
Detailed Molecular Mechanisms Involved in Drug-Induced Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: An Update

 ,
,  , and
, and
Abstract
:1. Introduction
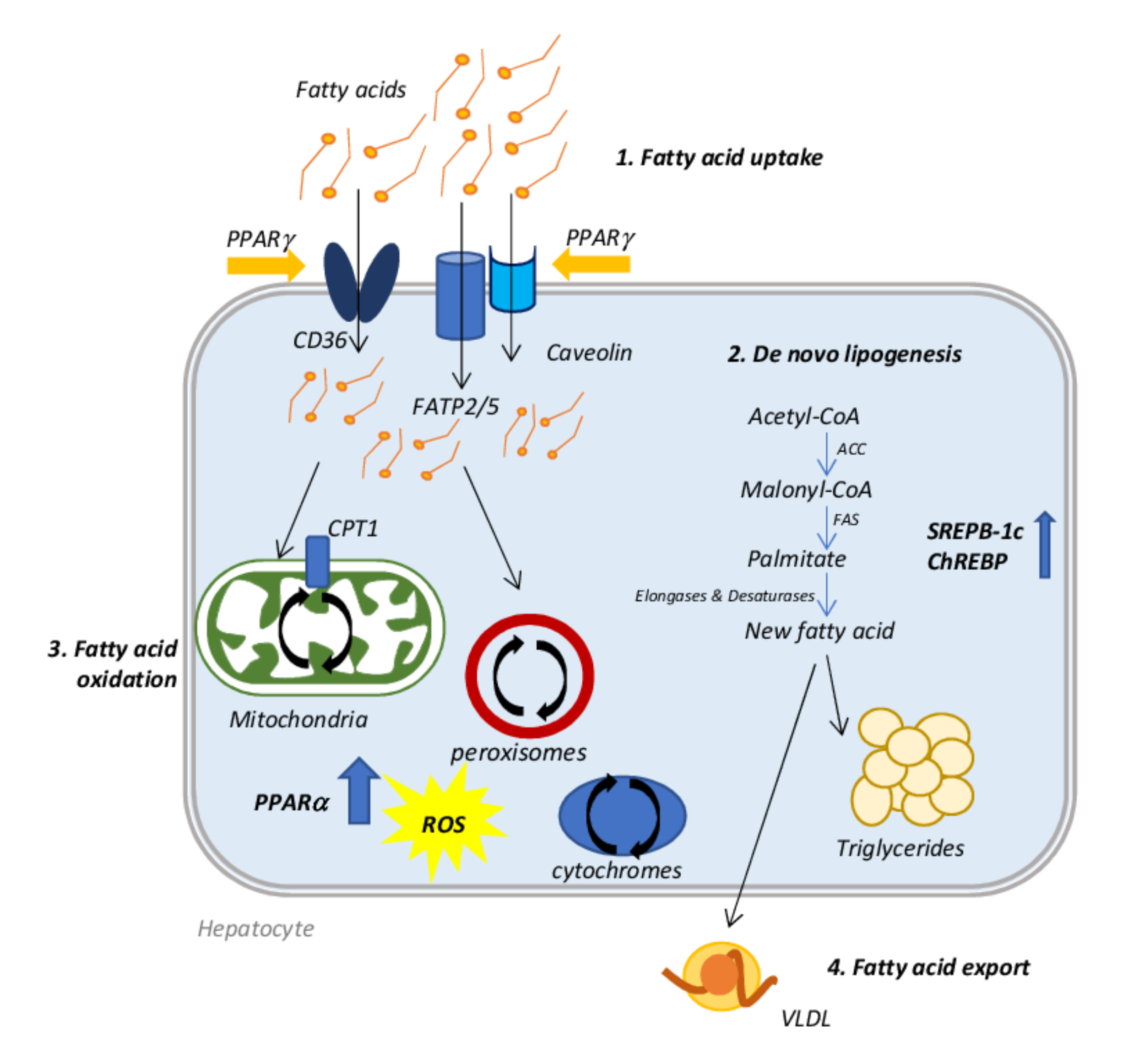
2. Molecular Mechanisms Involved in NAFLD Onset
3. Drugs Inducing Hepatic Steatosis
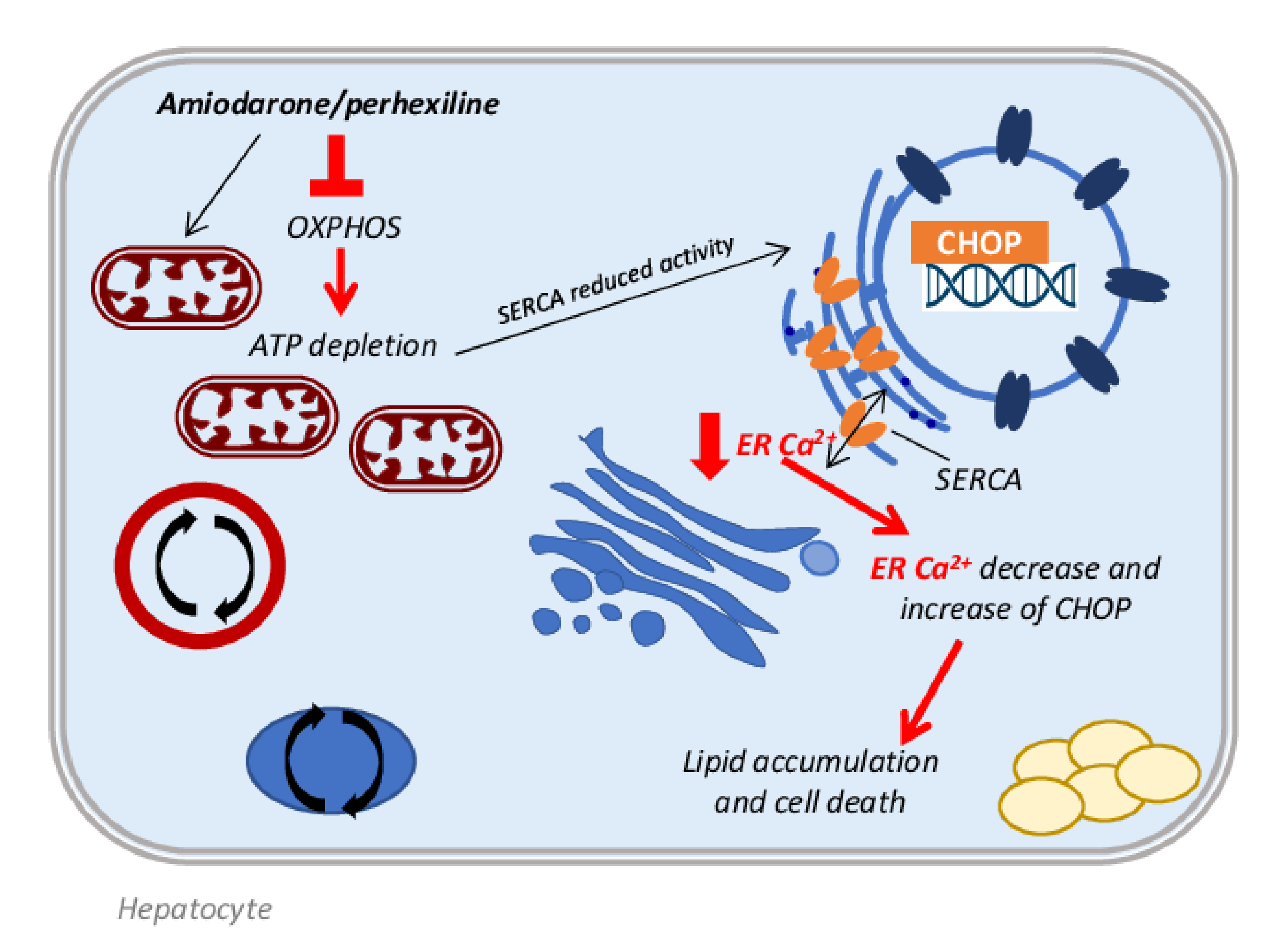
3.1. Antiarrhythmic Drugs
3.2. Antihypertensive Drugs
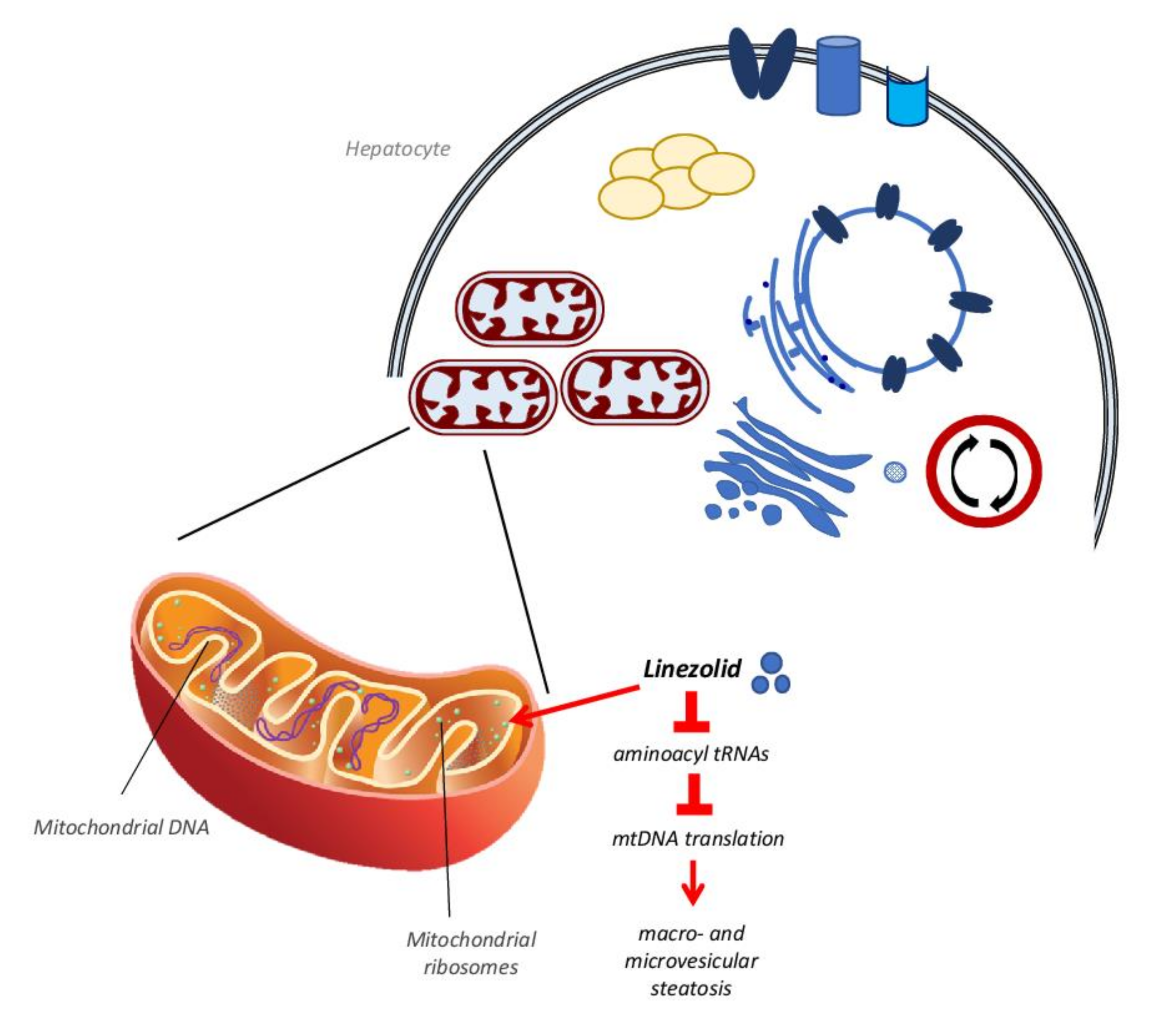
3.3. Antibiotics
3.4. Antineoplastic Drugs
3.5. Antiepileptic Drugs
3.6. Glucocorticoids
3.7. Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
3.8. Nucleoside Reverse Transcriptase Inhibitors (NRTIs)
3.9. Zonal Heterogeneity in Drug-Induced Lipid Accumulation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Acetyl CoA carboxylase |
| ACE | Angiotensin-converting enzyme |
| ACLY | ATP-citrate synthase |
| ACOX1 | Acyl-CoA oxidase 1 |
| AIDS | Acquired immune deficiency syndrome |
| ALP | Alkaline phosphatase |
| ALT | Alanine aminotransferase |
| ANGPTL4 | Angiopoietin-like 4 |
| Ang II | Angiontensin II |
| APAP | Acetaminophen |
| ApoB100 | Apolipoprotein B100 |
| AST | Aspartate transaminase |
| ATF4 | Activating transcription factor 4 |
| AZT | Zidovudine (3-azido-3′-deoxythymidine) |
| Bix | 1-(3,4-dihydroxyphenyl)-2-thiocyanate-ethanone |
| BSO | L-buthionine-(S,R)-sulfoximine |
| CASH | Chemotherapy-associated steatohepatitis |
| CCl4 | Carbon tetrachloride |
| CD36 | Cluster of differentiation 36 |
| CDAA | L-amino acid-defined |
| CHOP | CCAAT-enhancer-binding protein homologous protein |
| ChREBP | Carbohydrate regulatory element-binding protein |
| Cidea | Lipid droplet proteins cell death activator |
| Cidec | Cell death inducing DFFA like effector C |
| COX II | Cytochrome c oxidase subunit II |
| COX-2 | Cyclooxygenase-2 |
| CPT1 | carnitine palmitoyltransferase 1 |
| CYP2E1 | Cytochrome P450 2E1 |
| DEX | Dexamethasone |
| DGAT2 | Diacylglycerol acyltransferase 2 |
| dGK | Deoxyguanosine kinase |
| DIFLD | Drug-induced fatty liver disease |
| DIHS | Drug-induced hepatic steatosis |
| DILI | Drug-induced liver injury |
| DISH | Drug-induced steatohepatitis |
| DISH | Drug-induced steatohepatitis |
| DNL | De novo lipogenesis |
| DPD | Dihydropyrimidine dehydrogenase |
| eIF2α | Eukaryotic translation initiation factor-2α |
| ELP | Enalapril |
| eNOS | Endothelial nitric oxide synthase |
| ER | Endoplasmic reticulum |
| FAO | Fatty acid oxidation |
| FAS | Fatty acid synthase |
| FATP | Fatty acid transport protein |
| FOXO1 | Forkhead box protein O1 |
| 5-FU | 5-fluorouracil |
| FXR | Farnesoid X receptor |
| GC | Glucocorticoid |
| GR | Glucocorticoid receptor |
| GRP78 | Glucose-regulated protein 78 |
| GSH | Reduced glutathione |
| HAART | Highly active antiretroviral therapy |
| HADHβ | Beta-3-hydroxyacyl CoA dehydrogenase |
| HCV | Hepatitis C virus |
| Hepa1–6 | Mouse hepatoma cells |
| HSC | Hepatic stellate cells |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| HFHC | High-fat, high calorie, high-fructose |
| HIV | Human immunodeficiency virus |
| HMOX1 | Heme oxygenase 1 |
| Sabi∆Hep | Inducible hepatocyte specific SAB deletion |
| iNOS | Inducible nitric oxide synthase |
| JNK | c-Jun N-terminal kinases |
| LPO | Lipid hydroperoxides |
| MAFLD | Metabolic associated fatty liver disease |
| MAPK | Mitogen-activated protein kinase |
| MCD | Methionine and choline deficient |
| MCR | Mitochondrial respiratory chain |
| MKP3 | Mitogen-activated protein kinase phosphatase-3 |
| mTOR | Mammalian target of rapamycin |
| MTP | Microsomal triglyceride transfer protein |
| MTTP | Microsomal triglyceride transfer protein |
| MTX | Methotrexate |
| NAFLD | Non-alcoholic fatty liver disease |
| NAPQI | N-acetyl-p-bemzoquinone imine |
| NASH | Non-alcoholic steatohepatitis |
| NLRP3 | Nod-like receptor protein 3 |
| NRTIs | Nucleoside reverse transcriptase inhibitors |
| NSAIDs | Nonsteroidal anti-inflammatory agents |
| OCA | Obeticholic acid |
| OXPHOS | Oxidative phosphorylation |
| PDGF-C | Platelet-derived growth factor-C |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PERK | Protein kinase-like endoplasmic reticulum kinase |
| PGC-1α | PPARγ coactivator 1α |
| PPAR | Peroxisome roliferator-activated receptor |
| RAAS | renin–angiotensin–aldosterone system |
| ROS | Reactive oxygen species |
| SAB | SH3BP5 protein |
| (GalNAc-Sab-ASO) | SAB N-acetylgalactosamine antisense oligonucleotide |
| SREBP-1c | Sterol regulatory element-binding protein 1C |
| sXBP1 | spliced X-box binding protein 1 |
| TAG | Triacylglycerol |
| TGFβ1 | Transforming growth factor-β1 |
| TGH | Triacylglycerol hydrolase |
| TK2 | Thymidine kinase 2 |
| TNF-α | Tumor necrosis factor alpha |
| TXNIP | Thioredoxin-interacting protein |
| VEGF | Vascular endothelial growth factor |
| VLDL | Very low density lipoprotein |
| VPA | Valproic acid |
| WT | Wild type |
| α-SMA | α-smooth muscle actin |
| γ-GT | Gamma-glutamyltransferase |
References
- Palladini, G.; Di Pasqua, L.G.; Berardo, C.; Siciliano, V.; Richelmi, P.; Mannucci, B.; Croce, A.C.; Rizzo, V.; Perlini, S.; Vairetti, M.; et al. Fatty acid desaturase involvement in non-alcoholic fatty liver disease rat models: Oxidative stress versus metalloproteinases. Nutrients 2019, 11, 799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Vairetti, M.; Ferrigno, A. Nonalcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: Current Issues and Future Perspectives in Preclinical and Clinical Research. Int. J. Mol. Sci. 2020, 21, 9646. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Häring, H.U.; Cusi, K. Non-alcoholic fatty liver disease: Causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019, 7, 313–324. [Google Scholar] [CrossRef]
- Liu, D.J.; Peloso, G.M.; Yu, H.; Butterworth, A.S.; Wang, X.; Mahajan, A.; Saleheen, D.; Emdin, C.; Alam, D.; Alves, A.C.; et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat. Genet. 2017, 49, 1758–1766. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Cipolla, U.; Cassader, M.; Pinach, S.; Saba, F.; De Michieli, F.; Paschetta, E.; Bongiovanni, D.; Framarin, L.; Leone, N.; et al. TM6SF2 rs58542926 variant affects postprandial lipoprotein metabolism and glucose homeostasis in NAFLD. J. Lipid Res. 2017, 58, 1221–1229. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.J.; Harrison, S.A.; Brunt, E.M.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a Premature Change in Terminology. Hepatology 2021, 73, 1194–1198. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Jinjuvadia, R.; Antaki, F.; Lohia, P.; Liangpunsakul, S. The Association between Nonalcoholic Fatty Liver Disease and Metabolic Abnormalities in United States Population. J. Clin. Gastroenterol. 2017, 51, 160. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Ferrigno, A.; Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Siciliano, V.; Richelmi, P.; Vairetti, M. The selective blockade of metabotropic glutamate receptor-5 attenuates fat accumulation in an in vitro model of benign steatosis. Eur. J. Histochem. 2020, 64, 3175. [Google Scholar]
- Rabinowich, L.; Shibolet, O. Drug induced steatohepatitis: An uncommon culprit of a common disease. Biomed Res. Int. 2015, 2015, 168905. [Google Scholar] [CrossRef] [Green Version]
- Amacher, D.E.; Chalasani, N. Drug-induced hepatic steatosis. Semin. Liver Dis. 2014, 34, 205–214. [Google Scholar]
- Freiman, J.P.; Helfert, K.E.; Hamrell, M.R.; Stein, D.S. Hepatomegaly with severe steatosis in HIV-seropositive patients. AIDS 1993, 7, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Olano, J.P.; Borucki, M.J.; Wen, J.W.; Haque, A.K. Massive hepatic steatosis and lactic acidosis in a patient with AIDS who was receiving zidovudine. Clin. Infect. Dis. 1995, 21, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Chariot, P.; Drogou, I.; De Lacroix-Szmania, I.; Eliezer-Vanerot, M.C.; Chazaud, B.; Lombès, A.; Schaeffer, A.; Zafrani, E.S. Zidovudine-induced mitochondrial disorder with massive liver steatosis, myopathy, lactic acidosis, and mitochondrial DNA depletion. J. Hepatol. 1999, 30, 156–160. [Google Scholar] [CrossRef]
- Acosta, B.S.; Grimsley, E.W. Zidovudine-associated type B lactic acidosis and hepatic steatosis in an HIV-infected patient. South. Med. J. 1999, 92, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.D.; Guo, G.L. Mechanistic review of drug-induced steatohepatitis. Toxicol. Appl. Pharmacol. 2015, 289, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Esler, W.P.; Bence, K.K. Metabolic Targets in Nonalcoholic Fatty Liver Disease. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 247–267. [Google Scholar] [CrossRef] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Falcon, A.; Doege, H.; Fluitt, A.; Tsang, B.; Watson, N.; Kay, M.A.; Stahl, A. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am. J. Physiol.-Endocrinol. Metab. 2010, 299, E384–E393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doege, H.; Grimm, D.; Falcon, A.; Tsang, B.; Storm, T.A.; Xu, H.; Ortegon, A.M.; Kazantzis, M.; Kay, M.A.; Stahl, A. Silencing of Hepatic Fatty Acid Transporter Protein 5 in Vivo Reverses Diet-induced Non-alcoholic Fatty Liver Disease and Improves Hyperglycemia. J. Biol. Chem. 2008, 283, 22186–22192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [Green Version]
- Miquilena-Colina, M.E.; Lima-Cabello, E.; Sánchez-Campos, S.; García-Mediavilla, M.V.; Fernández-Bermejo, M.; Lozano-Rodríguez, T.; Vargas-Castrillón, J.; Xabier Buqué, B.O.; Aspichueta, P.; González-Gallego, J.; et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 2011, 60, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Diraison, F.; Moulin, P.H.; Beylot, M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003, 29, 478–485. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, S.H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2015, 15, 8713–8742. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Jia, Y.; Yang, G.; Zhang, X.; Boddu, P.C.; Petersen, B.; Narsingam, S.; Zhu, Y.J.; Thimmapaya, B.; Kanwar, Y.S.; et al. PPARα-deficient ob/ob obese mice become more obese and manifest severe hepatic steatosis due to decreased fatty acid oxidation. Am. J. Pathol. 2015, 185, 1396–1408. [Google Scholar] [CrossRef] [Green Version]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Rao, M.S.; Reddy, J.K. Peroxisomal β-oxidation and steatohepatitis. Semin. Liver Dis. 2001, 21, 43–55. [Google Scholar] [CrossRef]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddoway, L.A.; Sloan, R.W. Amiodarone: Guidelines for Use and Monitoring. Am. Fam. Physician 2003, 68, 2189–2196. [Google Scholar]
- Narayana, D.S.K.; Woods, D.D.R.; Boos, D.C.J. Management of Amiodarone-Related Thyroid Problems. Ther. Adv. Endocrinol. Metab. 2011, 2, 115. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Horowitz, J.D.; Frenneaux, M.P. Perhexiline. Cardiovasc. Drug Rev. 2007, 25, 76–97. [Google Scholar] [CrossRef] [PubMed]
- Inglis, S.; Stewart, S. Metabolic therapeutics in angina pectoris: History revisited with perhexiline. Eur. J. Cardiovasc. Nurs. 2006, 5, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Pessayre, D. Impaired mitochondrial function in microvesicular steatosis: Effects of drugs, ethanol, hormones and cytokines. J. Hepatol. 1997, 26, 43–53. [Google Scholar] [CrossRef]
- Fromenty, B.; Pessayre, D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol. Ther. 1995, 67, 101–154. [Google Scholar] [CrossRef]
- Lewis, J.H.; Ranard, R.C.; Caruso, A.; Jackson, L.K.; Mullick, F.; Ishak, K.G.; Seeff, L.B.; Zimmerman, H.J. Amiodarone hepatotoxicity: Prevalence and clinicopathologic correlations among 104 patients. Hepatology 1989, 9, 679–685. [Google Scholar] [CrossRef]
- Lewis, J.H.; Mullick, F.; Ishak, K.G.; Ranard, R.C.; Ragsdale, B.; Perse, R.M.; Rusnock, E.J.; Wolke, A.; Benjamin, S.B.; Seeff, L.B.; et al. Histopathologic analysis of suspected amiodarone hepatotoxicity. Hum. Pathol. 1990, 21, 59–67. [Google Scholar] [CrossRef]
- Anthérieu, S.; Rogue, A.; Fromenty, B.; Guillouzo, A.; Robin, M.A. Induction of vesicular steatosis by amiodarone and tetracycline is associated with up-regulation of lipogenic genes in heparg cells. Hepatology 2011, 53, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.A.; Borgne-Sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, D.; DeBeco, V.; Fisch, C.; Fromenty, B.; Guillouzo, A.; Pessayre, D. Inhibition by perhexiline of oxidative phosphorylation and the β-oxidation of fatty acids: Possible role in pseudoalcoholic liver lesions. Hepatology 1994, 19, 948–961. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Fisch, C.; Berson, A.; Letteron, P.; Larrey, D.; Pessayre, D. Dual effect of amiodarone on mitochondrial respiration. Initial protonophoric uncoupling effect followed by inhibition of the respiratory chain at the levels of complex I and complex II. J. Pharmacol. Exp. Ther. 1990, 255, 1377–1384. [Google Scholar]
- Spaniol, M.; Bracher, R.; Ha, H.R.; Follath, F.; Krähenbühl, S. Toxicity of amiodarone and amiodarone analogues on isolated rat liver mitochondria. J. Hepatol. 2001, 35, 628–636. [Google Scholar] [CrossRef]
- Fromenty, B. Inhibition of mitochondrial fatty acid oxidation in drug-induced hepatic steatosis. Liver Res. 2019, 3, 157–169. [Google Scholar] [CrossRef]
- Berson, A.; De Beco, V.; Letteron, P.; Robin, M.A.; Moreau, C.; El Kahwaji, J.; Verthier, N.; Feldmann, G.; Fromenty, B.; Pessayre, D. Steatohepatitis-inducing drugs cause mitochondrial dysfunction and lipid peroxidation in rat hepatocytes. Gastroenterology 1998, 114, 764–774. [Google Scholar] [CrossRef]
- Barclay, M.L.; Sawyers, S.M.; Begg, E.J.; Zhang, M.; Roberts, R.L.; Kennedy, M.A.; Elliott, J.M. Correlation of CYP2D6 genotype with perhexiline phenotypic metabolizer status. Pharmacogenetics 2003, 13, 627–632. [Google Scholar] [CrossRef]
- Erez, N.; Hubel, E.; Avraham, R.; Cohen, R.; Fishman, S.; Bantel, H.; Manns, M.; Tirosh, B.; Zvibel, I.; Shibolet, O. Hepatic amiodarone lipotoxicity is ameliorated by genetic and pharmacological inhibition of endoplasmatic reticulum stress. Toxicol. Sci. 2017, 159, 402–412. [Google Scholar] [CrossRef]
- Kudo, T.; Kanemoto, S.; Hara, H.; Morimoto, N.; Morihara, T.; Kimura, R.; Tabira, T.; Imaizumi, K.; Takeda, M. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008, 15, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Cataldi, M.; Citro, V.; Resnati, C.; Manco, F.; Tarantino, G. New Avenues for Treatment and Prevention of Drug-Induced Steatosis and Steatohepatitis: Much More Than Antioxidants. Adv. Ther. 2021, 38, 2094–2113. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhang, Y.; Chen, J.; Hu, Y.; Xu, Y. Verapamil ameliorates hepatic metaflammation by inhibiting thioredoxin-interacting protein/NLRP3 pathways. Front. Endocrinol. 2018, 9, 640. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Wu, Y.; Liao, Z.X.; Wang, H. Protective effect of verapamil on multiple hepatotoxic factors-induced liver fibrosis in rats. Pharmacol. Res. 2007, 55, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Brookes, M.J.; Cooper, B.T. Hypertension and fatty liver: Guilty by association? J. Hum. Hypertens. 2007, 21, 264–270. [Google Scholar] [CrossRef]
- Olivares-Reyes, J.A.; Arellano-Plancarte, A.; Castillo-Hernandez, J.R. Angiotensin II and the development of insulin resistance: Implications for diabetes. Mol. Cell. Endocrinol. 2009, 302, 128–139. [Google Scholar] [CrossRef]
- Bataller, R.; Sancho-Bru, P.; Ginès, P.; Lora, J.M.; Al-Garawi, A.; Solé, M.; Colmenero, J.; Nicolás, J.M.; Jiménez, W.; Weich, N.; et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 2003, 125, 117–125. [Google Scholar] [CrossRef]
- Warner, F.J.; Lubel, J.S.; McCaughan, G.W.; Angus, P.W. Liver fibrosis: A balance of ACEs? Clin. Sci. 2007, 113, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Bataller, R.; Gäbele, E.; Parsons, C.J.; Morris, T.; Yang, L.; Schoonhoven, R.; Brenner, D.A.; Rippe, R.A. Systemic infusion of angiotensin II exacerbates liver fibrosis in bile duct-ligated rats. Hepatology 2005, 41, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Hirose, A.; Ono, M.; Saibara, T.; Nozaki, Y.; Masuda, K.; Yoshioka, A.; Takahashi, M.; Akisawa, N.; Iwasaki, S.; Oben, J.A.; et al. Angiotensin II type 1 receptor blocker inhibits fibrosis in rat nonalcoholic steatohepatitis. Hepatology 2007, 45, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Kurita, S.; Takamura, T.; Ota, T.; Matsuzawa-Nagata, N.; Kita, Y.; Uno, M.; Nabemoto, S.; Ishikura, K.; Misu, H.; Ando, H.; et al. Olmesartan ameliorates a dietary rat model of non-alcoholic steatohepatitis through its pleiotropic effects. Eur. J. Pharmacol. 2008, 588, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaji, K.; Yoshiji, H.; Kitade, M.; Ikenaka, Y.; Noguchi, R.; Shirai, Y.; Aihara, Y.; Namisaki, T.; Yoshii, J.; Yanase, K.; et al. Combination treatment of angiotensin II type I receptor blocker and new oral iron chelator attenuates progression of nonalcoholic steatohepatitis in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G1094–G1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokohama, S.; Yoneda, M.; Haneda, M.; Okamoto, S.; Okada, M.; Aso, K.; Hasegawa, T.; Tokusashi, Y.; Miyokawa, N.; Nakamura, K. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology 2004, 40, 1222–1225. [Google Scholar] [CrossRef]
- Shirai, Y.; Oda, S.; Makino, S.; Tsuneyama, K.; Yokoi, T. Establishment of a mouse model of enalapril-induced liver injury and investigation of the pathogenesis. Lab. Investig. 2017, 97, 833–842. [Google Scholar] [CrossRef]
- da Silva, G.H.; Ribeiro Alves, A.V.F.; Duques, P.; Sevá-Pereira, T.; Soares, E.C.; Fazzio Escanhoela, C.A. View of Acute Hepatotoxicity Caused by Enalapril: A Case Report. Available online: https://www.jgld.ro/jgld/index.php/jgld/article/view/2010.2.10/925 (accessed on 7 September 2021).
- Babany, G.; Uzzan, F.; Larrey, D.; Degott, C.; Bourgeois, P.; René, E.; Vissuzaine, C.; Erlinger, S.; Benhamou, J.-P. Alcoholic-like liver lesions induced by nifedipine. J. Hepatol. 1989, 9, 252–255. [Google Scholar] [CrossRef]
- Nakagami, H.; Shimamura, M.; Miyake, T.; Shimosato, T.; Minobe, N.; Moritani, T.; Osako, M.K.; Nakagami, F.; Koriyama, H.; Kyutoku, M.; et al. Nifedipine prevents hepatic fibrosis in a non-alcoholic steatohepatitis model induced by an L-methionine-and choline-deficient diet. Mol. Med. Rep. 2012, 5, 37–40. [Google Scholar]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as metabolic regulators in the liver: Lessons from liver-specific PPAR-null mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [Green Version]
- Miyahara, T.; Schrum, L.; Rippe, R.; Xiong, S.; Yee, J.; Motomura, K.; Anania, F.A.; Willson, T.M.; Tsukamoto, H. Peroxisome Proliferator-activated Receptors and Hepatic Stellate Cell Activation *. J. Biol. Chem. 2000, 275, 35715–35722. [Google Scholar] [CrossRef] [Green Version]
- Bae, M.A.; Rhee, S.D.; Jung, W.H.; Ahn, J.H.; Song, B.J.; Cheon, H.G. Selective inhibition of activated stellate cells and protection from carbon tetrachloride-induced liver injury in rats by a new PPARgamma agonist KR62776. Arch. Pharm. Res. 2010, 33, 433–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westphal, J.F.; Vetter, D.; Brogard, J.M. Hepatic side-effects of antibiotics. J. Antimicrob. Chemother. 1994, 33, 387–401. [Google Scholar] [CrossRef]
- Fromenty, B. Alteration of mitochondrial DNA homeostasis in drug-induced liver injury. Food Chem. Toxicol. 2020, 135, 110916. [Google Scholar] [CrossRef]
- Fréneaux, E.; Labbe, G.; Letteron, P.; Le Dinh, T.; Degott, C.; Genève, J.; Larrey, D.; Pessayre, D. Inhibition of the mitochondrial oxidation of fatty acids by tetracycline in mice and in man: Possible role in microvesicular steatosis induced by this antibiotic. Hepatology 1988, 8, 1056–1062. [Google Scholar] [CrossRef]
- Szalowska, E.; van der Burg, B.; Man, H.-Y.; Hendriksen, P.J.M.; Peijnenburg, A.A.C.M. Model Steatogenic Compounds (Amiodarone, Valproic Acid, and Tetracycline) Alter Lipid Metabolism by Different Mechanisms in Mouse Liver Slices. PLoS ONE 2014, 9, e86795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breen, K.; Schenker, S.; Heimberg, M. The effect of tetracycline on the hepatic secretion of triglyceride. Biochim. Biophys. Acta-Lipids Lipid Metab. 1972, 270, 74–80. [Google Scholar] [CrossRef]
- Lettéron, P.; Sutton, A.; Mansouri, A.; Fromenty, B.; Pessayre, D. Inhibition of microsomal triglyceride transfer protein: Another mechanism for drug-induced steatosis in mice. Hepatology 2003, 38, 133–140. [Google Scholar] [CrossRef]
- Yin, H.Q.; Kim, M.; Kim, J.H.; Kong, G.; Lee, M.O.; Kang, K.S.; Yoon, B.I.L.; Kim, H.L.; Lee, B.H. Hepatic gene expression profiling and lipid homeostasis in mice exposed to seatogenic drug, tetracycline. Toxicol. Sci. 2006, 94, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.-J.; Lee, C.-H.; Lee, K.-Y.; Jung, S.-H.; Lee, B.-H. Increased Hepatic Fatty Acid Uptake and Esterification Contribute to Tetracycline-Induced Steatosis in Mice. Toxicol. Sci. 2015, 145, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Brüning, A.; Brem, G.J.; Vogel, M.; Mylonas, I. Tetracyclines cause cell stress-dependent ATF4 activation and mTOR inhibition. Exp. Cell Res. 2014, 320, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Yan, S.; Hu, H.; Duan, Z.; Yin, L.; Liao, S.; Sun, Y.; Yin, D.; Li, G. Proteomic profile of carbonylated proteins in rat liver: Discovering possible mechanisms for tetracycline-induced steatosis. Proteomics 2015, 15, 148–159. [Google Scholar] [CrossRef]
- Santini, A.; Ronchi, D.; Garbellini, M.; Piga, D.; Protti, A. Linezolid-induced lactic acidosis: The thin line between bacterial and mitochondrial ribosomes. Expert Opin. Drug Saf. 2017, 16, 833–843. [Google Scholar] [CrossRef]
- Vinh, D.C.; Rubinstein, E. Linezolid: A review of safety and tolerability. J. Infect. 2009, 59, S59–S74. [Google Scholar] [CrossRef]
- Leach, K.L.; Swaney, S.M.; Colca, J.R.; McDonald, W.G.; Blinn, J.R.; Thomasco, L.M.M.; Gadwood, R.C.; Shinabarger, D.; Xiong, L.; Mankin, A.S. The Site of Action of Oxazolidinone Antibiotics in Living Bacteria and in Human Mitochondria. Mol. Cell 2007, 26, 393–402. [Google Scholar] [CrossRef] [PubMed]
- De Vriese, A.S.; Van Coster, R.; Smet, J.; Seneca, S.; Lovering, A.; Van Haute, L.L.; Vanopdenbosch, L.J.; Martin, J.-J.; Ceuterick-de Groote, C.; Vandecasteele, S.; et al. Linezolid-Induced Inhibition of Mitochondrial Protein Synthesis. Clin. Infect. Dis. 2006, 42, 1111–1117. [Google Scholar] [CrossRef]
- Garrabou, G.; Soriano, A.; López, S.; Guallar, J.P.; Giralt, M.; Villarroya, F.; Martínez, J.A.; Casademont, J.; Cardellach, F.; Mensa, J.; et al. Reversible inhibition of mitochondrial protein synthesis during linezolid-related hyperlactatemia. Antimicrob. Agents Chemother. 2007, 51, 962–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Guillou, D.; Bucher, S.; Begriche, K.; Hoët, D.; Lombès, A.; Labbe, G.; Fromenty, B. Drug-Induced Alterations of Mitochondrial DNA Homeostasis in Steatotic and Nonsteatotic HepaRG Cells. J. Pharmacol. Exp. Ther. 2018, 365, 711–726. [Google Scholar] [CrossRef]
- De Bus, L.; Depuydt, P.; Libbrecht, L.; Vandekerckhove, L.; Nollet, J.; Benoit, D.; Vogelaers, D.; Van Vlierberghe, H. Severe Drug-induced Liver Injury Associated with Prolonged Use of Linezolid. J. Med. Toxicol. 2010, 6, 322–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.-H.; Zhang, C.; Zhang, D.-G.; Li, L.; Chen, X.; Xu, D.-X. Rifampicin-Induced Hepatic Lipid Accumulation: Association with Up-Regulation of Peroxisome Proliferator-Activated Receptor γ in Mouse Liver. PLoS ONE 2016, 11, e0165787. [Google Scholar] [CrossRef] [PubMed]
- Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2012, 1821, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Pieper-Fürst, U.; Lammert, F. Low-density lipoprotein receptors in liver: Old acquaintances and a newcomer. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2013, 1831, 1191–1198. [Google Scholar] [CrossRef]
- Allard, J.; Bucher, S.; Massart, J.; Ferron, P.-J.; Le Guillou, D.; Loyant, R.; Daniel, Y.; Launay, Y.; Buron, N.; Begriche, K.; et al. Drug-induced hepatic steatosis in absence of severe mitochondrial dysfunction in HepaRG cells: Proof of multiple mechanism-based toxicity. Cell Biol. Toxicol. 2020, 37, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Tamoxifen: A most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2003, 2, 205–213. [Google Scholar] [CrossRef]
- Meunier, L.; Larrey, D. Chemotherapy-associated steatohepatitis. Ann. Hepatol. 2020, 19, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Saphner, T.; Triest-Robertson, S.; Li, H.; Holzman, P. The association of nonalcoholic steatohepatitis and tamoxifen in patients with breast cancer. Cancer 2009, 115, 3189–3195. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Maisonneuve, P.; Castellana, P.; Rotmensz, N.; Rossi, S.; Maggioni, M.; Persico, M.; Colombo, A.; Monasterolo, F.; Casadei-Giunchi, D.; et al. Incidence and risk factors for non-alcoholic steatohepatitis: Prospective study of 5408 women enrolled in Italian tamoxifen chemoprevention trial. BMJ 2005, 330, 932. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.T.; Pan, H.J.; Lee, C.H. Prevention of Tamoxifen-related Nonalcoholic Fatty Liver Disease in Breast Cancer Patients. Clin. Breast Cancer 2018, 18, e677–e685. [Google Scholar] [CrossRef]
- Andrade, R.J.; Aithal, G.P.; Björnsson, E.S.; Kaplowitz, N.; Kullak-Ublick, G.A.; Larrey, D.; Karlsen, T.H. EASL Clinical Practice Guidelines: Drug-induced liver injury. J. Hepatol. 2019, 70, 1222–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, S.; Holmäng, A.; Björntorp, P. The effects of oestrogen and progesterone on insulin sensitivity in female rats. Acta Physiol. Scand. 1993, 149, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Riant, E.; Waget, A.; Cogo, H.; Arnal, J.F.; Burcelin, R.; Gourdy, P. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology 2009, 150, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Margolis, K.L.; Bonds, D.E.; Rodabough, R.J.; Tinker, L.; Phillips, L.S.; Allen, C.; Bassford, T.; Burke, G.; Torrens, J.; Howard, B.V. Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: Results from the Women’s Health Initiative Hormone Trial. Diabetologia 2004, 47, 1175–1187. [Google Scholar] [CrossRef]
- McKenzie, J.; Fisher, B.M.; Jaap, A.J.; Stanley, A.; Paterson, K.; Sattar, N. Effects of HRT on liver enzyme levels in women with type 2 diabetes: A randomized placebo-controlled trial. Clin. Endocrinol. 2006, 65, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Venetsanaki, V.; Polyzos, S.A. Menopause and Non-Alcoholic Fatty Liver Disease: A Review Focusing on Therapeutic Perspectives. Curr. Vasc. Pharmacol. 2018, 17, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; Nguyen, M.T.A.; Henstridge, D.C.; Nguyen, A.K.; Beaven, S.W.; Watt, M.J.; Hevener, A.L. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERα-deficient mice. Am. J. Physiol.-Endocrinol. Metab. 2010, 298, E304–E319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, J.D.Y.; Jones, M.E.E.; Prelle, K.; Simpson, E.R.; Boon, W.C. A selective estrogen receptor α agonist ameliorates hepatic steatosis in the male aromatase knockout mouse. J. Endocrinol. 2011, 210, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Ponnusamy, S.; Tran, Q.T.; Thiyagarajan, T.; Miller, D.D.; Bridges, D.; Narayanan, R. An estrogen receptor β-selective agonist inhibits non-alcoholic steatohepatitis in preclinical models by regulating bile acid and xenobiotic receptors. Exp. Biol. Med. 2017, 242, 606–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Q.; Delannoy, M.; Cooke, C.; Yager, J.D. Mitochondrial localization of ERα and ERβ in human MCF7 cells. American J. Physiol.-Endocrinol. Metab. 2004, 286, 1011–1022. [Google Scholar] [CrossRef]
- Maher, A.C.; Akhtar, M.; Tarnopolsky, M.A. Men supplemented with 17β-estradiol have increased β-oxidation capacity in skeletal muscle. Physiol. Genom. 2010, 42, 342–347. [Google Scholar] [CrossRef]
- Campbell, S.E.; Mehan, K.A.; Tunstall, R.J.; Febbraio, M.A.; Cameron-Smith, D. 17β-Estradiol upregulates the expression of peroxisome proliferator-activated receptor α and lipid oxidative genes in skeletal muscle. J. Mol. Endocrinol. 2003, 31, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zhou, J.; Du, Y. Estrogen receptor alpha interacts with mitochondrial protein HADHB and affects beta-oxidation activity. Mol. Cell. Proteomics 2012, 11, M111-011056. [Google Scholar] [CrossRef] [Green Version]
- Larosche, I.; Lettéron, P.; Fromenty, B.; Vadrot, N.; Abbey-Toby, A.; Feldmann, G.; Pessayre, D.; Mansouri, A. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J. Pharmacol. Exp. Ther. 2007, 321, 526–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelliott, C.J.; López, M.; Curtis, R.K.; Parker, N.; Laudes, M.; Yeo, G.; Jimenez-Liñan, M.; Grosse, J.; Saha, A.K.; Wiggins, D.; et al. Transcript and metabolite analysis of the effects of tamoxifen in rat liver reveals inhibition of fatty acid synthesis in the presence of hepatic steatosis. FASEB J. 2005, 19, 1108–1119. [Google Scholar] [CrossRef]
- Zhao, F.; Xie, P.; Jiang, J.; Zhang, L.; An, W.; Zhan, Y. The Effect and Mechanism of Tamoxifen-Induced Hepatocyte Steatosis in Vitro. Int. J. Mol. Sci. 2014, 15, 4019–4030. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, T.; Ogawa, Y.; Saibara, T.; Nishioka, A.; Kariya, S.; Fukumoto, M.; Onishi, S.; Yoshida, S. CYP17 polymorphism and tamoxifen-induced hepatic steatosis. Hepatol. Res. 2005, 33, 178–180. [Google Scholar] [CrossRef]
- Yang, Y.J.; Kim, K.M.; An, J.H.; Lee, D.B.; Shim, J.H.; Lim, Y.S.; Lee, H.C.; Lee, Y.S.; Ahn, J.H.; Jung, K.H.; et al. Clinical significance of fatty liver disease induced by tamoxifen and toremifene in breast cancer patients. Breast 2016, 28, 67–72. [Google Scholar] [CrossRef]
- Hamada, N.; Ogawa, Y.; Saibara, T.; Murata, Y.; Kariya, S.; Nishioka, A.; Terashima, M.; Inomata, T.; Yoshida, S. Toremifene-induced fatty liver and NASH in breast cancer patients with breast-conservation treatment. Int. J. Oncol. 2000, 17, 1119–1142. [Google Scholar] [CrossRef]
- Robinson, S.M.; Wilson, C.H.; Burt, A.D.; Manas, D.M.; White, S.A. Chemotherapy-Associated Liver Injury in Patients with Colorectal Liver Metastases: A Systematic Review and Meta-analysis. Ann. Surg. Oncol. 2012, 19, 4287–4299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vauthey, J.-N.; Pawlik, T.M.; Ribero, D.; Wu, T.-T.; Zorzi, D.; Hoff, P.M.; Xiong, H.Q.; Eng, C.; Lauwers, G.Y.; Mino-Kenudson, M.; et al. Chemotherapy Regimen Predicts Steatohepatitis and an Increase in 90-Day Mortality After Surgery for Hepatic Colorectal Metastases. J. Clin. Oncol. 2016, 24, 2065–2072. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Laurent, A.; Maru, D.; Vauthey, J.N. Management of chemotherapy-associated hepatotoxicity in colorectal liver metastases. Lancet Oncol. 2009, 10, 278–286. [Google Scholar] [CrossRef]
- Fernandez, F.G.; Ritter, J.; Goodwin, J.W.; Linehan, D.C.; Hawkins, W.G.; Strasberg, S.M. Effect of steatohepatitis associated with irinotecan or oxaliplatin pretreatment on resectability of hepatic colorectal metastases. J. Am. Coll. Surg. 2005, 200, 845–853. [Google Scholar] [CrossRef]
- Pilgrim, C.H.C.; Thomson, B.N.; Banting, S.; Phillips, W.A.; Michael, M. The developing clinical problem of chemotherapy-induced hepatic injury. ANZ J. Surg. 2012, 82, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, K.; Rickenbacher, A.; Weber, A.; Pestalozzi, B.C.; Clavien, P.A. Chemotherapy before liver resection of colorectal metastases: Friend or foe? Ann. Surg. 2012, 255, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Sommer, J.; Mahli, A.; Freese, K.; Schiergens, T.S.; Kuecuekoktay, F.S.; Teufel, A.; Thasler, W.E.; Müller, M.; Bosserhoff, A.K.; Hellerbrand, C. Analysis of molecular mechanisms of 5-fluorouracil-induced steatosis and inflammation in vitro and in mice. Oncotarget 2017, 8, 13059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, S.; Than, T.A.; Le, B.H.A.; García-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Win, S.; Than, T.A.; Zhang, J.; Oo, C.; Min, R.W.M.; Kaplowitz, N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 2018, 67, 2013–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, S.; Than, T.A.; Min, R.W.M.; Aghajan, M.; Kaplowitz, N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 2016, 63, 1987–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, J.W.; LoGrasso, P.V. Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem. 2011, 286, 16052–16062. [Google Scholar] [CrossRef] [Green Version]
- Win, S.; Min, R.W.M.; Zhang, J.; Kanel, G.; Wanken, B.; Chen, Y.; Li, M.; Wang, Y.; Suzuki, A.; Aung, F.W.M.; et al. Hepatic Mitochondrial SAB Deletion or Knockdown Alleviates Diet-Induced Metabolic Syndrome, Steatohepatitis, and Hepatic Fibrosis. Hepatology 2021, 74, 3127–3145. [Google Scholar] [CrossRef]
- Pilgrim, C.H.C.; Brettingham-Moore, K.; Pham, A.; Murray, W.; Link, E.; Smith, M.; Usatoff, V.; Evans, P.M.; Banting, S.; Thomson, B.N.; et al. MRNA gene expression correlates with histologically diagnosed chemotherapy-induced hepatic injury. HPB 2011, 13, 811–816. [Google Scholar] [CrossRef] [Green Version]
- McWhirter, D.; Kitteringham, N.; Jones, R.P.; Malik, H.; Park, K.; Palmer, D. Chemotherapy induced hepatotoxicity in metastatic colorectal cancer: A review of mechanisms and outcomes. Crit. Rev. Oncol. Hematol. 2013, 88, 404–415. [Google Scholar] [CrossRef]
- Horowitz, B.; Madras, B.K.; Meister, A.; Old, L.J.; Boyse, E.A.; Stockert, E. Asparagine synthetase activity of mouse leukemias. Science 1968, 160, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Oettgen, H.F.; Tallal, L.; Tan, C.C.; Murphy, M.L.; Clarkson, B.D.; Golbey, R.D.; Krakoff, I.H.; Karnofsky, D.A.; Burchenal, H.J. Clinical experience with L-asparaginase. Recent Results Cancer Res. 1970, 33, 219–235. [Google Scholar] [PubMed]
- Biggs, J.C.; Chestermant, C.N.; Holliday, J. L-asparaginase—Clinical Experience in Leukaemia, Lymphoma and Carcinoma. Aust. N. Z. J. Med. 1971, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Distasio, J.A.; Salazar, A.M.; Nadji, M.; Durden, D.L. Glutaminase-free asparaginase from vibrio succinogenes: An antilymphoma enzyme lacking hepatotoxicity. Int. J. Cancer 1982, 30, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Hart, J. Hstopathological features of L-asparaginase-induced liver disease. Semin. Liver Dis. 2003, 23, 295–299. [Google Scholar]
- Bodmer, M.; Sulz, M.; Stadlmann, S.; Droll, A.; Terracciano, L.; Krähenbühl, S. Fatal liver failure in an adult patient with acute lymphoblastic leukemia following treatment with L-asparaginase. Digestion 2006, 74, 28–32. [Google Scholar] [CrossRef]
- Leonard, J.V.; Kay, J.D.S. Acute encephalopathy and hyperammonaemia complicating treatment of acute lymphoblastic leukaemia with asparaginase. Lancet 1986, 327, 162–163. [Google Scholar] [CrossRef]
- Lee, A.U.; Farrell, G.C. Mechanism of azathioprine-induced injury to hepatocytes: Roles of glutathione depletion and mitochondrial injury. J. Hepatol. 2001, 35, 756–764. [Google Scholar] [CrossRef]
- Vairetti, M.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Ferrigno, A.; Berardo, C. Changes in Glutathione Content in Liver Diseases: An Update. Antioxidants 2021, 10, 364. [Google Scholar] [CrossRef] [PubMed]
- Raabe, M.; Véniant, M.M.; Sullivan, M.A.; Zlot, C.H.; Björkegren, J.; Nielsen, L.B.; Wong, J.S.; Hamilton, R.L.; Young, S.G. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J. Clin. Investig. 1999, 103, 1287–1298. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.H.; Schiff, E. Methotrexate-induced chronic liver injury: Guidelines for detection and prevention. The ACG Committee on FDA-related matters. American College of Gastroenterology. Am. J. Gastroenterol. 1988, 83, 1337–13345. [Google Scholar] [PubMed]
- Yamamoto, N.; Oliveira, M.B.M.; Campello, A.D.P.; Lopes, L.C.V.; Klüppel, M.L.W. Methotrexate: Studies on the cellular metabolism. I. Effect on mitochondrial oxygen uptake and oxidative phosphorylation. Cell Biochem. Funct. 1988, 6, 61–66. [Google Scholar] [CrossRef]
- Caetano, N.N.; Campello, A.P.; Carnieri, E.G.S.; Kluppel, M.L.W.; Oliveira, M.B.M. Effect of methotrexate (MTX) on NAD(P)+ dehydrogenases of HeLa cells: Malic enzyme, 2-oxoglutarate and isocitrate dehydrogenases. Cell Biochem. Funct. 1997, 15, 259–264. [Google Scholar] [CrossRef]
- Kim, J.; Lowe, K.; Chemistry, B.S.-J. Regulation of folate and one-carbon metabolism in mammalian cells. IV. Role of folylpoly-gamma-glutamate synthetase in methotrexate metabolism and cytotoxicity. J. Biol. Chem. 1993, 268, 21680–21685. [Google Scholar] [CrossRef]
- Huang, C.C.; Hsu, P.C.; Hung, Y.C.; Liao, Y.F.; Liu, C.C.; Hour, C.T.; Kao, M.C.; Tsay, G.J.; Hung, H.C.; Liu, G.Y. Ornithine decarboxylase prevents methotrexate-induced apoptosis by reducing intracellular reactive oxygen species production. Apoptosis 2005, 10, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, H.; Parvez, S.; Pasha, S.T.; Banerjee, B.D.; Raisuddin, S. Protective effect of lipoic acid against methotrexate-induced oxidative stress in liver mitochondria. Food Chem. Toxicol. 2010, 48, 1973–1979. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Shi, B.; Xue, H.; Li, Y.; Yang, X.; Yu, B.; Xu, Z.; Liu, F.; Li, J. Confirmation and Prevention of Intestinal Barrier Dysfunction and Bacterial Translocation Caused by Methotrexate. Dig. Dis. Sci. 2006, 51, 1549–1556. [Google Scholar] [CrossRef]
- Cronstein, B.N.; Eberle, M.A.; Gruber, H.E.; Levin, R.I. Methotrexate inhibits neutrophil function by stimulating adenosine release from connective tissue cells. Proc. Natl. Acad. Sci. USA 1991, 88, 2441–2445. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.S.L.; Montesinos, M.C.; Fernandez, P.; Desai, A.; Delano, D.L.; Yee, H.; Reiss, A.B.; Pillinger, M.H.; Chen, J.F.; Schwarzschild, M.A.; et al. Adenosine A 2A receptors play a role in the pathogenesis of hepatic cirrhosis. Br. J. Pharmacol. 2006, 148, 1144–1155. [Google Scholar] [CrossRef] [Green Version]
- Romoli, M.; Mazzocchetti, P.; D’Alonzo, R.; Siliquini, S.; Rinaldi, V.E.; Verrotti, A.; Calabresi, P.; Costa, C. Valproic Acid and Epilepsy: From Molecular Mechanisms to Clinical Evidences. Curr. Neuropharmacol. 2018, 17, 926–946. [Google Scholar] [CrossRef]
- Rakitin, A. Does valproic acid have potential in the treatment of diabetes mellitus? Front. Endocrinol. 2017, 8, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, B.L.; Craycraft, L.K.; Justice, S.B. Valproic Acid in the Treatment of Migraines. Adv. Emerg. Nurs. J. 2020, 42, 243–253. [Google Scholar] [CrossRef]
- Elshafay, A.; Hieu, T.H.; Doheim, M.F.; Kassem, M.A.M.; ELdoadoa, M.F.; Holloway, S.K.; Abo-elghar, H.; Hirayama, K.; Huy, N.T. Efficacy and Safety of Valproic Acid for Spinal Muscular Atrophy: A Systematic Review and Meta-Analysis. CNS Drugs 2019, 33, 239–250. [Google Scholar] [CrossRef]
- Laengle, J.; Kabiljo, J.; Hunter, L.; Homola, J.; Prodinger, S.; Egger, G.; Bergmann, M. Histone deacetylase inhibitors valproic acid and vorinostat enhance trastuzumab-mediated antibody-dependent cell-mediated phagocytosis. J. Immunother. Cancer 2020, 8, e000195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caponigro, F.; Di Gennaro, E.; Ionna, F.; Longo, F.; Aversa, C.; Pavone, E.; Maglione, M.G.; Di Marzo, M.; Muto, P.; Cavalcanti, E.; et al. Phase II clinical study of valproic acid plus cisplatin and cetuximab in recurrent and/or metastatic squamous cell carcinoma of Head and Neck-V-CHANCE trial. BMC Cancer 2016, 16, 918. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.; Ren, Q.; Zuo, W.; Jia, R.; Xie, L.; Lin, R.; Zhao, H.; Chen, J.; Lei, Y.; Wang, P.; et al. Valproic acid exhibits anti-tumor activity selectively against EGFR/ErbB2/ErbB3-coexpressing pancreatic cancer via induction of ErbB family members-targeting microRNAs. J. Exp. Clin. Cancer Res. 2019, 38, 150. [Google Scholar] [CrossRef]
- Farinelli, E.; Giampaoli, D.; Cenciarini, A.; Cercado, E.; Verrotti, A. Valproic acid and nonalcoholic fatty liver disease: A possible association? World J. Hepatol. 2015, 7, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-L.; Jing, X.; Sun, J.-Y.; Hu, Y.; Xu, Z.-J.; Ni, M.-M.; Chen, F.; Lu, X.-P.; Qiu, J.-C.; Wang, T. Valproic Acid and the Liver Injury in Patients with Epilepsy: An Update. Curr. Pharm. Des. 2019, 25, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Bowden, C. Valproate. Natl. Inst. Diabetes Dig. Kidney Dis. 2003, 5, 189–202. [Google Scholar] [CrossRef]
- Aires, C.C.P.; IJlst, L.; Stet, F.; Prip-Buus, C.; de Almeida, I.T.; Duran, M.; Wanders, R.J.A.; Silva, M.F.B. Inhibition of hepatic carnitine palmitoyl-transferase I (CPT IA) by valproyl-CoA as a possible mechanism of valproate-induced steatosis. Biochem. Pharmacol. 2010, 79, 792–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aires, C.C.P.; Soveral, G.; Luís, P.B.M.; ten Brink, H.J.; de Almeida, I.T.; Duran, M.; Wanders, R.J.A.; Silva, M.F.B. Pyruvate uptake is inhibited by valproic acid and metabolites in mitochondrial membranes. FEBS Lett. 2008, 582, 3359–3366. [Google Scholar] [CrossRef] [Green Version]
- Caiment, F.; Wolters, J.; Smit, E.; Schrooders, Y.; Kleinjans, J.; van den Beucken, T. Valproic acid promotes mitochondrial dysfunction in primary human hepatocytes in vitro; impact of C/EBPα-controlled gene expression. Arch. Toxicol. 2020, 94, 3463–3473. [Google Scholar] [CrossRef]
- van Breda, S.G.J.; Claessen, S.M.H.; van Herwijnen, M.; Theunissen, D.H.J.; Jennen, D.G.J.; de Kok, T.M.C.M.; Kleinjans, J.C.S. Integrative omics data analyses of repeated dose toxicity of valproic acid in vitro reveal new mechanisms of steatosis induction. Toxicology 2018, 393, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Pourahmad, J.; Eskandari, M.R.; Kaghazi, A.; Shaki, F.; Shahraki, J.; Fard, J.K. A new approach on valproic acid induced hepatotoxicity: Involvement of lysosomal membrane leakiness and cellular proteolysis. Toxicol. Vitr. 2012, 26, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Palsamy, P.; Bidasee, K.R.; Shinohara, T. Valproic acid suppresses Nrf2/Keap1 dependent antioxidant protection through induction of endoplasmic reticulum stress and Keap1 promoter DNA demethylation in human lens epithelial cells. Exp. Eye Res. 2014, 121, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahangar, N.; Naderi, M.; Noroozi, A.; Ghasemi, M.; Zamani, E.; Shaki, F. Zinc Deficiency and Oxidative Stress Involved in Valproic Acid Induced Hepatotoxicity: Protection by Zinc and Selenium Supplementation. Biol. Trace Elem. Res. 2017, 179, 102–109. [Google Scholar] [CrossRef]
- Gai, Z.; Krajnc, E.; Samodelov, S.L.; Visentin, M.; Kullak-Ublick, G.A. Obeticholic acid ameliorates valproic acid-induced hepatic steatosis and oxidative stress. Mol. Pharmacol. 2020, 97, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Grieco, A.; Alfei, B.; Di Rocco, P.; Miele, L.; Biolcati, G.; Griso, D.; Vecchio, F.M.; Bianco, A.; Gasbarrini, G. Non-alcoholic steatohepatitis induced by carbamazepine and variegate porphyria. Eur. J. Gastroenterol. Hepatol. 2001, 13, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Lillibridge, J.H.; Amore, B.M.; Slattery, J.T.; Kalhorn, T.F.; Nelson, S.D.; Finnell, R.H.; Bennett, G.D. Protein-reactive metabolites of carbamazepine in mouse liver microsomes. Drug Metab. Dispos. 1996, 24, 509–514. [Google Scholar]
- Hamed, S.A.; Fathy, R.A.; Radwan, M.E.; Abdellah, M.M. Fatty liver in adults receiving antiepileptic medications: Relationship to the metabolic risks. Expert Rev. Clin. Pharmacol. 2016, 9, 617–624. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory Action of Glucocorticoids—New Mechanisms for Old Drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [Green Version]
- Schäcke, H.; Döcke, W.D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids-Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15. [Google Scholar] [CrossRef] [Green Version]
- Rahimi, L.; Rajpal, A.; Ismail-Beigi, F. Glucocorticoid-Induced Fatty Liver Disease. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 1133. [Google Scholar] [CrossRef] [Green Version]
- Shibli-Rahhal, A.; Van Beek, M.; Schlechte, J.A. Cushing’s syndrome. Clin. Dermatol. 2006, 24, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Chanson, P.; Salenave, S. Metabolic syndrome in Cushing’s syndrome. Neuroendocrinology 2010, 92, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Uddén, J.; Björntorp, P.; Arner, P.; Barkeling, B.; Meurling, L.; Rössner, S. Effects of glucocorticoids on leptin levels and eating behaviour in women. J. Intern. Med. 2003, 253, 225–231. [Google Scholar] [CrossRef]
- Solano, J.M.; Jacobson, L. Glucocorticoids reverse leptin effects on food intake and body fat in mice without increasing NPY mRNA. Am. J. Physiol.-Endocrinol. Metab. 1999, 277, E708–E716. [Google Scholar] [CrossRef]
- Perry, R.J.; Resch, J.M.; Douglass, A.M.; Madara, J.C.; Rabin-Court, A.; Kucukdereli, H.; Wu, C.; Song, J.D.; Lowell, B.B.; Shulman, G.I. Leptin’s hunger-suppressing effects are mediated by the hypothalamic–pituitary–adrenocortical axis in rodents. Proc. Natl. Acad. Sci. USA 2019, 116, 13670–13679. [Google Scholar] [CrossRef] [Green Version]
- Ishida-Takahashi, R.; Uotani, S.; Abe, T.; Degawa-Yamauchi, M.; Fukushima, T.; Fujita, N.; Sakamaki, H.; Yamasaki, H.; Yamaguchi, Y.; Eguchi, K. Rapid Inhibition of Leptin Signaling by Glucocorticoids in Vitro and in Vivo. J. Biol. Chem. 2004, 279, 19658–19664. [Google Scholar] [CrossRef] [Green Version]
- Weickert, M.O.; Pfeiffer, A.F.H. Signalling mechanisms linking hepatic glucose and lipid metabolism. Diabetologia 2006, 49, 1732–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Dentin, R.; Pégorier, J.P.; Benhamed, F.; Foufelle, F.; Ferré, P.; Fauveau, V.; Magnuson, M.A.; Girard, J.; Postic, C. Hepatic Glucokinase Is Required for the Synergistic Action of ChREBP and SREBP-1c on Glycolytic and Lipogenic Gene Expression. J. Biol. Chem. 2004, 279, 20314–20326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; He, Q.; Xu, H. FOXO1-dependent up-regulation of MAP kinase phosphatase 3 (MKP-3) mediates glucocorticoid-induced hepatic lipid accumulation in mice. Mol. Cell. Endocrinol. 2014, 393, 46–55. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, A.M.; Beaudry, J.L.; Szigiato, A.A.; Trumble, S.J.; Snook, L.A.; Bonen, A.; Giacca, A.; Riddell, M.C. Consumption of a high-fat diet rapidly exacerbates the development of fatty liver disease that occurs with chronically elevated glucocorticoids. Am. J. Physiol.-Gastrointest. Liver Physiol. 2012, 302, G850–G863. [Google Scholar] [CrossRef] [Green Version]
- Dolinsky, V.W.; Douglas, D.N.; Lehner, R.; Vance, D.E. Regulation of the enzymes of hepatic microsomal triacylglycerol lipolysis and re-esterification by the glucocorticoid dexamethasone. Biochem. J. 2004, 378, 967–974. [Google Scholar] [CrossRef] [Green Version]
- Marino, J.S.; Stechschulte, L.A.; Stec, D.E.; Nestor-Kalinoski, A.; Coleman, S.; Hinds, T.D., Jr. Glucocorticoid Receptor β Induces Hepatic Steatosis by Augmenting Inflammation and Inhibition of the Peroxisome Proliferator-activated Receptor (PPAR) α. J. Biol. Chem. 2016, 291, 25776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Viswakarma, N.; Fu, T.; Yu, S.; Rao, M.S.; Borensztajn, J.; Reddy, J.K. Conditional ablation of mediator subunit MED1 (MED1/PPARBP) gene in mouse liver attenuates glucocorticoid receptor agonist dexamethasone-induced hepatic steatosis. Gene Expr. 2009, 14, 291–306. [Google Scholar] [CrossRef]
- Chen, T.-C.; Lee, R.A.; Tsai, S.L.; Kanamaluru, D.; Gray, N.E.; Yiv, N.; Cheang, R.T.; Tan, J.H.; Lee, J.Y.; Fitch, M.D.; et al. An ANGPTL4–ceramide–protein kinase Cζ axis mediates chronic glucocorticoid exposure–induced hepatic steatosis and hypertriglyceridemia in mice. J. Biol. Chem. 2019, 294, 9213–9224. [Google Scholar] [CrossRef]
- Utzeri, E.; Usai, P. Role of non-steroidal anti-inflammatory drugs on intestinal permeability and nonalcoholic fatty liver disease. World J. Gastroenterol. 2017, 23, 3954. [Google Scholar] [CrossRef]
- Bjarnason, I.; Scarpignato, C.; Holmgren, E.; Olszewski, M.; Rainsford, K.D.; Lanas, A. Mechanisms of Damage to the Gastrointestinal Tract from Nonsteroidal Anti-Inflammatory Drugs. Gastroenterology 2018, 154, 500–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adebayo, D.; Bjarnason, I. Is non-steroidal anti-inflammaory drug (NSAID) enteropathy clinically more important than NSAID gastropathy? Postgrad. Med. J. 2006, 82, 186–191. [Google Scholar] [CrossRef]
- Lanas, A.; Panes, J.; Pique, J. Clinical Implications of COX-1 and / or COX-2 Inhibition for the Distal Gastrointestinal Tract. Curr. Pharm. Des. 2005, 9, 2253–2266. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.Y.; Opekun, A.R.; Willingham, F.F.; Qureshi, W.A. Visible small-intestinal mucosal injury in chronic NSAID users. Clin. Gastroenterol. Hepatol. 2005, 3, 55–59. [Google Scholar] [CrossRef]
- Lanas, A.; Sopeña, F. Nonsteroidal Anti-Inflammatory Drugs and Lower Gastrointestinal Complications. Gastroenterol. Clin. N. Am. 2009, 38, 333–352. [Google Scholar] [CrossRef]
- Bjarnason, I.; Hayllar, J.; Macpherson, A.N.d.J.; Russell, A.N.t.S. Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology 1993, 104, 1832–1847. [Google Scholar] [CrossRef]
- Bessone, F. Non-steroidal anti-inflammatory drugs: What is the actual risk of liver damage? World J. Gastroenterol. 2010, 16, 5651. [Google Scholar] [CrossRef]
- Doi, H.; Horie, T. Salicylic acid-induced hepatotoxicity triggered by oxidative stress. Chem. Biol. Interact. 2010, 183, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nozaki, Y.; Wada, K.; Yoneda, M.; Endo, H.; Takahashi, H.; Iwasaki, T.; Inamori, M.; Abe, Y.; Kobayashi, N.; et al. Effectiveness of antiplatelet drugs against experimental non-alcoholic fatty liver disease. Gut 2008, 57, 1583–1591. [Google Scholar] [CrossRef]
- Ibrahim, M.; Farghaly, E.; Gomaa, W.; Kelleni, M.; Abdelrahman, A.M. Nitro-aspirin is a potential therapy for non alcoholic fatty liver disease. Eur. J. Pharmacol. 2011, 659, 289–295. [Google Scholar] [CrossRef]
- Shen, H.; Shahzad, G.; Jawairia, M.; Bostick, R.M.; Mustacchia, P. Association between aspirin use and the prevalence of nonalcoholic fatty liver disease: A cross-sectional study from the Third National Health and Nutrition Examination Survey. Aliment. Pharmacol. Ther. 2014, 40, 1066–1073. [Google Scholar] [CrossRef]
- Murohara, T.; Horowitz, J.R.; Silver, M.; Tsurumi, Y.; Chen, D.; Sullivan, A.; Isner, J.M. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation 1998, 97, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Sánchez De Miguel, L.; De Frutos, T.; González-Fernández, F.; Del Pozo, V.; Lahoz, C.; Jiménez, A.; Rico, L.; García, R.; Aceituno, E.; Millás, I.; et al. Aspirin inhibits inducible nitric oxide synthase expression and tumour necrosis factor-α release by cultured smooth muscle cells. Eur. J. Clin. Investig. 1999, 29, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.S.; Hughes, S.D.; Gilbertson, D.G.; Palmer, T.E.; Holdren, M.S.; Haran, A.C.; Odell, M.M.; Bauer, R.L.; Ren, H.P.; Haugen, H.S.; et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 3389–3394. [Google Scholar] [CrossRef] [Green Version]
- Tarantino, G.; Caputi, A. JNKs, insulin resistance and inflammation: A possible link between NAFLD and coronary artery disease. World J. Gastroenterol. 2011, 17, 3785–3794. [Google Scholar] [CrossRef]
- Simon, T.G.; Henson, J.; Osganian, S.; Masia, R.; Chan, A.T.; Chung, R.T.; Corey, K.E. Daily Aspirin Use Associated With Reduced Risk For Fibrosis Progression In Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2019, 17, 2776–2784. [Google Scholar] [CrossRef] [Green Version]
- García-Román, R.; Francés, R. Acetaminophen-Induced Liver Damage in Hepatic Steatosis. Clin. Pharmacol. Ther. 2020, 107, 1068–1081. [Google Scholar] [CrossRef]
- Kučera, O.; Al-Dury, S.; Lotková, H.; Roǔar, T.; Rychtrmoc, D.; Červinková, Z. Steatotic rat hepatocytes in primary culture are more susceptible to the acute toxic effect of acetaminophen. Physiol. Res. 2012, 61, S93. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Krausz, K.W.; Shah, Y.M.; Idle, J.R.; Gonzalez, F.J. Serum Metabolomics Reveals Irreversible Inhibition of Fatty Acid β-Oxidation through the Suppression of PPARα Activation as a Contributing Mechanism of Acetaminophen-Induced Hepatotoxicity. Chem. Res. Toxicol. 2009, 22, 699–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Pence, L.; Beger, R.; Chaudhuri, S.; McCullough, S.; Yan, K.; Simpson, P.; Hennings, L.; Hinson, J.; James, L. Acylcarnitine Profiles in Acetaminophen Toxicity in the Mouse: Comparison to Toxicity, Metabolism and Hepatocyte Regeneration. Metabolites 2013, 3, 606–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, M.R.; Li, F.; Sharpe, M.R.; Williams, C.D.; Curry, S.C.; Ma, X.; Jaeschke, H. Circulating acylcarnitines as biomarkers of mitochondrial dysfunction after acetaminophen overdose in mice and humans. Arch. Toxicol. 2013, 88, 391–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, K.G.; Carpenter, K.; Hammond, J.; Christodoulou, J.; Wilcken, B. Acylcarnitine profiles in fibroblasts from patients with respiratory chain defects can resemble those from patients with mitochondrial fatty acid [beta ]-oxidation disorders. Metabolism 2002, 51, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Aubert, J.; Begriche, K.; Delannoy, M.; Morel, I.; Pajaud, J.; Ribault, C.; Lepage, S.; McGill, M.R.; Lucas-Clerc, C.; Turlin, B.; et al. Differences in Early Acetaminophen Hepatotoxicity between Obese ob/ob and db/db Mice. J. Pharmacol. Exp. Ther. 2012, 342, 676–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Du, Y.; Yuan, X.; Han, X.; Dong, Z.; Chen, X.; Wu, H.; Zhang, J.; Xu, L.; Han, C.; et al. Hepatic hypoxia-inducible factors inhibit PPARα expression to exacerbate acetaminophen induced oxidative stress and hepatotoxicity. Free Radic. Biol. Med. 2017, 110, 102–116. [Google Scholar] [CrossRef]
- Geneve, J.; Hayat-Bonan, B.; Labbe, G.; Degott, C.; Letteron, P.; Freneaux, E.; Dinh, T.L.; Larrey, D.; Pessayre, D. Inhibition of mitochondrial beta-oxidation of fatty acids by pirprofen. Role in microvesicular steatosis due to this nonsteroidal anti-inflammatory drug. J. Pharmacol. Exp. Ther. 1987, 242, 1133–1137. [Google Scholar]
- Freneaux, E.; Fromenty, B.; Berson, A.; Labbe, G.; Degott, C.; Letteron, P.; Larrey, D.; Pessayre, D. Stereoselective and nonstereoselective effects of ibuprofen enantiomers on mitochondrial beta-oxidation of fatty acids. J. Pharmacol. Exp. Ther. 1990, 255. [Google Scholar]
- Caldwell, J.; Hutt, A.J.; Fournel-Gigleux, S. The metabolic chiral inversion and dispositional enantioselectivity of the 2-arylpropionic acids and their biological consequences. Biochem. Pharmacol. 1988, 37, 105–114. [Google Scholar] [CrossRef]
- Caldwell, J. Xenobiotic acyl-coenzymes A: Critical intermediates in the biochemical pharmacology and toxicology of carboxylic acids. Biochem. Soc. Trans. 1984, 12, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.; Day, R.; Knihinicki, R.; Duffield, A. The stereoselective uptake of ibuprofen enantiomers into adipose tissue. Biochem. Pharmacol. 1986, 35, 3403–3405. [Google Scholar] [CrossRef]
- Lu, W.; Cheng, F.; Jiang, J.; Zhang, C.; Deng, X.; Xu, Z.; Zou, S.; Shen, X.; Tang, Y.; Huang, J. FXR antagonism of NSAIDs contributes to drug-induced liver injury identified by systems pharmacology approach. Sci. Rep. 2015, 5, 8114. [Google Scholar] [CrossRef] [Green Version]
- Massart, J.; Begriche, K.; Buron, N.; Porceddu, M.; Borgne-Sanchez, A.; Fromenty, B. Drug-Induced Inhibition of Mitochondrial Fatty Acid Oxidation and Steatosis. Curr. Pathobiol. Rep. 2013, 1, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.; Sanyal, A.J. Drug-induced steatohepatitis. Clin. Liver Dis. 2013, 17, 533–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Famularo, G.; Gasbarrone, L.; Minisola, G. Probable ketoprofen-associated nonalcoholic fatty liver disease and steatohepatitis. Ann. Pharmacother. 2011, 45, 423. [Google Scholar] [CrossRef] [PubMed]
- Montessori, V.; Harris, M.; Montaner, J.S.G. Hepatotoxicity of nucleoside reverse transcriptase inhibitors. Semin. Liver Dis. 2003, 23, 167–171. [Google Scholar]
- Guaraldi, G.; Squillace, N.; Stentarelli, C.; Orlando, G.; D’Amico, R.; Ligabue, G.; Fiocchi, F.; Zona, S.; Loria, P.; Esposito, R.; et al. Nonalcoholic fatty liver disease in HIV-infected patients referred to a metabolic clinic: Prevalence, characteristics, and predictors. Clin. Infect. Dis. 2008, 47, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crum-Cianflone, N.; Dilay, A.; Collins, G.; Asher, D.; Campin, R.; Medina, S.; Goodman, Z.; Parker, R.; Lifson, A.; Capozza, T.; et al. Nonalcoholic Fatty Liver Disease (NAFLD) among HIV-Infected Persons. J. Acquir. Immune Defic. Syndr. 2009, 50, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furman, P.A.; Fyfe, J.A.; St Clair, M.H.; Weinhold, K.; Rideout, J.L.; Freeman, G.A.; Lehrman, S.N.; Bolognesi, D.P.; Broder, S.; Mitsuya, H. Phosphorylation of 3’-azido-3’-deoxythymidine and selective interaction of the 5’-triphosphate with human immunodeficiency virus reverse transcriptase. Proc. Natl. Acad. Sci. USA 1986, 83, 8333–8337. [Google Scholar] [CrossRef] [Green Version]
- Lewis, W.; Dalakas, M.C. Mitochondrial toxicity of antiviral drugs. Nat. Med. 1995, 1, 417–422. [Google Scholar] [CrossRef]
- Johnson, A.A.; Ray, A.S.; Hanes, J.; Suo, Z.; Colacino, J.M.; Anderson, K.S.; Johnson, K.A. Toxicity of Antiviral Nucleoside Analogs and the Human Mitochondrial DNA Polymerase. J. Biol. Chem. 2001, 276, 40847–40857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, J.C.; Thio, C.L. Liver Disease in the HIV-Infected Individual. Clin. Gastroenterol. Hepatol. 2010, 8, 1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Abdelmegeed, M.A.; Jang, S.; Song, B.-J. Zidovudine (AZT) and Hepatic Lipid Accumulation: Implication of Inflammation, Oxidative and Endoplasmic Reticulum Stress Mediators. PLoS ONE 2013, 8, e76850. [Google Scholar]
- Stankov, M.V.; Panayotova-Dimitrova, D.; Leverkus, M.; Vondran, F.W.R.; Bauerfeind, R.; Binz, A.; Behrens, G.M.N. Autophagy inhibition due to thymidine analogues as novel mechanism leading to hepatocyte dysfunction and lipid accumulation. AIDS 2012, 26, 1995–2006. [Google Scholar] [CrossRef] [PubMed]
- Coronel-Castillo, C.E.; Qi, X.; Contreras-Carmona, J.; Ramírez-Pérez, O.L.; Méndez-Sánchez, N. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in HIV infection: A metabolic approach of an infectious disease. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Eriksson, S.; Wang, L. Down-regulation of mitochondrial thymidine kinase 2 and deoxyguanosine kinase by didanosine: Implication for mitochondrial toxicities of anti-HIV nucleoside analogs. Biochem. Biophys. Res. Commun. 2014, 450, 1021–1026. [Google Scholar] [CrossRef]
- Hall, Z.; Bond, N.J.; Ashmore, T.; Sanders, F.; Ament, Z.; Wang, X.; Murray, A.J.; Bellafante, E.; Virtue, S.; Vidal-Puig, A.; et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology 2017, 65, 1165–1180. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Tiniakos, D.G. Histopathology of nonalcoholic fatty liver disease. World J. Gastroenterol. 2010, 16, 5286–5296. [Google Scholar] [CrossRef]
- Gasmi, B.; Kleiner, D.E. Liver Histology Diagnostic and Prognostic Features. Clin. Liver Dis. 2020, 24, 61. [Google Scholar] [CrossRef]
- Kleiner, D.E. Histopathological challenges in suspected drug-induced liver injury. Liver Int. 2018, 38, 198–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, S.; Arima, N.; Ito, M.; Fujiyama, S.; Kamo, Y.; Ueki, Y. Non-alcoholic steatohepatitis-like pattern in liver biopsy of rheumatoid arthritis patients with persistent transaminitis during low-dose methotrexate treatment. PLoS ONE 2018, 13, e0203084. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Category | Mechanisms |
|---|---|---|
| Amiodarone | antiarrhythmic | Inhibition of OXPHOS, FAO, CPT1 Inhibition of MRC complex I and III Induction of ER stress Upregulation of SREBP-1c, ACLY, FAS, SCD1 |
| Perhexiline | antiarrhythmic | Inhibition of OXPHOS, FAO, CPT1 Inhibition of MRC complex I and III |
| Diltiazem | antiarrhythmic | Conflicting data: observed steatosis with no described molecular mechanism |
| Verapamil | antiarrhythmic | Conflicting data: observed steatosis but also reduced inflammation, collagen deposition, lipid peroxidation, α-SMA and TGFβ1 |
| Losartan | antihypertensive | Amelioration of NAFLD: reduced markers of hepatic fibrosis |
| Enalapril | antihypertensive | Macro- and microvesicular steatosis with increased inflammation |
| Nifedipine | antihypertensive | Conflicting data are reported: increased enzyme release, macro- and microsteatosis with fibrosis; upregulation of PPARγ receptor with consequent reduction in NASH, fibrosis and AST release |
| Tetracyclines | antibiotic | Inhibition of FAO (PPARα, CPT1), inhibition of lipid export, Upregulation of fatty acid transport, Upregulation of CYP2E1 and ROS production |
| Linezolid | antibiotic | Inhibition of mtDNA translation and OXPHOS activity |
| Rifampicin | antibiotic | Upregulation of de novo lipogenesis (SCD1, ACC, FAS), Upregulation of fatty acid uptake (PXR, PPARγ, CD36), Inhibition of FAO |
| Tamoxifen | antineoplastic | Inhibition of FAO through ERα/β receptors, Inhibition of mtDNA synthesis, Conflicting data: downregulation of FAS, but also induction of SREBP-1c |
| Toremifene | antineoplastic | Few cases of steatosis |
| Irinotecan | antineoplastic | Decrease in mtDNA synthesis, Inhibition of OXPHOS, Mitochondrial dysfunction, inflammation and fibrosis |
| 5-Fluorouracil | antineoplastic | Increase in triglyceride accumulation, Mitochondrial dysfunction, Increase in peroxisomal beta-oxidation and ROS generation, Induction of JNK, IL-8 and ICAM.-1 |
| L-Asparaginase | antineoplastic | Mitochondrial dysfunction, Alteration in VLDL secretion and metabolism, |
| Methotrexate | antineoplastic a | Decrease in OXPHOS, Downregulation of mitochondrial enzymes, Depletion of folate mitochondrial stores, Activation of stellate cell A2A receptor, leading to fibrosis |
| Valproic acid | antiepileptic | Acyl CoA sequestration and mitochondrial FAO inhibition, Reduction in citric acid cycle flux, Inhibition of OXPHOS, Inhibition of ATP production, Upregulation of CD36, Increase in oxidative stress and decrease in antioxidant defenses |
| Carbamazepine | antiepileptic | Decrease in microsomal cytochrome P-450 dependent enzyme activity |
| Dexamethasone | glucocorticoid | Hyperphagia (inhibition of leptin signaling pathway), Increase in ChREBP and SREBP-1c activity, Increase in de novo lipogenesis, upregulation of CD36, decreased secretion of VLDL, decreased FAO |
| Betamethasone | glucocorticoid | |
| Prednisolone | glucocorticoid | |
| Triamcinolone | glucocorticoid | |
| Salicylic acid (Aspirin) | NSAID | Conflicting data: mitochondrial dysfunction, increase in eNOS and decrease in iNOS and TNF-α, reduction in JNK activity |
| Acetaminophen | NSAID | Increase in ROS generation, Mitochondrial damage, GSH depletion, Accumulation of long-chain acylcarnitines, Reduction in PPARα expression, Alteration of FAO |
| Pirprofen | NSAID | Inhibition of FAO and natural CoA activity |
| Ibuprofen | NSAID | Inhibition of FXR transcriptional activity |
| Diclofenac | NSAID | Inhibition of mtFAO |
| Naproxen | NSAID | Inhibition of mtFAO |
| Ketoprofen | NSAID | Inhibition of mitochondrial function, ROS accumulation |
| Zidovudine (AZT) | NRTI | Inhibition of DNA polymerase γ, ER stress induction, Increase in SREBP-1c, Decrease in PPARα, phospho-AMP kinase and 3-keto-acyl-CoA thiolase, Inhibition of autophagy |
| Stavudine | NRTI | Inhibition of DNA polymerase γ, Inhibition of autophagy |
| Didanosine | NRTI | Inhibition of DNA polymerase γ, Inhibition of oxygen consumption and complex I and III activity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Pasqua, L.G.; Cagna, M.; Berardo, C.; Vairetti, M.; Ferrigno, A. Detailed Molecular Mechanisms Involved in Drug-Induced Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: An Update. Biomedicines 2022, 10, 194. https://doi.org/10.3390/biomedicines10010194
Di Pasqua LG, Cagna M, Berardo C, Vairetti M, Ferrigno A. Detailed Molecular Mechanisms Involved in Drug-Induced Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: An Update. Biomedicines. 2022; 10(1):194. https://doi.org/10.3390/biomedicines10010194
Chicago/Turabian StyleDi Pasqua, Laura Giuseppina, Marta Cagna, Clarissa Berardo, Mariapia Vairetti, and Andrea Ferrigno. 2022. "Detailed Molecular Mechanisms Involved in Drug-Induced Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: An Update" Biomedicines 10, no. 1: 194. https://doi.org/10.3390/biomedicines10010194