3D multicellular microenvironment. World J. Stem Cells 2019, 11, 705. [Google Scholar] [CrossRef] [PubMed]
- Ferdousi, F.; Kondo, S.; Sasaki, K.; Uchida, Y.; Ohkohchi, N.; Zheng, Y.-W.; Isoda, H. Microarray analysis of verbenalin-treated human amniotic epithelial cells reveals therapeutic potential for Alzheimer’s Disease. Aging 2020, 12, 5516. [Google Scholar] [CrossRef] [PubMed]
- Aonuma, K.; Ferdousi, F.; Xu, D.; Tominaga, K.; Isoda, H. Effects of isorhamnetin in human amniotic epithelial stem cells in vitro and its cardioprotective effects in vivo. Front. Cell Dev. Biol. 2020, 8, 980. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Ferdousi, F.; Zheng, Y.-W.; Oda, T.; Isoda, H. Human Amniotic Epithelial Cells as a Tool to Investigate the Effects of Cyanidin 3-O-Glucoside on Cell Differentiation. Int. J. Mol. Sci. 2021, 22, 3768. [Google Scholar] [CrossRef]
- Bejaoui, M.; Ferdousi, F.; Zheng, Y.-W.; Oda, T.; Isoda, H. Regulating cell fate of human amnion epithelial cells using natural compounds: An example of enhanced neural and pigment differentiation by 3,4,5-tri-O-caffeoylquinic acid. Cell Commun. Signal. 2021, 19, 26. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Ferdousi, F.; Zheng, Y.-W.; Oda, T.; Isoda, H. Global Gene Expression Profiling Reveals Isorhamnetin Induces Hepatic-Lineage Specific Differentiation in Human Amniotic Epithelial Cells. Front. Cell Dev. Biol. 2020, 8, 1260. [Google Scholar] [CrossRef]
- Narayan Bhilwade, H.; Tatewaki, N.; Nishida, H.; Konishi, T. Squalene as novel food factor. Curr. Pharm. Biotechnol. 2010, 11, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Rajaratnam, R.A.; Gylling, H.; Miettinen, T.A. Independent association of serum squalene and noncholesterol sterols with coronary artery disease in postmenopausal women. J. Am. Coll. Cardiol. 2000, 35, 1185–1191. [Google Scholar] [CrossRef] [Green Version]
- Popa, O.; Băbeanu, N.E.; Popa, I.; Niță, S.; Dinu-Pârvu, C.E. Methods for obtaining and determination of squalene from natural sources. BioMed Res. Int. 2015, 2015, 367202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spanova, M.; Daum, G. Squalene–biochemistry, molecular biology, process biotechnology, and applications. Eur. J. Lipid Sci. Technol. 2011, 113, 1299–1320. [Google Scholar] [CrossRef]
- Patel, A.; Rova, U.; Christakopoulos, P.; Matsakas, L. Simultaneous production of DHA and squalene from Aurantiochytrium sp. grown on forest biomass hydrolysates. Biotechnol. Biofuels 2019, 12, 1–12. [Google Scholar] [CrossRef]
- Watanabe, K.; Arafiles, K.H.V.; Higashi, R.; Okamura, Y.; Tajima, T.; Matsumura, Y.; Nakashimada, Y.; Matsuyama, K.; Aki, T. Isolation of high carotenoid-producing Aurantiochytrium sp. mutants and improvement of astaxanthin productivity using metabolic information. J. Oleo Sci. 2018, 67, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Kaya, K.; Nakazawa, A.; Matsuura, H.; Honda, D.; Inouye, I.; Watanabe, M.M. Thraustochytrid Aurantiochytrium sp. 18W-13a accummulates high amounts of squalene. Biosci. Biotechnol. Biochem. 2011, 75, 2246–2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Sakamaki, M.; Ferdousi, F.; Yoshida, M.; Demura, M.; Watanabe, M.M.; Isoda, H. Ethanol extract of Aurantiochytrium mangrovei 18w-13a strain possesses anti-inflammatory effects on murine macrophage RAW264 Cells. Front. Physiol. 2018, 9, 1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Yoshida, M.; Watanabe, M.M.; Isoda, H. Anti-inflammatory effects of Aurantiochytrium Limacinum 4W-1b ethanol extract on murine macrophage RAW264 Cells. BioMed Res. Int. 2019, 2019, 3104057. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, K.; Othman, M.B.; Ferdousi, F.; Yoshida, M.; Watanabe, M.; Tominaga, K.; Isoda, H. Modulation of the neurotransmitter systems through the anti-inflammatory and antidepressant-like effects of squalene from Aurantiochytrium sp. PLoS ONE 2019, 14, e0218923. [Google Scholar] [CrossRef]
- Sasaki, K.; Geribaldi-Doldan, N.; Wu, Q.; Davies, J.; Szele, F.G.; Isoda, H. The microalgae Aurantiochytrium Sp. increases neurogenesis and improves spatial learning and memory in senescence-accelerated prone 8 mice. Front. Cell Dev. Biol. 2020, 8, 1877. [Google Scholar] [CrossRef]
- Dougherty, J.D.; Schmidt, E.F.; Nakajima, M.; Heintz, N. Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res. 2010, 38, 4218–4230. [Google Scholar] [CrossRef]
- Xu, X.; Wells, A.B.; O’Brien, D.R.; Nehorai, A.; Dougherty, J.D. Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J. Neurosci. 2014, 34, 1420–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Greene, C.S.; Krishnan, A.; Wong, A.K.; Ricciotti, E.; Zelaya, R.A.; Himmelstein, D.S.; Zhang, R.; Hartmann, B.M.; Zaslavsky, E.; Sealfon, S.C. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative toxicogenomics database (CTD): Update 2021. Nucleic Acids Res. 2021, 49, D1138–D1143. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Asthana, M.; Sharma, S.; Roy, P.; Amdekar, S.; Singh, V.; Deval, H.; Parkash, O.; Sharma, R. Importance of Using DNA Microarray in Studying Medicinal Plant. WebmedCent. Mol. Biol. 2012, 3, WMC003876. [Google Scholar]
- Li, C.-C.; Lo, H.-Y.; Hsiang, C.-Y.; Ho, T.-Y. DNA microarray analysis as a tool to investigate the therapeutic mechanisms and drug development of Chinese medicinal herbs. BioMedicine 2012, 2, 10–16. [Google Scholar] [CrossRef]
- Young, R.A. Biomedical discovery with DNA arrays. Cell 2000, 102, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Kilian, K.A. Bridging the gap: From 2D cell culture to 3D microengineered extracellular matrices. Adv. Healthc. Mater. 2015, 4, 2780–2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, G.M.; Zhu, Q.; Perez-Garcia, C.G.; Chou, S.-J.; Suh, H.; Gage, F.H.; O’Leary, D.D.; Verma, I.M. Role of BRCA1 in brain development. Proc. Natl. Acad. Sci. USA 2014, 111, E1240–E1248. [Google Scholar] [CrossRef] [Green Version]
- Autry, A.E.; Monteggia, L.M. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [Green Version]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-derived neurotrophic factor: A key molecule for memory in the healthy and the pathological brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Woodbury, M.E.; Ikezu, T. Fibroblast growth factor-2 signaling in neurogenesis and neurodegeneration. J. Neuroimmune Pharmacol. 2014, 9, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskenazi, B.; Huen, K.; Marks, A.; Harley, K.G.; Bradman, A.; Barr, D.B.; Holland, N. PON1 and neurodevelopment in children from the CHAMACOS study exposed to organophosphate pesticides in utero. Environ. Health Perspect. 2010, 118, 1775–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menini, T.; Gugliucci, A. Paraoxonase 1 in neurological disorders. Redox Rep. 2014, 19, 49–58. [Google Scholar] [CrossRef]
- Pape, H.-C.; Jüngling, K.; Seidenbecher, T.; Lesting, J.; Reinscheid, R.K. Neuropeptide S: A transmitter system in the brain regulating fear and anxiety. Neuropharmacology 2010, 58, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.M.; Staels, B. Peroxisome proliferator-activated receptor γ and adipose tissue—understanding obesity-related changes in regulation of lipid and glucose metabolism. J. Clin. Endocrinol. Metab. 2007, 92, 386–395. [Google Scholar] [CrossRef]
- Hida, K.; Wada, J.; Eguchi, J.; Zhang, H.; Baba, M.; Seida, A.; Hashimoto, I.; Okada, T.; Yasuhara, A.; Nakatsuka, A. Visceral adipose tissue-derived serine protease inhibitor: A unique insulin-sensitizing adipocytokine in obesity. Proc. Natl. Acad. Sci. USA 2005, 102, 10610–10615. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Volat, F.; Sandhow, L.; Galitzky, J.; Nguyen, T.; Esteve, D.; Åström, G.; Mejhert, N.; Ledoux, S.; Thalamas, C. CD36 is a marker of human adipocyte progenitors with pronounced adipogenic and triglyceride accumulation potential. Stem Cells 2017, 35, 1799–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Silva, C.; Rotellar, F.; Gil, M.J.; Cienfuegos, J.A.; Salvador, J.; Frühbeck, G. Expression of caveolin-1 in human adipose tissue is upregulated in obesity and obesity-associated type 2 diabetes mellitus and related to inflammation. Clin. Endocrinol. 2008, 68, 213–219. [Google Scholar] [CrossRef]
- Hu, E.; Liang, P.; Spiegelman, B.M. AdipoQ Is a Novel Adipose-specific Gene Dysregulated in Obesity. J. Biol. Chem. 1996, 271, 10697–10703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Li, P.; Ye, J. Lipid homeostasis and the formation of macrophage-derived foam cells in atherosclerosis. Protein Cell 2012, 3, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Itabe, H.; Yamaguchi, T.; Nimura, S.; Sasabe, N. Perilipins: A diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ganbold, M.; Ferdousi, F.; Arimura, T.; Tominaga, K.; Isoda, H. New amphiphilic squalene derivative improves metabolism of adipocytes differentiated from diabetic adipose-derived stem cells and prevents excessive lipogenesis. Front. Cell Dev. Biol. 2020, 8, 577259. [Google Scholar] [CrossRef]
- Quentin, E.; Gladen, A.; Roden, L.; Kresse, H. A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: Galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome. Proc. Natl. Acad. Sci. USA 1990, 87, 1342–1346. [Google Scholar] [CrossRef] [Green Version]
- Westergren-Thorsson, G.; Cöster, L.; Akesson, A.; Wollheim, F.A. Altered dermatan sulfate proteoglycan synthesis in fibroblast cultures established from skin of patients with systemic sclerosis. J. Rheumatol. 1996, 23, 1398–1406. [Google Scholar] [PubMed]
- An, Y.; Lin, S.; Tan, X.; Zhu, S.; Nie, F.; Zhen, Y.; Gu, L.; Zhang, C.; Wang, B.; Wei, W. Exosomes from adipose-derived stem cells and application to skin wound healing. Cell Prolif. 2021, 54, e12993. [Google Scholar] [CrossRef]
- Carrasco, E.; Soto-Heredero, G.; Mittelbrunn, M. The role of extracellular vesicles in cutaneous remodeling and hair follicle dynamics. Int. J. Mol. Sci. 2019, 20, 2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicero, A.L.; Delevoye, C.; Gilles-Marsens, F.; Loew, D.; Dingli, F.; Guéré, C.; André, N.; Vié, K.; Van Niel, G.; Raposo, G. Exosomes released by keratinocytes modulate melanocyte pigmentation. Nature Commun. 2015, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.H.; Lee, Y.B.; Kang, D.; Choi, E.; Nam, Y.; Lee, K.H.; You, H.-J.; Kang, H.J.; An, S.H.; Jeon, H. Overcome the barriers of the skin: Exosome therapy. Biomater. Res. 2021, 25, 1–13. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Overlapped Genes (pSI Threshold 0.0001) | Functions |
|---|---|---|
| Adipose Tissue | PLIN1, TUSC5 | Negative regulation of lipid catabolic process, insulin-stimulated glucose uptake in adipocytes |
| Adrenal Gland | KCNK3, GSTA3 | potassium channel activity, Glutathione metabolism |
| Blood | none | |
| Blood Vessel | ACAN | Integrin Pathway, extracellular signal-regulated kinase (ERK) Signaling, mitogen-activated protein kinase (MAPK) Signaling |
| Brain | HTR5A, BARHL1, GAP43, MBP, SNAP25, HTR2C, PLP1, RPH3A, DIRAS2, GABRA6, GFAP, CACNG3, OPCML, GAD2, RESP18, SLC4A10, SLCO1A2, JPH3, LRTM2, STMN2, LIX1, SLC1A2, NEFL, SLC6A7, ZIC5, OLFM3, PRKCG, GRM4, CACNG7, HS3ST4, OTP | Chemical synaptic transmission, axon development, neurotransmitter secretion, response to wounding, axon ensheathment, locomotion, negative regulation of neuron apoptotic process, astrocyte development, glutamate secretion, long-term synaptic potentiation, positive regulation of neurogenesis |
| Breast | FDCSP | Secreted protein in follicular dendritic cells |
| Colon | LRRC31, MS4A12, CDH17, FXYD3, CEACAM7, CEACAM5, MUC13 | Homophilic cell adhesion via plasma membrane adhesion molecules, integral component of membrane |
| Esophagus | none | |
| Fallopian Tube | none | |
| Heart | BMP10, MYBPC3 | Ventricular cardiac muscle tissue morphogenesis, heart contraction, regulation of striated muscle contraction, blood circulation |
| Kidney | SLC5A12, SLC12A1, PAX2, SLC13A1, FOXI1, SLC34A1, ATP6V0D2 | Inorganic anion transmembrane transport, Na-ion transmembrane transporter activity |
| Liver | PLG, FGB, FGG, UGT1A1 | Negative regulation of blood coagulation, negative regulation of extrinsic apoptotic signaling pathway, response to Interleukin-1 (IL-1) |
| Lung | SFTPA2 | Involved in respiratory gaseous exchange and toll-like receptor signaling pathway |
| Muscle | MYH2, ASB12 | Enables microfilament motor activity, p21-activated kinase (PAK) Pathway, ubiquitin protein ligase activity, Innate Immune System |
| Nerve | SLC25A48, OR6B2 | Integral component of membrane, signal transduction, involved in G protein-coupled receptor (GPCR) signaling pathway |
| Ovary | WFIKKN2 | Negative regulation of Transforming growth factor-beta (TGF- β) receptor signaling pathway |
| Pancreas | RBPJL | Development and maintenance of the exocrine pancreas, Notch Signaling Pathway |
| Pituitary | none | |
| Prostate | KLK2 | Prostate-specific antigenproduction |
| Skin | LCE1C, ALOXE3, ALOX12B, LCE2B, LCE1F, CA6, PLA2G2F, KRT74, KRT82, KRT75, CYP4F8, FAM83C | Skin development, fatty acid metabolic process, keratinocyte differentiation, lipoxygenase pathway, linoleic acid metabolic process, regulation of water loss via skin |
| Stomach | GIF | Transportation and absorption of the vital micronutrient vitamin B12 |
| Testis | RNF151, CTCFL, RNASE11, RNF17, DCAF4L2, C7ORF72, EFCAB3, SSX2B, FAM170A, C10ORF53, APOBEC4, MAGEC1, SIRPD, MAEL, UBQLN3, RFX4, DAZL, DEFB121, SLCO6A1, PABPC3, C5ORF60, OR8B12, SLC36A3, PRSS54, TKTL1, SYCP1, OPN5, VWA3B | Spermatogenesis, DNA methylation involved in gamete generation, male gamete generation |
| Thyroid | IYD, TSHR, LIPG, ZNF804B, SLC26A4 | Thyroxine biosynthesis, GPCR downstream signalling, Lipoprotein metabolism |
| Uterus | PGR | |
| Vagina | TMPRSS11A, SERPINB13 |
| Gene Symbol | Description | Fold Change | p-Value | Biological Process * |
|---|---|---|---|---|
| CLEC18A | C-type lectin domain family 18, member A | 2.53 | 0.026 | |
| TSPAN1 | tetraspanin 1 | 2.48 | 0.039 | Regulation of vesicle-mediated transport, positive regulation of endocytosis |
| OR13H1 | olfactory receptor, family 13, subfamily H, member 1 | 2.43 | 0.048 | |
| PPP1CA | protein phosphatase 1, catalytic subunit, alpha isozyme | 2.06 | 0.023 | Circadian rhythm, positive regulation of ubiquitin-protein transferase activity, regulation of immune response, regulation of ERK1 and ERK2 cascade, positive regulation of MAPK cascade |
| SLC16A14 | solute carrier family 16, member 14 | 1.98 | 0.007 | Negative regulation of protein modification process |
| LDB1 | LIM domain binding 1 | 1.9 | 0.042 | Epigenetic regulation of gene expression, regulation of DNA binding transcription factor activity, positive regulation of cell differentiation, regulation of mitotic cell cycle |
| FASTK | Fas-activated serine/threonine kinase | 1.85 | 0.004 | Regulation of RNA splicing, Endoplasmic-reticulum-associated protein degradation (ERAD) pathway |
| NDST1 | N-deacetylase/N-sulfotransferase (heparan glucosaminyl) 1 | 1.84 | 0.014 | Cellular carbohydrate biosynthetic process, glycoprotein biosynthetic process |
| GLYCTK | glycerate kinase | 1.83 | 0.044 | Regulation of DNA binding transcription factor activity |
| POLRMT | polymerase (RNA) mitochondrial (DNA directed) | 1.8 | 0.049 | Mitochondrial transcription, RNA methylation, ribosome biogenesis |
| TUBGCP6 | tubulin, gamma complex associated protein 6 | 1.8 | 0.027 | Microtubule polymerization |
| ATP13A2 | ATPase type 13A2 | 1.79 | 0.047 | Cellular response to oxidative stress, cellular calcium ion homeostasis, regulation of macroautophagy, positive regulation of Notch signaling pathway, regulation of neurogenesis |
| RAB11FIP3 | RAB11 family interacting protein 3 (class II) | 1.78 | 0.028 | Cell division, negative regulation of adiponectin secretion |
| CTDSP2 | CTD small phosphatase 2 | 1.76 | 0.039 | Protein dephosphorylation, positive regulation of DNA binding transcription factor activity, steroid hormone mediated signaling pathway |
| ARHGEF17 | Rho guanine nucleotide exchange factor 17 | 1.75 | 0.038 | Actin cytoskeleton organization |
| PLEKHG6 | pleckstrin homology domain containing, family G (with RhoGef domain) member 6 | 1.75 | 0.017 | |
| SMARCD2 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily d, member 2 | 1.75 | 0.009 | Chromatin remodeling |
| SIRT2 | sirtuin 2 | 1.73 | 0.021 | Cellular response to hypoxia, tubulin deacetylation, histone H4 deacetylation, histone H3 deacetylation, regulation of muscle tissue development, phosphatidylinositol-mediated signaling, regulation of angiogenesis |
| AKR1B1 | aldo-keto reductase family 1, member B1 (aldose reductase) | 1.72 | 0.049 | Cellular ketone metabolic process, cellular hyperosmotic salinity response |
| Gene Symbol | Description | Fold Change | p-Value | Biological Process * |
|---|---|---|---|---|
| SMC4 | structural maintenance of chromosomes 4 | −4.89 | 0.019 | Nuclear chromosome segregation, mitotic nuclear division, DNA conformation change, positive regulation of cell cycle process, cell cycle checkpoint, regulation of DNA repair |
| ANXA5 | annexin A5 | −4.71 | 0.023 | Regulation of vesicle fusion, cellular response to TGF-β stimulus, regulation of type 2 immune response, integrin-mediated signaling pathway, apoptotic signaling pathway |
| DAPP1 | dual adaptor of phosphotyrosine and 3-phosphoinositides | −4.58 | 0.019 | Negative regulation of wound healing, cytokine-mediated signaling pathway, tyrosine phosphorylation of STAT protein, Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway (JAK-STAT) cascade |
| PHLDB2 | pleckstrin homology-like domain, family B, member 2 | −4.09 | 0.046 | Response to wounding, epithelial to mesenchymal transition, mesenchymal cell differentiation, regulation of focal adhesion assembly, negative regulation of adherens junction organization, positive regulation of apoptotic process, canonical Wnt signaling pathway, positive regulation of MAPK cascade |
| SDHD | succinate dehydrogenase complex subunit D, integral membrane protein | −3.83 | 0.038 | Tricarboxylic acid metabolic process, drug metabolic process, energy derivation by oxidation of organic compounds, cellular respiration |
| NBPF10 | neuroblastoma breakpoint family, member 10 | −3.73 | 0.012 | Negative regulation of hemopoiesis, negative regulation of cell cycle, negative regulation of cell differentiation |
| RAB18 | RAB18, member RAS oncogene family | −3.61 | 0.041 | Positive regulation of DNA-templated transcription, lipid particle organization, positive regulation of mRNA splicing |
| BCLAF1 | BCL2-associated transcription factor 1 | −3.47 | 0.004 | Positive regulation of apoptotic signaling pathway, regulation of mRNA processing, posttranscriptional regulation of gene expression, DNA repair, regulation of histone modification |
| HNRNPH3 | heterogeneous nuclear ribonucleoprotein H3 (2H9) | −3.44 | 0.019 | Epithelial cell differentiation, RNA splicing, regulation of mRNA processing, regulation of translational initiation, bone morphogenetic protein (BMP) signaling pathway, positive regulation of histone modification |
| TBC1D23 | TBC1 domain family, member 23 | −3.4 | 0.013 | Cytosolic transport, central nervous system development, brain development, head development |
| SH3YL1 | SH3 and SYLF domain containing 1 | −3.34 | 0.039 | |
| PAPD4 | PAP associated domain containing 4 | −3.13 | 0.042 | |
| MIS18BP1 | MIS18 binding protein 1 | −3.11 | 0.007 | Regulation of mitotic nuclear division, positive regulation of cell cycle, cell cycle checkpoint |
| SLMAP | sarcolemma associated protein | −3.09 | 0.005 | Regulation of action potential, membrane depolarization during cardiac muscle cell action potential, regulation of voltage-gated sodium channel activity, regulation of sprouting angiogenesis, positive regulation of stress-activated MAPK cascade |
| MITD1 | microtubule interacting and trafficking domain containing 1 | −3.08 | 0.048 | Wound healing, mitotic cytokinesis, negative regulation of protein binding |
| PDCD10 | programmed cell death 10 | −3.06 | 0.043 | Stress-activated MAPK cascade, response to reactive oxygen species (ROS), negative regulation of endothelial and epithelial cell proliferation, cardiovascular system development, sprouting angiogenesis, intrinsic apoptotic signaling pathway, positive regulation of Notch signaling pathway |
| OSBPL8 | oxysterol binding protein-like 8 | −3.05 | 0.009 | Lipid transport, localization and storage, regulation of sequestering of triglyceride, fat cell differentiation |
| SPCS2 | signal peptidase complex subunit 2 | −2.99 | 0.043 | Mitochondrial transcription, mitochondrial RNA metabolic process, purine ribonucleotide metabolic process, ATP metabolic process |
| CCDC186 | coiled-coil domain containing 186 | −2.98 | 0.01 | Negative regulation of protein phosphorylation and protein modification process |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferdousi, F.; Furuya, K.; Sasaki, K.; Zheng, Y.-W.; Oda, T.; Isoda, H. DNA Microarray-Based Global Gene Expression Profiling in Human Amniotic Epithelial Cells Predicts the Potential of Microalgae-Derived Squalene for the Nervous System and Metabolic Health. Biomedicines 2022, 10, 48. https://doi.org/10.3390/biomedicines10010048
Ferdousi F, Furuya K, Sasaki K, Zheng Y-W, Oda T, Isoda H. DNA Microarray-Based Global Gene Expression Profiling in Human Amniotic Epithelial Cells Predicts the Potential of Microalgae-Derived Squalene for the Nervous System and Metabolic Health. Biomedicines. 2022; 10(1):48. https://doi.org/10.3390/biomedicines10010048
Chicago/Turabian StyleFerdousi, Farhana, Kinji Furuya, Kazunori Sasaki, Yun-Wen Zheng, Tatsuya Oda, and Hiroko Isoda. 2022. "DNA Microarray-Based Global Gene Expression Profiling in Human Amniotic Epithelial Cells Predicts the Potential of Microalgae-Derived Squalene for the Nervous System and Metabolic Health" Biomedicines 10, no. 1: 48. https://doi.org/10.3390/biomedicines10010048