
Screening of the Promising Direct Thrombin Inhibitors from Haematophagous Organisms. Part I: Recombinant Analogues and Their Antithrombotic Activity In Vitro

 , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals, Enzymes, and Materials
2.2. Bacterial Strains and Medium
2.3. Construction of Expression Vectors Containing the Haemadin and Variegin Genes
2.4. Shake-Flask Fermentation of E. coli Producer Strains of Fusion Proteins GyrA-Hae and Var-GyrA
2.5. Purification and Cleavage of the GyrA-Hae and Var-GyrA Fusion Proteins
2.6. Batch Fermentation
2.7. Semi-Preparative Purification of Haemadin and Variegin
2.8. Measurement of Antithrombin Activity of the Direct Thrombin Inhibitor In Vitro
2.8.1. Inhibition of Thrombin Amidolytic Activity
2.8.2. Kinetic Analysis of Thrombin Inhibition
2.8.3. Coagulation Tests in Mouse Plasma
2.8.4. Coagulation Tests in Human Plasma
2.9. Analytical Methods
2.10. Statistical Analyses
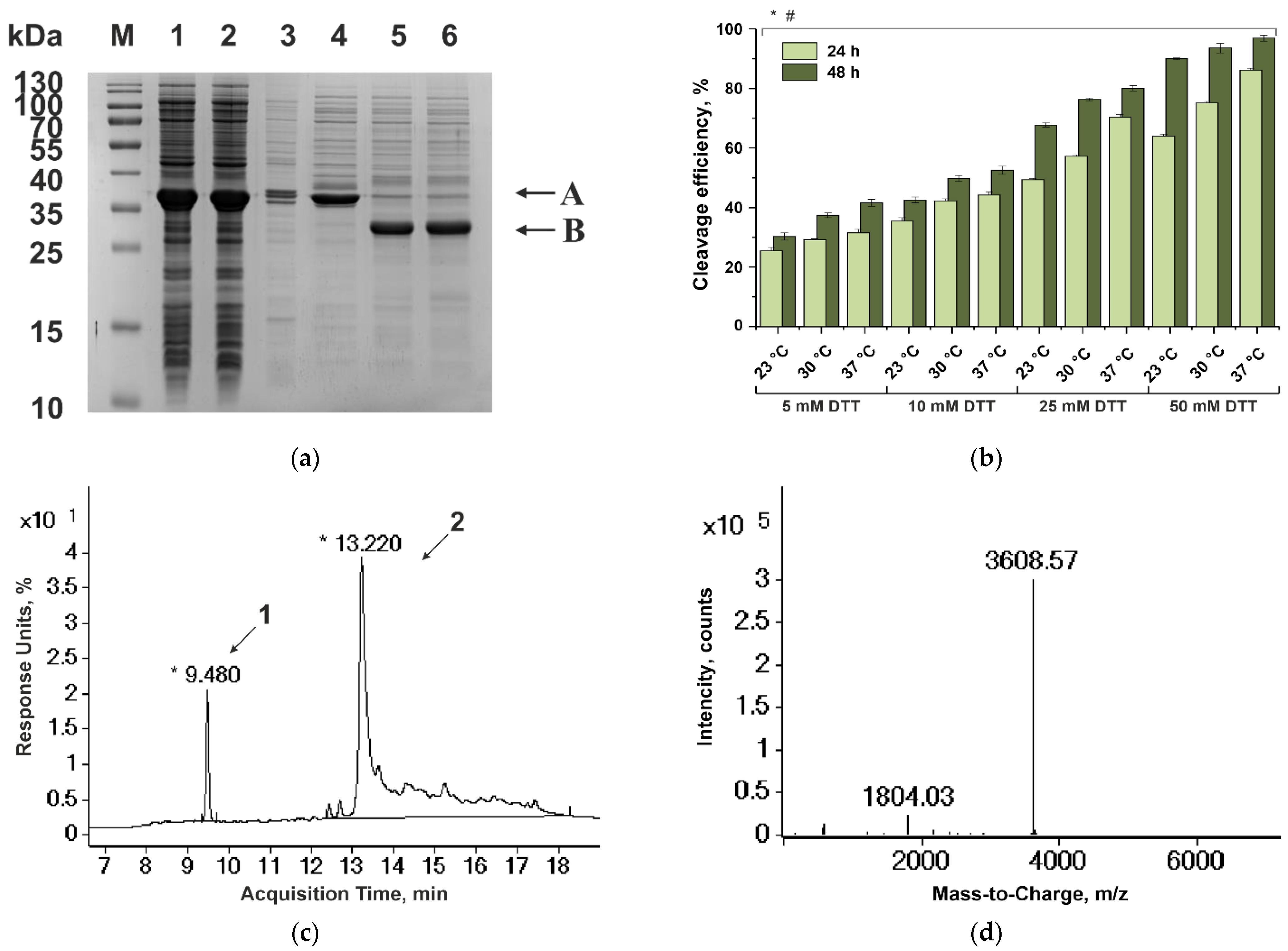
3. Results
3.1. Recombinant Haemadin and Variegin Production
3.1.1. Construction of Expression Vectors
3.1.2. Cultivation of Strains Producing the Fusion Proteins Containing Haemadin and Variegin
3.1.3. Purification of Haemadin
3.1.4. Purification of Variegin
3.2. Antithrombin Activity
3.2.1. Inhibition of the Amidolytic Thrombin Activity
3.2.2. Results of Clotting Assays on Mouse and Human Plasma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ali, M.R.; Salim Hossain, M.; Islam, M.A.; Saiful Islam Arman, M.; Sarwar Raju, G.; Dasgupta, P.; Noshin, T.F. Aspect of thrombolytic therapy: A review. Sci. World J. 2014, 2014, 586510. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J.; Eikelboom, J.W.; Chan, N.C. Fifty years of research on antithrombotic therapy: Achievements and disappointments. Eur. J. Intern. Med. 2019, 70, 1–7. [Google Scholar] [CrossRef]
- Bartholomew, J.R. Update on the management of venous thromboembolism. Cleveland Clin. J. Med. 2017, 84, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A. Antithrombotic therapy. Top. Companion Anim. Med. 2012, 27, 88–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badireddy, M.; Mudipalli, V.R. Deep Venous Thrombosis Prophylaxis; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Becattini, C.; Agnelli, G. Acute treatment of venous thromboembolism. Blood 2020, 135, 305–316. [Google Scholar] [CrossRef]
- Lee, C.J.; Ansell, J.E. Direct thrombin inhibitors. Br. J. Clin. Pharm. 2011, 72, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Almutairi, A.R.; Zhou, L.; Gellad, W.F.; Lee, J.K.; Slack, M.K.; Martin, J.R.; Lo-Ciganic, W.H. Effectiveness and Safety of Non-vitamin K Antagonist Oral Anticoagulants for Atrial Fibrillation and Venous Thromboembolism: A Systematic Review and Meta-analyses. Clin. Ther. 2017, 39, 1456–1478.e36. [Google Scholar] [CrossRef] [PubMed]
- Wardrop, D.; Keeling, D. The story of the discovery of heparin and warfarin. Br. J. Haematol. 2008, 141, 757–763. [Google Scholar] [CrossRef]
- Hoppensteadt, D.; Walenga, J.M.; Fareed, J.; Bick, R.L. Heparin, low-molecular-weight heparins, and heparin pentasaccharide: Basic and clinical differentiation. Hematol. Oncol. Clin. N. Am. 2003, 17, 313–341. [Google Scholar] [CrossRef]
- Mahat, K.C.; Sedhai, Y.R.; Krishnan, P. Argatroban; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Sun, Z.; Lan, X.; Li, S.; Zhao, H.; Tang, Z.; Xi, Y. Comparisons of argatroban to lepirudin and bivalirudin in the treatment of heparin-induced thrombocytopenia: A systematic review and meta-analysis. Int J. Hematol. 2017, 106, 476–483. [Google Scholar] [CrossRef]
- Kennedy, B.; Gargoum, F.S.; Kennedy, L.; Khan, F.; Curran, D.R.; O’Connor, T.M. Emerging anticoagulants. Curr. Med. Chem. 2012, 19, 3388–3416. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, B.I.; Ekman, S.; Lindbratt, S.; Baur, M.; Bach, D.; Torholm, C.; Kalebo, P.; Close, P. Prevention of thromboembolism with use of recombinant hirudin. Results of a double-blind, multicenter trial comparing the efficacy of desirudin (Revasc) with that of unfractionated heparin in patients having a total hip replacement. J. Bone Jt. Surg Am. 1997, 79, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Adkins, J.C.; Wilde, M.I. Lepirudin: A review of its potential place in the management of thrombotic disorders. BioDrugs 1998, 10, 227–255. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Janssens, U.; Berg, G.; Bock, M.; Kwasny, H.; Kemkes-Matthes, B.; Eichler, P.; Volpel, H.; Potzsch, B.; Luz, M. Lepirudin (recombinant hirudin) for parenteral anticoagulation in patients with heparin-induced thrombocytopenia. Heparin-Associated Thrombocytopenia Study (HAT) investigators. Circulation 1999, 100, 587–593. [Google Scholar] [CrossRef] [Green Version]
- Lubenow, N.; Eichler, P.; Lietz, T.; Greinacher, A.; Hit Investigators, G. Lepirudin in patients with heparin-induced thrombocytopenia—Results of the third prospective study (HAT-3) and a combined analysis of HAT-1, HAT-2, and HAT-3. J. Thromb. Haemost. 2005, 3, 2428–2436. [Google Scholar] [CrossRef] [PubMed]
- FDA Refuses to Approve Lepirudin as Anticoagulant in ACS Patients. Available online: https://www.medscape.com/viewarticle/783631 (accessed on 13 November 2021).
- Dear Health Care Professional Letter Refludan 2012 Final. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&cad=rja&uact=8&ved=2ahUKEwjypp_a8ZP0AhXoAxAIHe-CDngQFnoECAYQAw&url=https%3A%2F%2Fwww.hrsa.gov%2Fsites%2Fdefault%2Ffiles%2Fopa%2Fprogramrequirements%2Fmanufacturerletters%2F2012%2Frefludan05312012.pdf&usg=AOvVaw12m92aMwzhADLkJpQtEv-q (accessed on 13 November 2021).
- Graetz, T.J.; Tellor, B.R.; Smith, J.R.; Avidan, M.S. Desirudin: A review of the pharmacology and clinical application for the prevention of deep vein thrombosis. Expert Rev. Cardiovasc. 2011, 9, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Stone, G.W.; McLaurin, B.T.; Cox, D.A.; Bertrand, M.E.; Lincoff, A.M.; Moses, J.W.; White, H.D.; Pocock, S.J.; Ware, J.H.; Feit, F.; et al. Bivalirudin for patients with acute coronary syndromes. N. Engl. J. Med. 2006, 355, 2203–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubb, K.J.; Salehi, P.; Chedrawy, E.G. Bivalirudin: Alternative anticoagulation during cardiopulmonary bypass in patients with heparin-induced thrombocytopenia. Recent Pat. Cardiovasc. Drug Discov. 2010, 5, 20–24. [Google Scholar] [CrossRef]
- Jove, M.; Maslanka, M.; Minkowitz, H.S.; Jaffer, A.K.; Investigators, D.-A. Safety of desirudin in thrombosis prevention after total knee arthroplasty: The DESIR-ABLE study. Am. J. 2014, 21, 496–499. [Google Scholar] [CrossRef]
- Capranzano, P.; Dangas, G. Bivalirudin for primary percutaneous coronary intervention in acute myocardial infarction: The HORIZONS-AMI trial. Expert Rev. Cardiovasc. 2012, 10, 411–422. [Google Scholar] [CrossRef]
- El Khoury, M.; Karam, B.; Tabet, R.; Lafferty, J.C.; Snyder, S.T. Current Practice of Percutaneous Coronary Intervention in Patients With Coagulation Disorders. Cureus 2021, 13, e18284. [Google Scholar] [CrossRef]
- Taylor, T.; Campbell, C.T.; Kelly, B. A Review of Bivalirudin for Pediatric and Adult Mechanical Circulatory Support. Am. J. Cardiovasc Drugs 2021, 21, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Hamzah, M.; Jarden, A.M.; Ezetendu, C.; Stewart, R. Evaluation of Bivalirudin as an Alternative to Heparin for Systemic Anticoagulation in Pediatric Extracorporeal Membrane Oxygenation. Pediatr. Crit. Care Med. 2020, 21, 827–834. [Google Scholar] [CrossRef]
- Young, G. Anticoagulation Therapies in Children. Pediatr. Clin. N. Am. 2017, 64, 1257–1269. [Google Scholar] [CrossRef]
- Ryerson, L.; Lequier, L. Anticoagulation in Pediatric Extracorporeal Life Support-Time for a More Direct Approach? Pediatr. Crit. Care Med. 2020, 21, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.L. Bivalirudin as an Alternative to Heparin for Anticoagulation in Infants and Children. J. Pediatr. Pharmacol. Ther. 2015, 20, 408–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, C.Y.; Kini, R.M. Molecular diversity of anticoagulants from haematophagous animals. Thromb. Haemost. 2009, 102, 437–453. [Google Scholar] [CrossRef] [PubMed]
- Bock, P.E.; Panizzi, P.; Verhamme, I.M. Exosites in the substrate specificity of blood coagulation reactions. J. Thromb. Haemost. 2007, 5 (Suppl. S1), 81–94. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, A.C.; de Sanctis, D.; Gutierrez-Gallego, R.; Cereija, T.B.; Macedo-Ribeiro, S.; Fuentes-Prior, P.; Pereira, P.J. Unique thrombin inhibition mechanism by anophelin, an anticoagulant from the malaria vector. Proc. Natl. Acad. Sci. USA 2012, 109, E3649–E3658. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.Y.; Kumar, S.; Kazimirova, M.; Nuttall, P.A.; Radhakrishnan, U.P.; Kim, S.; Jagadeeswaran, P.; Imamura, T.; Mizuguchi, J.; Iwanaga, S.; et al. Crystal structure of thrombin in complex with S-variegin: Insights of a novel mechanism of inhibition and design of tunable thrombin inhibitors. PLoS ONE 2011, 6, e26367. [Google Scholar] [CrossRef] [Green Version]
- Richardson, J.L.; Fuentes-Prior, P.; Sadler, J.E.; Huber, R.; Bode, W. Characterization of the residues involved in the human alpha-thrombin-haemadin complex: An exosite II-binding inhibitor. Biochemistry 2002, 41, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Kostromina, M.A.; Esipov, R.S.; Miroshnikov, A.I. Biotechnological production of recombinant analogs of hirudin-1 from Hirudomedicinalis. Russ. J. Bioorg. Chem. 2012, 38, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Esipov, R.S.; Kostromina, M.A. Comparative analysis of the effectiveness of C-terminal cleavage intein-based constructs in producing a recombinant analog of anophelin, an anticoagulant from Anopheles albimanus. Appl. Biochem. Biotechnol. 2015, 175, 2468–2488. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kazimirova, M.; Trimnell, A.; Takac, P.; Labuda, M.; Nuttall, P.A.; Kini, R.M. Variegin, a novel fast and tight binding thrombin inhibitor from the tropical bont tick. J. Biol. Chem. 2007, 282, 29101–29113. [Google Scholar] [CrossRef] [Green Version]
- Strube, K.H.; Kroger, B.; Bialojan, S.; Otte, M.; Dodt, J. Isolation, sequence analysis, and cloning of haemadin. An anticoagulant peptide from the Indian leech. J. Biol. Chem. 1993, 268, 8590–8595. [Google Scholar] [CrossRef]
- Nakamura, Y.; Gojobori, T.; Ikemura, T. Codon usage tabulated from international DNA sequence databases: Status for the year 2000. Nucleic Acids Res. 2000, 28, 292. [Google Scholar] [CrossRef] [Green Version]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Mathys, S.; Evans, T.C.; Chute, I.C.; Wu, H.; Chong, S.; Benner, J.; Liu, X.Q.; Xu, M.Q. Characterization of a self-splicing mini-intein and its conversion into autocatalytic N- and C-terminal cleavage elements: Facile production of protein building blocks for protein ligation. Gene 1999, 231, 1–13. [Google Scholar] [CrossRef]
- Southworth, M.W.; Amaya, K.; Evans, T.C.; Xu, M.Q.; Perler, F.B. Purification of proteins fused to either the amino or carboxy terminus of the Mycobacterium xenopi gyrase A intein. Biotechniques 1999, 27, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Frottin, F.; Martinez, A.; Peynot, P.; Mitra, S.; Holz, R.C.; Giglione, C.; Meinnel, T. The proteomics of N-terminal methionine cleavage. Mol. Cell Proteom. 2006, 5, 2336–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenton, J.W., 2nd; Villanueva, G.B.; Ofosu, F.A.; Maraganore, J.M. Thrombin inhibition by hirudin: How hirudin inhibits thrombin. Haemostasis 1991, 21 (Suppl. S1), 27–31. [Google Scholar] [CrossRef] [PubMed]
- Karges, H.E.; Funk, K.A.; Ronneberger, H. Activity of coagulation and fibrinolysis parameters in animals. Arzneimittelforschung 1994, 44, 793–797. [Google Scholar]
- Siller-Matula, J.M.; Plasenzotti, R.; Spiel, A.; Quehenberger, P.; Jilma, B. Interspecies differences in coagulation profile. ThrombHaemost 2008, 100, 397–404. [Google Scholar] [CrossRef]
- Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers, U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) July 2005 Pharmacology and Toxicology. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estimating-maximum-safe-starting-dose-initial-clinical-trials-therapeutics-adult-healthy-volunteers (accessed on 13 November 2021).
- Jesty, J.; Beltrami, E. Positive feedbacks of coagulation: Their role in threshold regulation. Arter. Thromb. Vasc. Biol. 2005, 25, 2463–2469. [Google Scholar] [CrossRef] [PubMed]
- Bozic Mijovski, M. Advances in monitoring anticoagulant therapy. Adv. Clin. Chem. 2019, 90, 197–213. [Google Scholar] [CrossRef]
- Troisi, R.; Balasco, N.; Autiero, I.; Vitagliano, L.; Sica, F. Exosite Binding in Thrombin: A Global Structural/Dynamic Overview of Complexes with Aptamers and Other Ligands. Int. J. Mol. Sci. 2021, 22, 10803. [Google Scholar] [CrossRef]
- Segers, K.; Dahlback, B.; Bock, P.E.; Tans, G.; Rosing, J.; Nicolaes, G.A. The role of thrombin exosites I and II in the activation of human coagulation factor V. J Biol. Chem. 2007, 282, 33915–33924. [Google Scholar] [CrossRef] [Green Version]
- Myles, T.; Yun, T.H.; Leung, L.L. Structural req.quirements for the activation of human factor VIII by thrombin. Blood 2002, 100, 2820–2826. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.H.; Stafford, A.R.; Leslie, B.A.; Fredenburgh, J.C.; Weitz, J.I. Dabigatran and Argatroban Diametrically Modulate Thrombin Exosite Function. PLoS ONE 2016, 11, e0157471. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Stafford, A.R.; Wu, C.; Yeh, C.H.; Kim, P.Y.; Fredenburgh, J.C.; Weitz, J.I. Exosite 2-Directed Ligands Attenuate Protein C Activation by the Thrombin-Thrombomodulin Complex. Biochemistry 2017, 56, 3119–3128. [Google Scholar] [CrossRef]
- Chang, J.Y. Thrombin specificity. Requirement for apolar amino acids adjacent to the thrombin cleavage site of polypeptide substrate. Eur. J. Biochem. 1985, 151, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, T.; Rothe, M.; Dodt, J. Mechanism of the inhibition of alpha-thrombin by hirudin-derived fragments hirudin(1-47) and hirudin(45-65). Eur. J. Biochem. 1991, 195, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Maraganore, J.M.; Bourdon, P.; Jablonski, J.; Ramachandran, K.L.; Fenton, J.W., 2nd. Design and characterization of hirulogs: A novel class of bivalent peptide inhibitors of thrombin. Biochemistry 1990, 29, 7095–7101. [Google Scholar] [CrossRef]
- Witting, J.I.; Bourdon, P.; Brezniak, D.V.; Maraganore, J.M.; Fenton, J.W., 2nd. Thrombin-specific inhibition by and slow cleavage of hirulog-1. Biochem. J. 1992, 283, 737–743. [Google Scholar] [CrossRef] [Green Version]
- Richardson, J.L.; Kroger, B.; Hoeffken, W.; Sadler, J.E.; Pereira, P.; Huber, R.; Bode, W.; Fuentes-Prior, P. Crystal structure of the human alpha-thrombin-haemadin complex: An exosite II-binding inhibitor. EMBO J. 2000, 19, 5650–5660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tans, G.; Nicolaes, G.A.; Thomassen, M.C.; Hemker, H.C.; van Zonneveld, A.J.; Pannekoek, H.; Rosing, J. Activation of human factor V by meizothrombin. J. Biol. Chem. 1994, 269, 15969–15972. [Google Scholar] [CrossRef]
- Stojanovski, B.M.; Pelc, L.A.; Zuo, X.; Pozzi, N.; Cera, E.D. Enhancing the anticoagulant profile of meizothrombin. Biomol. Concepts 2018, 9, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Francischetti, I.M.; Valenzuela, J.G.; Ribeiro, J.M. Anophelin: Kinetics and mechanism of thrombin inhibition. Biochemistry 1999, 38, 16678–16685. [Google Scholar] [CrossRef] [PubMed]
- Wienen, W.; Stassen, J.M.; Priepke, H.; Ries, U.J.; Hauel, N. In-vitro profile and ex-vivo anticoagulant activity of the direct thrombin inhibitor dabigatran and its orally active prodrug, dabigatran etexilate. Thromb. Haemost. 2007, 98, 155–162. [Google Scholar]
- AAALAC. Directory of Accredited Organizations Search Result. Available online: https://www.aaalac.org/accreditation-program/directory/directory-of-accredited-organizations-search-result/?nocache=1#home_acc_dir_search (accessed on 13 November 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thrombin Inhibitor | Preincubation Time | IC50 * | Inhibition Constant Ki |
|---|---|---|---|
| Hirudin-1 | 0 min | 21.47 ± 0.49 pM | |
| 30 min | 28.01 ± 0.36 pM | 217 ± 25 fM | |
| Haemadin | 0 min | 22.03 ± 0.57 pM | |
| 30 min | 33.15 ± 0.68 pM | 237 ± 25 fM | |
| Anophelin | 0 min | 6.35 ± 0.14 nM | |
| 30 min | 6.78 ± 0.04 nM | 99.4 ± 11.54 pM | |
| Variegin | 0 min | 14.55 ± 1.88 nM | |
| 30 min | 18.39 ± 0.61 nM | 371.6 ± 22.5 pM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostromina, M.A.; Tukhovskaya, E.A.; Shaykhutdinova, E.R.; Slashcheva, G.A.; Ismailova, A.M.; Palikov, V.A.; Palikova, Y.A.; Dyachenko, I.A.; Kravchenko, I.N.; Sadovnikova, E.S.; et al. Screening of the Promising Direct Thrombin Inhibitors from Haematophagous Organisms. Part I: Recombinant Analogues and Their Antithrombotic Activity In Vitro. Biomedicines 2022, 10, 11. https://doi.org/10.3390/biomedicines10010011
Kostromina MA, Tukhovskaya EA, Shaykhutdinova ER, Slashcheva GA, Ismailova AM, Palikov VA, Palikova YA, Dyachenko IA, Kravchenko IN, Sadovnikova ES, et al. Screening of the Promising Direct Thrombin Inhibitors from Haematophagous Organisms. Part I: Recombinant Analogues and Their Antithrombotic Activity In Vitro. Biomedicines. 2022; 10(1):11. https://doi.org/10.3390/biomedicines10010011
Chicago/Turabian StyleKostromina, Maria A., Elena A. Tukhovskaya, Elvira R. Shaykhutdinova, Gulsara A. Slashcheva, Alina M. Ismailova, Victor A. Palikov, Yuliya A. Palikova, Igor A. Dyachenko, Irina N. Kravchenko, Elena S. Sadovnikova, and et al. 2022. "Screening of the Promising Direct Thrombin Inhibitors from Haematophagous Organisms. Part I: Recombinant Analogues and Their Antithrombotic Activity In Vitro" Biomedicines 10, no. 1: 11. https://doi.org/10.3390/biomedicines10010011