
Direct Electrochemical Analysis in Seawater: Evaluation of Chloride and Bromide Detection

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion | Molar Concentration/mM |
|---|---|
| Na+ | 4.81 × 102 |
| Mg2+ | 5.41 × 101 |
| Ca2+ | 1.05 × 101 |
| K+ | 1.05 × 101 |
| Sr2+ | 9.20 × 10−2 |
| Cl− | 5.59 × 102 |
| SO42− | 2.89 × 101 |
| HCO3− | 1.89 |
| CO32− | 1.89 × 10−1 |
| Br− | 8.63 × 10−1 |
| B(OH)4− | 8.39 × 10−2 |
| B(OH)3 | 3.42 × 10−1 |
| F− | 7.00 × 10−2 |
2. Experimental Section
2.1. Chemicals and Reagents
2.2. Electrochemical Apparatus and Methods
2.3. Sample Collection and Ion Chromatography Analysis
3. Results and Discussion
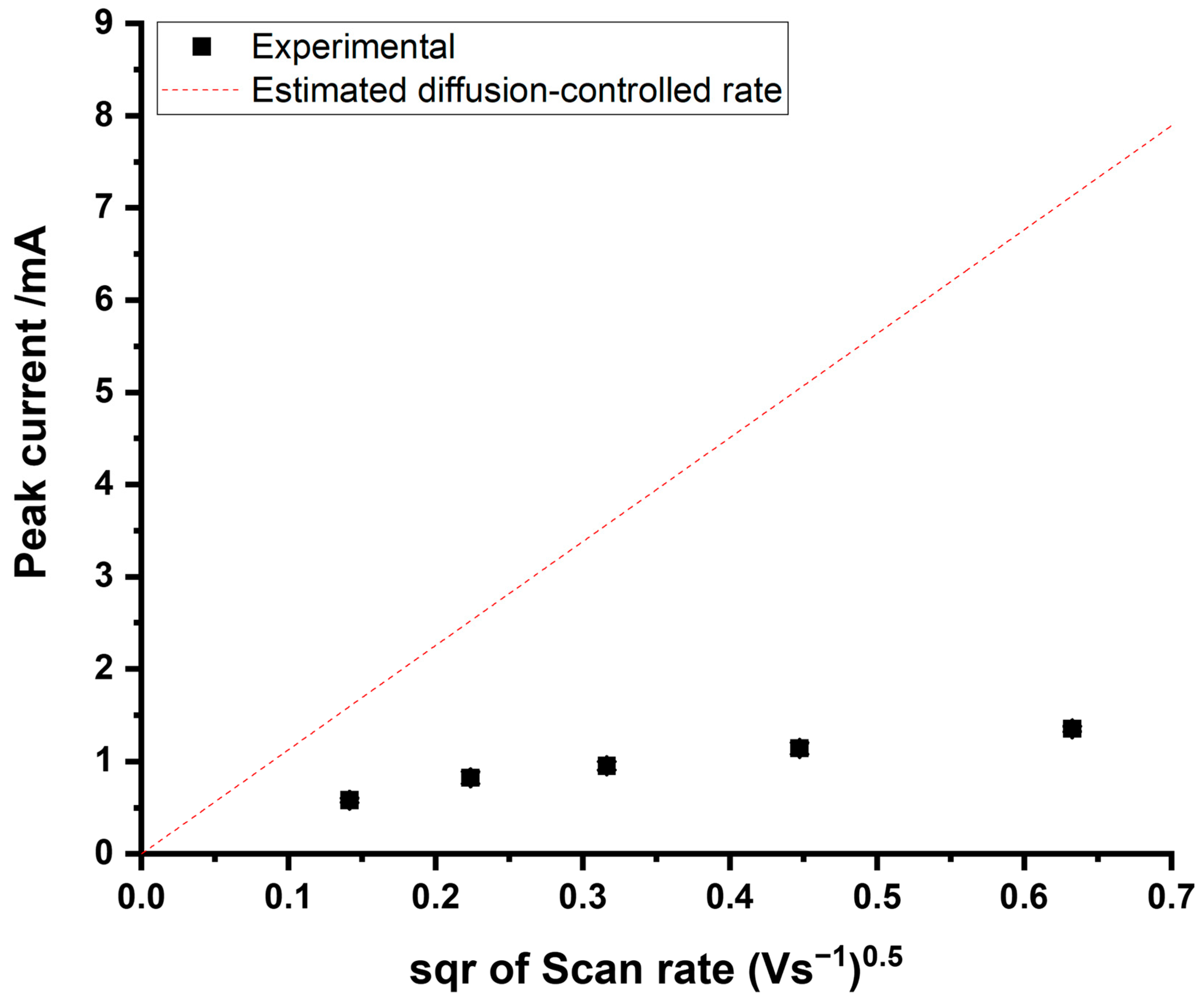
3.1. Cyclic Voltammetric Study of a Au Electrode in ASW
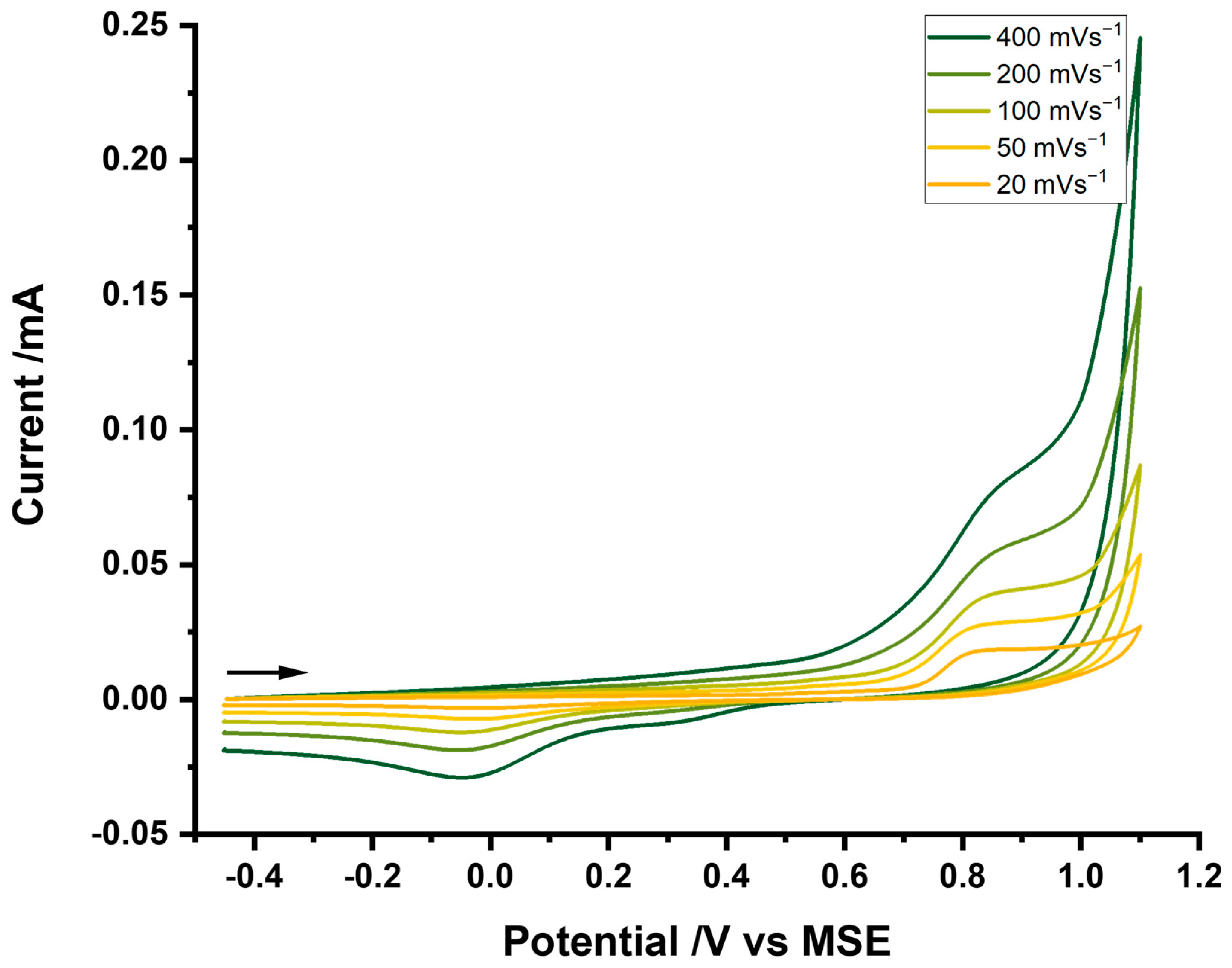
3.2. Cyclic Voltammetry at Glassy Carbon and Platinum Electrodes in ASW
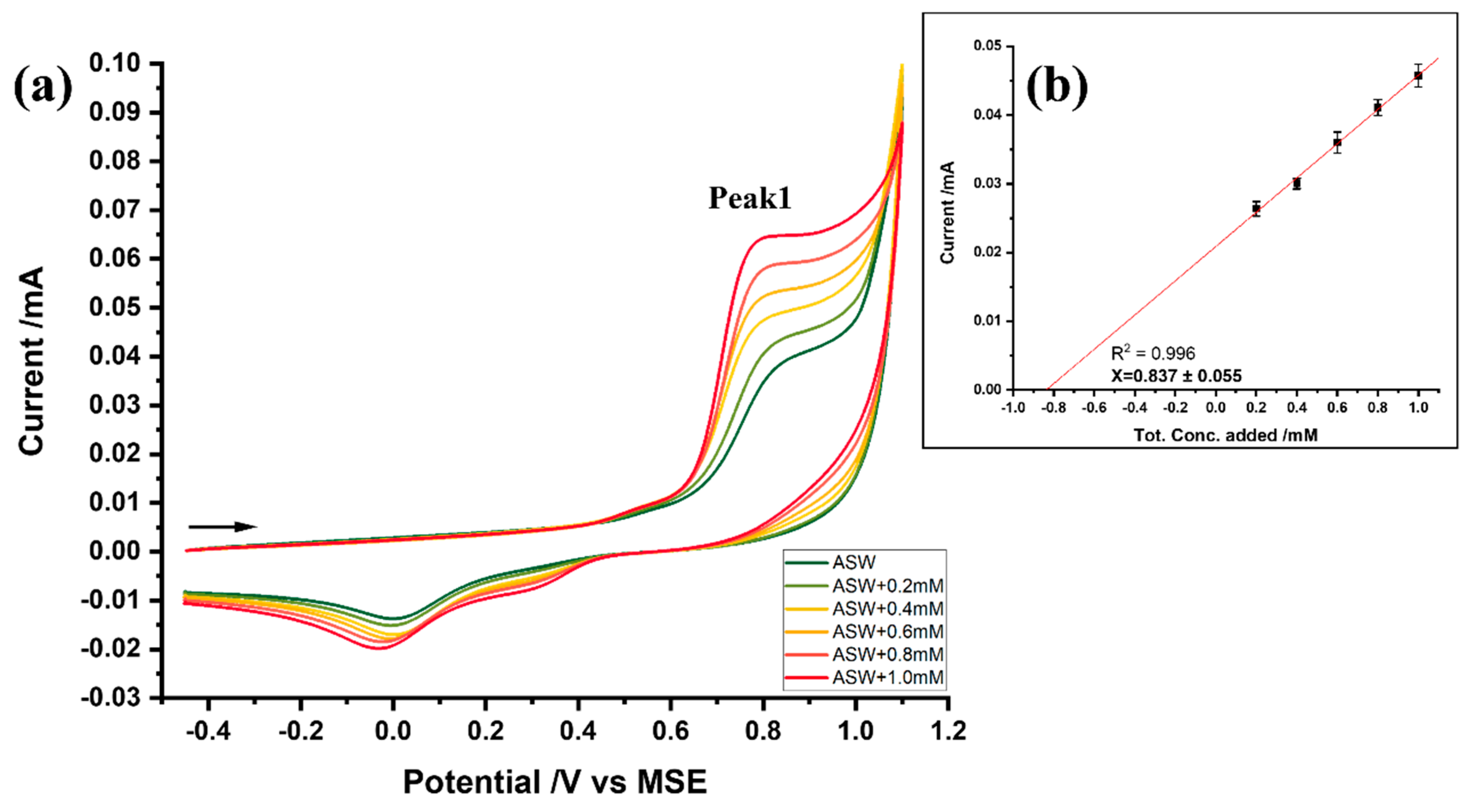
3.3. Analysis of Bromide at the Glassy Carbon Electrode
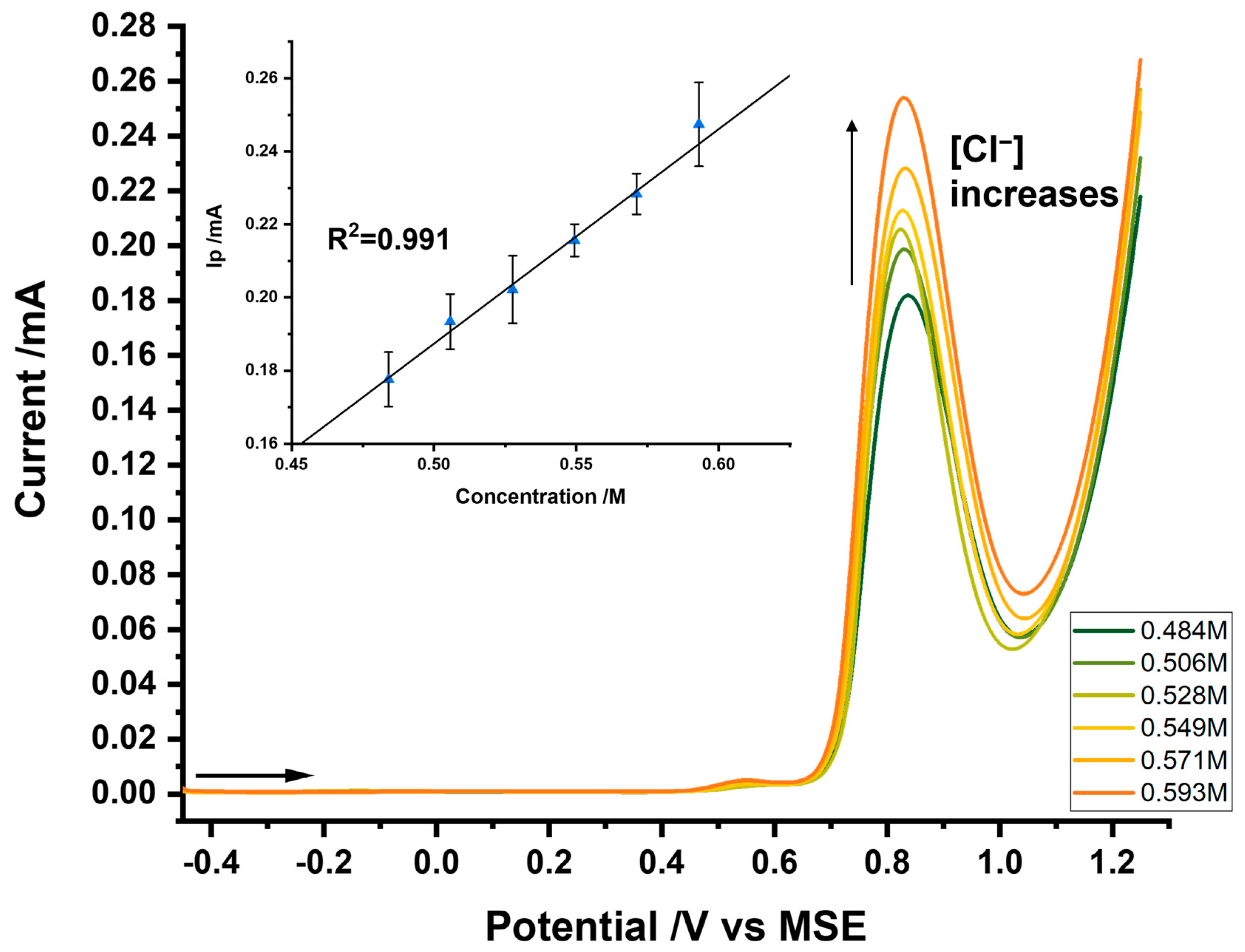
3.4. Analysis of Chloride at the Platinum Electrode
3.5. Real Sample Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Water Scarcity. Available online: https://www.unwater.org/water-facts/water-scarcity (accessed on 14 March 2023).
- Spellman, F.R. The Handbook of Nature; Bernan Press: Lanham, MD, USA, 2019. [Google Scholar]
- Bint El Hassan, H.E.S.; Guerrier, D.; Closas, A.; Demilecamps, C.; kassim, Y.; Lacirignola, C.; Lamaddalena, N.; Mateo-Sagasta, J.; Rivera, N. Water Around the Mediterranean; The Center for Mediterranean Integration (CMI): Marseille, France, 2018. [Google Scholar]
- Pérez-González, A.; Urtiaga, A.M.; Ibáñez, R.; Ortiz, I. State of the art and review on the treatment technologies of water reverse osmosis concentrates. Water Res. 2012, 46, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H. A review of desalting process techniques and economic analysis of the recovery of salts from retentates. Desalination 2011, 270, 1–8. [Google Scholar] [CrossRef]
- Wallace, W.J. The Development of the Chlorinity/Salinity Concept in Oceanography; Elsevier Science Ltd.: New York, NY, USA, 1974. [Google Scholar]
- Millero, F.J.; Feistel, R.; Wright, D.G.; McDougall, T.J. The composition of Standard Seawater and the definition of the Reference-Composition Salinity Scale. Deep Sea Res. 2008, 55, 50–72. [Google Scholar] [CrossRef]
- Stewart, R.H. Introduction to Physical Oceanography; Department of Oceanography, Texas A&M University: College Station, TX, USA, 2008. [Google Scholar]
- Shumway, S.E. Effect of salinity fluctuation on the osmotic pressure and Na+, Ca2+ and Mg2+ ion concentrations in the hemolymph of bivalve molluscs. Mar. Biol. 1977, 41, 153–177. [Google Scholar] [CrossRef]
- Guo, Y.; Compton, R.G. A bespoke chloride sensor for seawater: Simple and fast with a silver electrode. Talanta 2021, 232, 122502. [Google Scholar] [CrossRef]
- Yamamoto, M. Stimulation of elemental mercury oxidation in the presence of chloride ion in aquatic environments. Chemosphere 1996, 32, 1217–1224. [Google Scholar] [CrossRef]
- Levard, C.; Mitra, S.; Yang, T.; Jew, A.D.; Badireddy, A.R.; Lowry, G.V.; Brown, G.E., Jr. Effect of Chloride on the Dissolution Rate of Silver Nanoparticles and Toxicity to E. coli. Environ. Sci. Technol. 2013, 47, 5738–5745. [Google Scholar] [CrossRef] [PubMed]
- Batchelor-McAuley, C.; Tschulik, K.; Neumann, C.; Laborda, E.; Compton, R. Why are silver nanoparticles more toxic than bulk silver? Towards understanding the dissolution and toxicity of silver nanoparticles. Int. J. Electrochem. Sci. 2014, 9, 1132–1138. [Google Scholar]
- Gilberg, M.R.; Seeley, N.J. The identity of compounds containing chloride ions in marine iron corrosion products: A critical review. Stud. Conserv. 1981, 26, 50–56. [Google Scholar] [CrossRef]
- Otieno, M.; Beushausen, H.; Alexander, M. Chloride-induced corrosion of steel in cracked concrete—Part I: Experimental studies under accelerated and natural marine environments. Cem. Concr. Res. 2016, 79, 373–385. [Google Scholar] [CrossRef]
- Downs, A.J.; Adams, C.J. The Chemistry of Chlorine, Bromine, Iodine and Astatine: Pergamon Texts in Inorganic Chemistry, Volume 7; Elsevier: Amsterdam, The Netherlands, 2017; Volume 7. [Google Scholar]
- Leri, A.C.; Hakala, J.A.; Marcus, M.A.; Lanzirotti, A.; Reddy, C.M.; Myneni, S.C.B. Natural organobromine in marine sediments: New evidence of biogeochemical Br cycling. Glob. Biogeochem. Cycles 2010, 24, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Pilson, M.E. An Introduction to the Chemistry of the Sea; Cambridge University Press: Cambridge, UK, 2012. [Google Scholar]
- Gribble, G.W. Amazing Organohalogens: Although best known as synthetic toxicants, thousands of halogen compounds are, in fact, part of our natural enviornment. Am. Sci. 2004, 92, 342–349. [Google Scholar] [CrossRef]
- Dorji, P.; Kim, D.I.; Hong, S.; Phuntsho, S.; Shon, H.K. Pilot-scale membrane capacitive deionisation for effective bromide removal and high water recovery in seawater desalination. Desalination 2020, 479, 114309. [Google Scholar] [CrossRef]
- Lewis, E.L. The practical salinity scale of 1978 and its antecedents. Mar. Geod. 1982, 5, 350–357. [Google Scholar] [CrossRef]
- Lagerloef, G.S.; Swift, C.T.; Le Vine, D.M. Sea surface salinity: The next remote sensing challenge. Oceanog. 1995, 8, 44–50. [Google Scholar] [CrossRef]
- Anfält, T.; Twengström, S. The determination of bromide in natural waters by flow injection analysis. Anal. Chim. Acta 1986, 179, 453–457. [Google Scholar] [CrossRef]
- Vengosh, A.; Pankratov, I. Chloride/Bromide and Chloride/Fluoride Ratios of Domestic Sewage Effluents and Associated Contaminated Ground Water. Ground Water 1998, 36, 815–824. [Google Scholar] [CrossRef]
- Morgan, J.J.; Stumm, W. Aquatic Chemistry: An Introduction Emphasizing Chemical Equilibria in Natural Waters; John Wiley & Sons: New York, NY, USA, 1981. [Google Scholar]
- Salameh, E.; Tarawneh, A.; Al-Raggad, M. Origin of high bromide concentration in the water sources in Jordan and in the Dead Sea water. Arabian J. Geosci. 2016, 9, 414. [Google Scholar] [CrossRef]
- Jain, A.; Chaurasia, A.; Sahasrabuddhey, B.; Verma, K.K. Determination of bromide in complex matrices by pre-column derivatization linked to solid-phase extraction and high-performance liquid chromatography. J. Chromatogr. A 1996, 746, 31–41. [Google Scholar] [CrossRef]
- Borges, E.P.; Lavorante, A.F.; Reis, B.F.d. Determination of bromide ions in seawater using flow system with chemiluminescence detection. Anal. Chim. Acta 2005, 528, 115–119. [Google Scholar] [CrossRef]
- Grasshoff, K.; Wenck, A. A modern version of the Mohr-Knudsen titration for the chlorinity of sea water. ICES Mar. Sci. Symp. 1972, 34, 522–528. [Google Scholar] [CrossRef]
- Hong, T.-K.; Kim, M.-H.; Czae, M.-Z. Determination of chlorinity of water without the use of chromate indicator. Int. J. Anal. Chem. 2010, 2010, 602939. [Google Scholar] [CrossRef] [PubMed]
- Reitemeier, R. Semimicroanalysis of saline soil solutions. Ind. Eng. Chem. Anal. Ed. 1943, 15, 393–402. [Google Scholar] [CrossRef]
- Reeburgh, W.S.; Carpenter, J.H. Determination of chlorinity using A differential potentiometric. Limnol. Oceanogr. 1964, 9, 589–591. [Google Scholar] [CrossRef]
- Clesceri, L.S.; Greenberg, A.E.; Eaton, A.D. Standard Methods for the Examination of Water and Wastewater, 20th ed.; APHA American Public Health Association: Washington, DC, USA, 1998. [Google Scholar]
- Willard, H.H.; Merritt, L.L., Jr.; Dean, J.A.; Settle, F.A., Jr. Instrumental Methods of Analysis, 7th ed.; Wadsworth Publishing Company: Belmont, CA, USA, 1988. [Google Scholar]
- Lewis, E. The practical salinity scale 1978 and its antecedents. IEEE J. Ocean. 1980, 5, 3–8. [Google Scholar] [CrossRef]
- Lewis, E.; Perkin, R. The Practical Salinity Scale 1978: Conversion of existing data. Deep Sea Res. 1981, 28, 307–328. [Google Scholar] [CrossRef]
- Unesco. Tenth Report of the Joint Panel on Oceanographic Tables and Standards; Unesco Division of Marine Sciences: Sidney, BC, Canada, 1980. [Google Scholar]
- Uraisin, K.; Takayanagi, T.; Oshima, M.; Nacapricha, D.; Motomizu, S. Kinetic-spectrophotometric method for the determination of trace amounts of bromide in seawater. Talanta 2006, 68, 951–956. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.R. Applying the phenol red colorimetric method for bromide analysis to reducing waters. Talanta 1993, 40, 43–51. [Google Scholar] [CrossRef]
- Anagnostopoulou, P.I.; Koupparis, M.A. Automated flow-injection phenol red method for determination of bromide and bromide salts in drugs. Anal. Chem. 1986, 58, 322–326. [Google Scholar] [CrossRef]
- Emaus, W.J.M.; Henning, H.J. Determination of bromide in sodium chloride matrices by flow-injection analysis using blank peak elimination and kinetic discrimination. Anal. Chim. Acta 1993, 272, 245–250. [Google Scholar] [CrossRef]
- Uraisin, K.; Nacapricha, D.; Lapanantnoppakhun, S.; Grudpan, K.; Motomizu, S. Determination of trace amounts of bromide by flow injection/stopped-flow detection technique using kinetic-spectrophotometric method. Talanta 2005, 68, 274–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonehara, N.; Chaen, S.-i.; Tomiyasu, T.; Sakamoto, H. Flow injection-spectrophotometric determination of trace amounts of bromide by its catalytic effect on the 4,4′-bis(dimethylamino)diphenylmethane-chloramine T reaction. Anal. Sci. 1999, 15, 277–281. [Google Scholar] [CrossRef] [Green Version]
- Shishehbori, M.R.; Elah, J.R. Kinetic spectrophotometric method For trace amounts determination of bromide in pharmaceutical samples using Janus Green-Bromate system. Int. J. Ind. Chem. 2011, 2, 27–32. [Google Scholar]
- Sheibani, A.; Shishehbore, M.R.; Ardakani, Z.T. Kinetic spectrophotometric determination of bromide in clidinium-c drug. Chin. Chem. Lett. 2011, 22, 595–598. [Google Scholar] [CrossRef]
- Akaiwa, H.; Kawamoto, H.; Osumi, M. Simultaneous determination of bromide and chloride in natural waters by ion-exchange chromatography and direct potentiometry with an ion-selective electrode. Talanta 1982, 29, 689–690. [Google Scholar] [CrossRef]
- Seefeld, S.; Baltensperger, U. Determination of bromide in snow samples by ion chromatography with electrochemical detection. Anal. Chim. Acta 1993, 283, 246–250. [Google Scholar] [CrossRef]
- Fabre, H.; Blanchin, M.D.; Bosc, N. Capillary electrophoresis for the determination of bromide, chloride and sulfate as impurities in calcium acamprosate. Anal. Chim. Acta 1999, 381, 29–37. [Google Scholar] [CrossRef]
- Stålberg, O.; Sander, K.; Sänger-van de Griend, C. The determination of bromide in a local anaesthetic hydrochloride by capillary electrophoresis using direct UV detection. J. Chromatogr. A 2002, 977, 265–275. [Google Scholar] [CrossRef]
- Pascali, J.P.; Trettene, M.; Bortolotti, F.; Paoli, G.d.; Gottardo, R.; Tagliaro, F. Direct analysis of bromide in human serum by capillary electrophoresis. J. Chromatogr. B 2006, 839, 2–5. [Google Scholar] [CrossRef]
- Fukushi, K.; Watanabe, K.; Takeda, S.; Wakida, S.-I.; Yamane, M.; Higashi, K.; Hiiro, K. Determination of bromide ions in seawater by capillary zone electrophoresis using diluted artificial seawater as the buffer solution. J. Chromatogr. A 1998, 802, 211–217. [Google Scholar] [CrossRef]
- Kocatürk, N.; Öztekin, N.; Bedia Erim, F. Direct determination of bromide ions in seawater by capillary zone electrophoresis using polyethyleneimine-coated capillaries. Anal. Bioanal. Chem. 2003, 377, 1207–1211. [Google Scholar] [CrossRef]
- Rechnitz, G.A.; Kresx, M.R. Potentiometric measurements with choride-sensitive and bromide-sensitive membrane electrodes. Anal. Chem. 1966, 38, 1786–1788. [Google Scholar] [CrossRef]
- Zahran, E.M.; Hua, Y.; Li, Y.; Flood, A.H.; Bachas, L.G. Triazolophanes: A new class of halide-selective ionophores for potentiometric sensors. Anal. Chem. 2010, 82, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Isildak, Ö.; Özbek, O.; Yigit, K. A bromide-selective PVC membrane potentiometric sensor. Bulg. Chem. Commun. 2020, 52, 448–452. [Google Scholar] [CrossRef]
- Vlascici, D.; Plesu, N.; Fagadar-Cosma, G.; Lascu, A.; Petric, M.; Crisan, M.; Belean, A.; Fagadar-Cosma, E. Potentiometric sensors for iodide and bromide based on Pt(II)-Porphyrin. Sensors 2018, 18, 2297. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Compton, R.G. A bespoke reagent-free amperometric bromide sensor for seawater. Talanta 2023, 253, 124019. [Google Scholar] [CrossRef]
- Harris, D.C. Quantitative Chemical Analysis; Macmillan: London, UK, 2010. [Google Scholar]
- Sekerka, I.; Lechner, J.F. Ion Selective Electrode for Determination of Chloride Ion in Biological Materials, Food Products, Soils, and Waste Water. J Assoc Off Anal Chem. 2020, 61, 1493–1495. [Google Scholar] [CrossRef]
- Montemor, M.; Alves, J.; Simoes, A.; Fernandes, J.; Lourenço, Z.; Costa, A.; Appleton, A.; Ferreira, M. Multiprobe chloride sensor for in situ monitoring of reinforced concrete structures. Cem. Concr. Compos. 2006, 28, 233–236. [Google Scholar] [CrossRef]
- Arai, K.; Kusu, F.; Noguchi, N.; Takamura, K.; Osawa, H. Selective determination of chloride and bromide ions in serum by cyclic voltammetry. Anal. Biochem. 1996, 240, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Wang, H.; Miao, Z.; Wang, X.; Fang, Y.; Chen, Q.; Shao, X. Synthesis of silver nanowires and their applications in the electrochemical detection of halide. Talanta 2011, 84, 673–678. [Google Scholar] [CrossRef]
- Bujes-Garrido, J.; Izquierdo-Bote, D.; Heras, A.; Colina, A.; Arcos-Martínez, M.J. Determination of halides using Ag nanoparticles-modified disposable electrodes. A first approach to a wearable sensor for quantification of chloride ions. Anal. Chim. Acta 2018, 1012, 42–48. [Google Scholar] [CrossRef]
- Malongo, T.K.; Patris, S.; Macours, P.; Cotton, F.; Nsangu, J.; Kauffmann, J.-M. Highly sensitive determination of iodide by ion chromatography with amperometric detection at a silver-based carbon paste electrode. Talanta 2008, 76, 540–547. [Google Scholar] [CrossRef]
- Cunha-Silva, H.; Julia Arcos-Martinez, M. Development of a selective chloride sensing platform using a screen-printed platinum electrode. Talanta 2019, 195, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Millero, F.J. The Physical Chemistry of Seawater. Annu. Rev. Earth Planet. Sci. 1974, 2, 101–150. [Google Scholar] [CrossRef]
- Qi, P.H.; Hiskey, J.B. Electrochemical behavior of gold in iodide solutions. Hydrometallurgy 1993, 32, 161–179. [Google Scholar] [CrossRef]
- Nicol, M.J. The anodic behaviour of gold. Gold Bull. 1980, 13, 46–55. [Google Scholar] [CrossRef] [Green Version]
- Nicol, M.J. The anodic behaviour of gold. Gold Bull. 1980, 13, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Loo, B.H. In situ identification of halide complexes on gold electrode by surface-enhanced Raman spectroscopy. J. Phys. Chem. 1982, 86, 433–437. [Google Scholar] [CrossRef]
- Gaur, J.; Schmid, G. Electrochemical behavior of gold in acidic chloride solutions. J. Electroanal. Chem. Interfacial Electrochem. 1970, 24, 279–286. [Google Scholar] [CrossRef]
- Cadle, S.; Bruckenstein, S. A ring-disk study of the effect of trace chloride ion on the anodic behavior of gold in 0.2 M H2SO4. J. Electroanal. Chem. Interfacial Electrochem. 1973, 48, 325–331. [Google Scholar] [CrossRef]
- Nicol, M.J.; Schalch, E. Reports Nos. 1844 and 1848; National Institute for Metallurgy: Johannesburg, South Africa, 1976. [Google Scholar]
- Heumann, T.; Panesar, H.S. Beitrag zur Frage nach dem Auflösungsmechanismus von Gold zu Chlorkomplexen und nach seiner Passivierung. Z. Phys. Chem. 1965, 229O, 84–97. [Google Scholar] [CrossRef]
- Morel, F.M.M.; Rueter, J.G.; Anderson, D.M.; Guillard, R.R.L. Aquil: A chemically defined phytoplankton culture medium for trace metal studies. J. Phycol. 1979, 15, 135–141. [Google Scholar] [CrossRef]
- Price, N.M.; Harrison, G.I.; Hering, J.G.; Hudson, R.J.; Nirel, P.M.; Palenik, B.; Morel, F.M. Preparation and chemistry of the artificial algal culture medium Aquil. Biol. Oceanogr. 1989, 6, 443–461. [Google Scholar] [CrossRef]
- Yu, J.; Yang, M.; Batchelor-McAuley, C.; Barton, S.; Rickaby, R.E.M.; Bouman, H.A.; Compton, R.G. Rapid Opto-electrochemical Differentiation of Marine Phytoplankton. ACS Meas. Sci. Au 2022, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lin, C.; Batchelor-McAuley, C.; Chen, L.; Compton, R.G. Tafel analysis in practice. J. Electroanal. Chem. 2018, 826, 117–124. [Google Scholar] [CrossRef]
- Compton, R.G.; Banks, C.E. Understanding Voltammetry, 3rd ed.; World Scientific: Singapore, 2018. [Google Scholar]
- Guidelli, R.; Compton, R.G.; Feliu, J.M.; Gileadi, E.; Lipkowski, J.; Schmickler, W.; Trasatti, S. Defining the transfer coefficient in electrochemistry: An assessment (IUPAC Technical Report). Pure Appl. Chem. 2014, 86, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Guidelli, R.; Compton, R.G.; Feliu, J.M.; Gileadi, E.; Lipkowski, J.; Schmickler, W.; Trasatti, S. Definition of the transfer coefficient in electrochemistry (IUPAC Recommendations 2014). Pure Appl. Chem. 2014, 86, 259–262. [Google Scholar] [CrossRef] [Green Version]
- Poisson, A.; Papaud, A. Diffusion coefficients of major ions in seawater. Marine Chemistry 1983, 13, 265–280. [Google Scholar] [CrossRef]







| Composition | Molar Concentration (mol dm−3) |
|---|---|
| NaCl | 4.20 × 10−1 |
| KCl | 9.39 × 10−3 |
| MgCl2 6H2O | 5.46 × 10−2 |
| CaCl2 2H2O | 1.05 × 10−2 |
| SrCl2 6H2O | 6.38 × 10−5 |
| Na2SO4 | 2.88 × 10−2 |
| NaHCO3 | 2.38 × 10−3 |
| KBr | 8.40 × 10−4 |
| NaF | 7.15 × 10−5 |
| H3BO3 | 4.85 × 10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Compton, R.G. Direct Electrochemical Analysis in Seawater: Evaluation of Chloride and Bromide Detection. Chemosensors 2023, 11, 297. https://doi.org/10.3390/chemosensors11050297
Chen Y, Compton RG. Direct Electrochemical Analysis in Seawater: Evaluation of Chloride and Bromide Detection. Chemosensors. 2023; 11(5):297. https://doi.org/10.3390/chemosensors11050297
Chicago/Turabian StyleChen, Yuqi, and Richard G. Compton. 2023. "Direct Electrochemical Analysis in Seawater: Evaluation of Chloride and Bromide Detection" Chemosensors 11, no. 5: 297. https://doi.org/10.3390/chemosensors11050297