
The Diversity of Astrocyte Activation during Multiple Sclerosis: Potential Cellular Targets for Novel Disease Modifying Therapeutics
 ,
,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Astrocytes as Regulators of CNS Homeostasis and Metabolism
3. Astrocyte Subgroups: Moving Away from the A1-A2 Dipole
4. Astrocytes and Aging
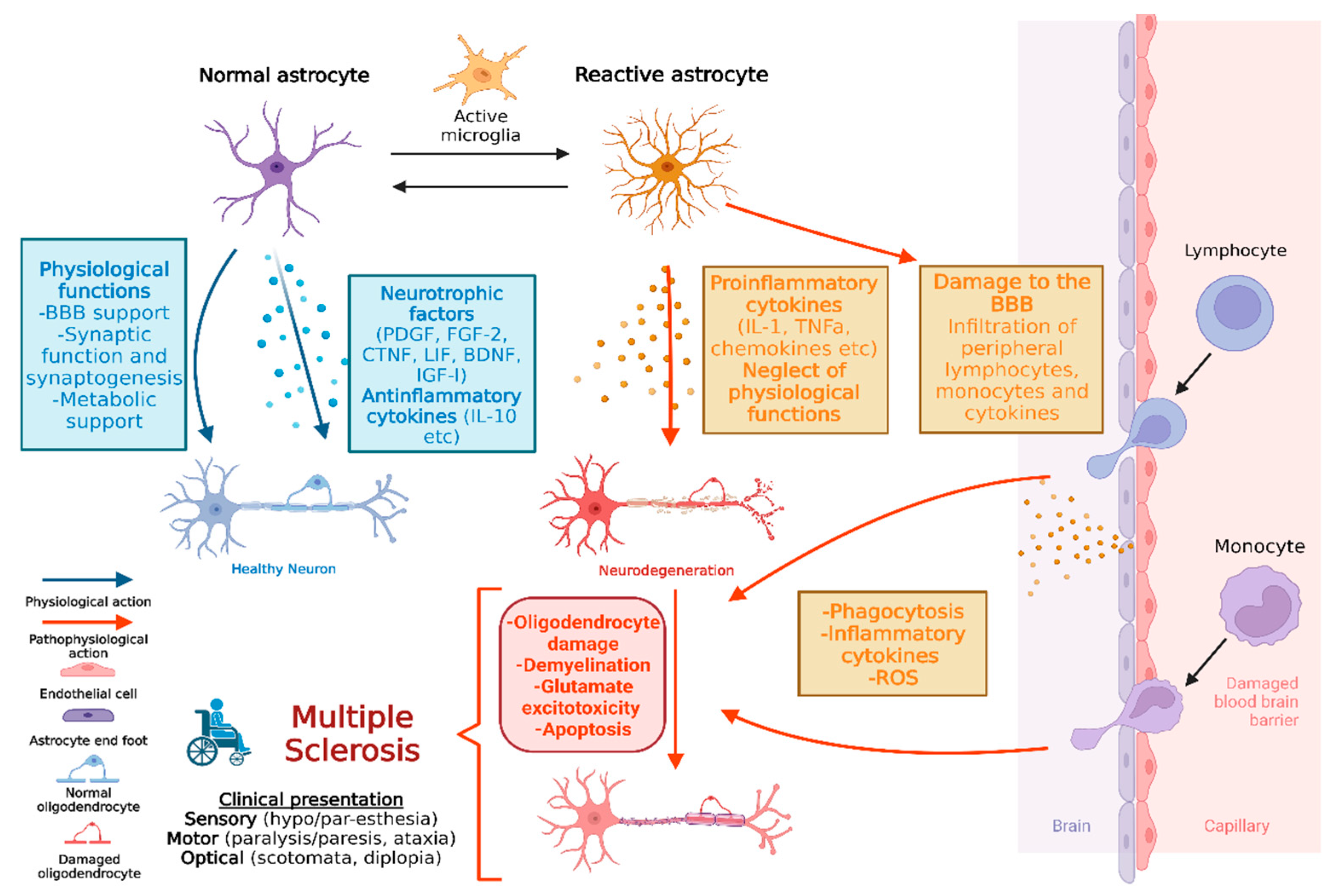
5. The Astrocytic Response during Multiple Sclerosis
6. Established Therapies for Multiple Sclerosis
7. Astrocyte-Selective Therapies: Challenges and Potential Solutions
8. Conclusions—Future Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| IGF-1 | Insulin-like growth factor 1 |
| GFAP | Glial fibrillary acidic protein |
| TNFα | Tumor necrosis factor a |
| BDNF | Brain-derived neurotrophic factor |
| CTNF | Ciliary neurotrophic factor |
| DMTs | Disease-modifying therapies |
| MBP | Myelin basic protein |
| NGF | Nerve growth factor |
| EAE | Experimental autoimmune encephalitis |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| MAVS | Mitochondrial antiviral signaling protein |
| MOG | Myelin oligodendrocyte glycoprotein |
| PLP | Proteolipid protein |
| MCT-1 | Monocarboxylate transporter 1 |
| GLUT-1 | Glucose transporter 1 |
| SREBP | Sterol regulatory element-binding protein |
| EAAT | Excitatory amino acid transporter |
| GLAST | Glutamate/aspartate transporter |
References
- Kıray, H.; Lindsay, S.L.; Hosseinzadeh, S.; Barnett, S.C. The multifaceted role of astrocytes in regulating myelination. Exp. Neurol. 2016, 283 Pt B, 541–549. [Google Scholar] [CrossRef]
- Li, J.; Zhang, L.; Chu, Y.; Namaka, M.; Deng, B.; Kong, J.; Bi, X. Astrocytes in Oligodendrocyte Lineage Development and White Matter Pathology. Front. Cell. Neurosci. 2016, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Ludwin, S.K.; Rao, V.T.; Moore, C.S.; Antel, J.P. Astrocytes in multiple sclerosis. Mult. Scler. 2016, 22, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Planas-Fontánez, T.M.; Sainato, D.M.; Sharma, I.; Dreyfus, C.F. Roles of astrocytes in response to aging, Alzheimer’s disease and multiple sclerosis. Brain Res. 2021, 1764, 147464. [Google Scholar] [CrossRef]
- Cragnolini, A.B.; Lampitella, G.; Virtuoso, A.; Viscovo, I.; Panetsos, F.; Papa, M.; Cirillo, G. Regional brain susceptibility to neurodegeneration: What is the role of glial cells? Neural Regen. Res. 2020, 15, 838–842. [Google Scholar] [CrossRef]
- Kimelberg, H.K.; Nedergaard, M. Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics 2010, 7, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Gómez, M.V.; Alberdi, E.; Pérez-Navarro, E.; Alberch, J.; Matute, C. Bax and calpain mediate excitotoxic oligodendrocyte death induced by activation of both AMPA and kainate receptors. J. Neurosci. 2011, 31, 2996–3006. [Google Scholar] [CrossRef]
- Gomolka, R.S.; Hablitz, L.M.; Mestre, H.; Giannetto, M.; Du, T.; Hauglund, N.L.; Xie, L.; Peng, W.; Martinez, P.M.; Nedergaard, M.; et al. Loss of aquaporin-4 results in glymphatic system dysfunction via brain-wide interstitial fluid stagnation. eLife 2023, 12, e82232. [Google Scholar] [CrossRef]
- Nagelhus, E.A.; Ottersen, O.P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef]
- Katada, R.; Akdemir, G.; Asavapanumas, N.; Ratelade, J.; Zhang, H.; Verkman, A.S. Greatly improved survival and neuroprotection in aquaporin-4-knockout mice following global cerebral ischemia. FASEB J. 2014, 28, 705–714. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Halsey, A.; Wang, M.X.; Törnroth-Horsefield, S.; Conner, A.C.; Badaut, J.; Iliff, J.J.; Bill, R.M. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain 2022, 145, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhong, L.; Geng, J. Neuromyelitis optica spectrum disorder: Pathogenesis, treatment, and experimental models. Mult. Scler. Relat. Disord. 2019, 27, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Paul, F.; Weinshenker, B.G.; Levy, M.; Kim, H.J.; Wildemann, B. Neuromyelitis optica. Nat. Rev. Dis. Primers 2020, 6, 85. [Google Scholar] [CrossRef]
- Carnero Contentti, E.; Correale, J. Neuromyelitis optica spectrum disorders: From pathophysiology to therapeutic strategies. J. Neuroinflammation 2021, 18, 208. [Google Scholar] [CrossRef] [PubMed]
- Höftberger, R.; Lassmann, H. Inflammatory demyelinating diseases of the central nervous system. Handb. Clin. Neurol. 2018, 145, 263–283. [Google Scholar] [CrossRef]
- Kofuji, P.; Newman, E.A. Potassium buffering in the central nervous system. Neuroscience 2004, 129, 1045–1056. [Google Scholar] [CrossRef]
- Wu, Y.; Dissing-Olesen, L.; MacVicar, B.A.; Stevens, B. Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. 2015, 36, 605–613. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur. J. Neurosci. 1998, 10, 2129–2142. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef]
- De Luca, C.; Colangelo, A.M.; Alberghina, L.; Papa, M. Neuro-Immune Hemostasis: Homeostasis and Diseases in the Central Nervous System. Front. Cell. Neurosci. 2018, 12, 459. [Google Scholar] [CrossRef]
- Ishibashi, T.; Dakin, K.A.; Stevens, B.; Lee, P.R.; Kozlov, S.V.; Stewart, C.L.; Fields, R.D. Astrocytes promote myelination in response to electrical impulses. Neuron 2006, 49, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Bellosta, S. Cholesterol: Its regulation and role in central nervous system disorders. Cholesterol 2012, 2012, 292598. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-P.; Tang, Y.; Zhou, S.; Toh, B.H.; McLean, C.; Li, H. Cholesterol involvement in the pathogenesis of neurodegenerative diseases. Mol. Cell. Neurosci. 2010, 43, 33–42. [Google Scholar] [CrossRef]
- Saher, G.; Stumpf, S.K. Cholesterol in myelin biogenesis and hypomyelinating disorders. Biochim. Biophys. Acta 2015, 1851, 1083–1094. [Google Scholar] [CrossRef]
- Theotokis, P.; Lourbopoulos, A.; Touloumi, O.; Lagoudaki, R.; Kofidou, E.; Nousiopoulou, E.; Poulatsidou, K.-N.; Kesidou, E.; Tascos, N.; Spandou, E.; et al. Time course and spatial profile of Nogo-A expression in experimental autoimmune encephalomyelitis in C57BL/6 mice. J. Neuropathol. Exp. Neurol. 2012, 71, 907–920. [Google Scholar] [CrossRef]
- Theotokis, P.; Touloumi, O.; Lagoudaki, R.; Nousiopoulou, E.; Kesidou, E.; Siafis, S.; Tselios, T.; Lourbopoulos, A.; Karacostas, D.; Grigoriadis, N.; et al. Nogo receptor complex expression dynamics in the inflammatory foci of central nervous system experimental autoimmune demyelination. J. Neuroinflamm. 2016, 13, 265. [Google Scholar] [CrossRef]
- Skripuletz, T.; Hackstette, D.; Bauer, K.; Gudi, V.; Pul, R.; Voss, E.; Berger, K.; Kipp, M.; Baumgärtner, W.; Stangel, M. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain 2013, 136 Pt 1, 147–167. [Google Scholar] [CrossRef]
- Traiffort, E.; Kassoussi, A.; Zahaf, A.; Laouarem, Y. Astrocytes and Microglia as Major Players of Myelin Production in Normal and Pathological Conditions. Front. Cell. Neurosci. 2020, 14, 79. [Google Scholar] [CrossRef]
- Itoh, N.; Itoh, Y.; Tassoni, A.; Ren, E.; Kaito, M.; Ohno, A.; Ao, Y.; Farkhondeh, V.; Johnsonbaugh, H.; Burda, J.; et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl. Acad. Sci. USA 2018, 115, E302–E309. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer-Guglielmi, B.; Fleckenstein, B.; Jung, G.; Hamprecht, B. Immunocytochemical localization of glycogen phosphorylase isozymes in rat nervous tissues by using isozyme-specific antibodies. J. Neurochem. 2003, 85, 73–81. [Google Scholar] [CrossRef]
- Baltan, S. Can lactate serve as an energy substrate for axons in good times and in bad, in sickness and in health? Metab. Brain Dis. 2015, 30, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Tekkök, S.B.; Brown, A.M.; Westenbroek, R.; Pellerin, L.; Ransom, B.R. Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J. Neurosci. Res. 2005, 81, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Rinholm, J.E.; Hamilton, N.B.; Kessaris, N.; Richardson, W.D.; Bergersen, L.H.; Attwell, D. Regulation of oligodendrocyte development and myelination by glucose and lactate. J. Neurosci. 2011, 31, 538–548. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.; Nouri, N.; Zhou, L.; Giffard, R.; Barres, B. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Jasmin, L.; Ohara, P.T. Remyelination within the CNS: Do schwann cells pave the way for oligodendrocytes? Neuroscientist 2002, 8, 198–203. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Fonken, L.K. Glial Cells Shape Pathology and Repair After Spinal Cord Injury. Neurotherapeutics 2018, 15, 554–577. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Reactive astrocytes in neural repair and protection. Neuroscientist 2005, 11, 400–407. [Google Scholar] [CrossRef]
- Dugast, E.; Shah, S.; Laplaud, D.-A. Single-Cell Analysis to Better Understand the Mechanisms Involved in MS. Int. J. Mol. Sci. 2022, 23, 12142. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.A.; Clark, I.C.; Tjon, E.C.; Li, Z.; Zandee, S.E.J.; Couturier, C.P.; Watson, B.R.; Scalisi, G.; Alkwai, S.; Rothhammer, V.; et al. MAFG-driven astrocytes promote CNS inflammation. Nature 2020, 578, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Kihara, Y.; Zhu, Y.; Jonnalagadda, D.; Romanow, W.; Palmer, C.; Siddoway, B.; Rivera, R.; Dutta, R.; Trapp, B.D.; Chun, J. Single-Nucleus RNA-seq of Normal-Appearing Brain Regions in Relapsing-Remitting vs. Secondary Progressive Multiple Sclerosis: Implications for the Efficacy of Fingolimod. Front. Cell. Neurosci. 2022, 16, 918041. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-G.; Wheeler, M.A.; Quintana, F.J. Function and therapeutic value of astrocytes in neurological diseases. Nat. Rev. Drug Discov. 2022, 21, 339–358. [Google Scholar] [CrossRef]
- Hassanzadeh, S.; Jalessi, M.; Jameie, S.B.; Khanmohammadi, M.; Bagher, Z.; Namjoo, Z.; Davachi, S.M. More attention on glial cells to have better recovery after spinal cord injury. Biochem. Biophys. Rep. 2021, 25, 100905. [Google Scholar] [CrossRef]
- Cichorek, M.; Kowiański, P.; Lietzau, G.; Lasek, J.; Moryś, J. Neuroglia—Development and role in physiological and pathophysiological processes. Folia. Morphol. 2021, 80, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Torres, C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18, e12937. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Arellano, J.J.; Parpura, V.; Zorec, R.; Verkhratsky, A. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 2016, 323, 170–182. [Google Scholar] [CrossRef]
- Fabricius, K.; Jacobsen, J.S.; Pakkenberg, B. Effect of age on neocortical brain cells in 90+ year old human females—A cell counting study. Neurobiol. Aging 2013, 34, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef]
- Kawano, H.; Katsurabayashi, S.; Kakazu, Y.; Yamashita, Y.; Kubo, N.; Kubo, M.; Okuda, H.; Takasaki, K.; Kubota, K.; Mishima, K.; et al. Long-term culture of astrocytes attenuates the readily releasable pool of synaptic vesicles. PLoS ONE 2012, 7, e48034. [Google Scholar] [CrossRef]
- Bitto, A.; Sell, C.; Crowe, E.; Lorenzini, A.; Malaguti, M.; Hrelia, S.; Torres, C. Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 2010, 316, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Souza, D.G.; Bellaver, B.; Souza, D.O.; Quincozes-Santos, A. Characterization of adult rat astrocyte cultures. PLoS ONE 2013, 8, e60282. [Google Scholar] [CrossRef]
- Nichols, N.R.; Day, J.R.; Laping, N.J.; Johnson, S.A.; Finch, C.E. GFAP mRNA increases with age in rat and human brain. Neurobiol. Aging 1993, 14, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Safaiyan, S.; Kannaiyan, N.; Snaidero, N.; Brioschi, S.; Biber, K.; Yona, S.; Edinger, A.L.; Jung, S.; Rossner, M.J.; Simons, M. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 2016, 19, 995–998. [Google Scholar] [CrossRef]
- Lalo, U.; Rasooli-Nejad, S.; Pankratov, Y. Exocytosis of gliotransmitters from cortical astrocytes: Implications for synaptic plasticity and aging. Biochem. Soc. Trans. 2014, 42, 1275–1281. [Google Scholar] [CrossRef]
- Potokar, M.; Vardjan, N.; Stenovec, M.; Gabrijel, M.; Trkov, S.; Jorgačevski, J.; Kreft, M.; Zorec, R. Astrocytic vesicle mobility in health and disease. Int. J. Mol. Sci. 2013, 14, 11238–11258. [Google Scholar] [CrossRef]
- Boisvert, M.M.; Erikson, G.A.; Shokhirev, M.N.; Allen, N.J. The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 2018, 22, 269–285. [Google Scholar] [CrossRef]
- Mangold, C.A.; Masser, D.R.; Stanford, D.R.; Bixler, G.V.; Pisupati, A.; Giles, C.B.; Wren, J.D.; Ford, M.M.; Sonntag, W.E.; Freeman, W.M. CNS-wide Sexually Dimorphic Induction of the Major Histocompatibility Complex 1 Pathway with Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 16–29. [Google Scholar] [CrossRef]
- Nag, T.C.; Wadhwa, S. Accumulation of lipid inclusions in astrocytes of aging human optic nerve. Acta Biol. Hung 2012, 63 (Suppl. S1), 54–64. [Google Scholar] [CrossRef]
- Palmer, A.L.; Ousman, S.S. Astrocytes and Aging. Front. Aging Neurosci. 2018, 10, 337. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-M.; Han, Y.-W.; Han, X.-H.; Zhang, K.; Chang, Y.-N.; Hu, Z.-M.; Qi, H.-X.; Ting, C.; Zhen, Z.; Hong, W. Upstream regulators and downstream effectors of NF-κB in Alzheimer’s disease. J. Neurol. Sci. 2016, 366, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Soreq, L.; UK Brain Expression Consortium; North American Brain Expression Consortium; Rose, J.; Soreq, E.; Hardy, J.; Trabzuni, D.; Cookson, M.R.; Smith, C.; Ryten, M.; et al. Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep. 2017, 18, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Moccia, M.; Coetzee, T.; Cohen, J.A.; Correale, J.; Graves, J.; Marrie, R.A.; Montalban, X.; Yong, V.W.; Thompson, A.J.; et al. Multiple sclerosis progression: Time for a new mechanism-driven framework. Lancet Neurol. 2023, 22, 78–88. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Jaronen, M.; Covacu, R.; Zandee, S.E.J.; Scalisi, G.; Rothhammer, V.; Tjon, E.C.; Chao, C.-C.; Kenison, J.E.; Blain, M.; et al. Environmental Control of Astrocyte Pathogenic Activities in CNS Inflammation. Cell 2019, 176, 581–596.e18. [Google Scholar] [CrossRef]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNFalpha: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef]
- Chao, C.-C.; Gutiérrez-Vázquez, C.; Rothhammer, V.; Mayo, L.; Wheeler, M.A.; Tjon, E.C.; Zandee, S.E.J.; Blain, M.; de Lima, K.A.; Takenaka, M.C.; et al. Metabolic Control of Astrocyte Pathogenic Activity via cPLA2-MAVS. Cell 2019, 179, 1483–1498.e22. [Google Scholar] [CrossRef]
- Stadelmann, C.; Kerschensteiner, M.; Misgeld, T.; Brück, W.; Hohlfeld, R.; Lassmann, H. BDNF and gp145trkB in multiple sclerosis brain lesions: Neuroprotective interactions between immune and neuronal cells? Brain 2002, 125 Pt 1, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Frederick, T.J.; Miller, S.D. Astrocytes in multiple sclerosis: A product of their environment. Cell. Mol. Life Sci. 2008, 65, 2702–2720. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, M.; Merola, A.; Piacentino, C.; Votta, B.; Capello, E.; Mancardi, G.L.; Mutani, R.; Giordana, M.T.; Cavalla, P. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: Correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J. Neuropathol. Exp. Neurol. 2007, 66, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, C.F.; Raine, C.S. The astrocyte in multiple sclerosis revisited. Glia 2013, 61, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, R.T.; Habib, S.; Sangaraju, S.L.; Yepez, D.; Grandes, X.A. Multiple Sclerosis: Therapeutic Strategies on the Horizon. Cureus 2022, 14, e24895. [Google Scholar] [CrossRef]
- De Angelis, F.; John, N.A.; Brownlee, W.J. Disease-modifying therapies for multiple sclerosis. BMJ 2018, 363, k4674. [Google Scholar] [CrossRef]
- Frohman, E.M.; Shah, A.; Eggenberger, E.; Metz, L.; Zivadinov, R.; Stüve, O. Corticosteroids for multiple sclerosis: I. Application for treating exacerbations. Neurotherapeutics 2007, 4, 618–626. [Google Scholar] [CrossRef]
- Keegan, M.; König, F.; McClelland, R.; Brück, W.; Morales, Y.; Bitsch, A.; Panitch, H.; Lassmann, H.; Weinshenker, B.; Rodriguez, M.; et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet 2005, 366, 579–582. [Google Scholar] [CrossRef]
- Piehl, F. Current and emerging disease-modulatory therapies and treatment targets for multiple sclerosis. J. Intern. Med. 2021, 289, 771–791. [Google Scholar] [CrossRef]
- Aharoni, R.; Teitelbaum, D.; Leitner, O.; Meshorer, A.; Sela, M.; Arnon, R. Specific Th2 cells accumulate in the central nervous system of mice protected against experimental autoimmune encephalomyelitis by copolymer 1. Proc. Natl. Acad. Sci. USA 2000, 97, 11472–11477. [Google Scholar] [CrossRef]
- Bar-Or, A.; Pachner, A.; Menguy-Vacheron, F.; Kaplan, J.; Wiendl, H. Teriflunomide and its mechanism of action in multiple sclerosis. Drugs 2014, 74, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Dimethyl Fumarate: A Review in Relapsing-Remitting MS. Drugs 2016, 76, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, B.Z.; Cohen, J.A.; Conway, D.S. Sphingosine 1-Phosphate Receptor Modulators for the Treatment of Multiple Sclerosis. Neurotherapeutics 2017, 14, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.M.; Ammoscato, F.; Giovannoni, G.; Baker, D.; Schmierer, K. Cladribine: Mechanisms and mysteries in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1266–1271. [Google Scholar] [CrossRef]
- Neuhaus, O.; Kieseier, B.C.; Hartung, H.-P. Mechanisms of mitoxantrone in multiple sclerosis--what is known? J. Neurol. Sci. 2004, 223, 25–27. [Google Scholar] [CrossRef]
- Outteryck, O. Natalizumab in relapsing-remitting multiple sclerosis. Expert Rev. Neurother. 2016, 16, 471–481. [Google Scholar] [CrossRef]
- Havrdova, E.; Horakova, D.; Kovarova, I. Alemtuzumab in the treatment of multiple sclerosis: Key clinical trial results and considerations for use. Ther. Adv. Neurol. Disord. 2015, 8, 31–45. [Google Scholar] [CrossRef]
- Syed, Y.Y. Ocrelizumab: A Review in Multiple Sclerosis. CNS Drugs 2018, 32, 883–890. [Google Scholar] [CrossRef]
- Leslie, C.C. Cytosolic phospholipase A2: Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef]
- Venier, R.E.; Igdoura, S.A. Miglustat as a therapeutic agent: Prospects and caveats. J. Med. Genet. 2012, 49, 591–597. [Google Scholar] [CrossRef]
- Jin, M.-H.; Lee, Y.-H.; Kim, J.-M.; Sun, H.-N.; Moon, E.-Y.; Shong, M.H.; Kim, S.-U.; Lee, S.H.; Lee, T.-H.; Yu, D.-Y.; et al. Characterization of neural cell types expressing peroxiredoxins in mouse brain. Neurosci. Lett. 2005, 381, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.-M.; Park, K.-R.; Kim, E.-C.; Hong, J.T. PRDX6 controls multiple sclerosis by suppressing inflammation and blood brain barrier disruption. Oncotarget 2015, 6, 20875–20884. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Fernández, M.; de la Cuesta, F.; Tallón, A.; Cuesta, I.; Fernández-Fournier, M.; Laso-García, F.; Gómez-de Frutos, M.C.; Díez-Tejedor, E.; Otero-Ortega, L. Potential Roles of Extracellular Vesicles as Biomarkers and a Novel Treatment Approach in Multiple Sclerosis. Int. J. Mol. Sci. 2021, 22, 9011. [Google Scholar] [CrossRef]
- Sáenz-Cuesta, M.; Alberro, A.; Muñoz-Culla, M.; Osorio-Querejeta, I.; Fernandez-Mercado, M.; Lopetegui, I.; Tainta, M.; Prada, Á.; Castillo-Triviño, T.; Falcón-Pérez, J.M.; et al. The First Dose of Fingolimod Affects Circulating Extracellular Vesicles in Multiple Sclerosis Patients. Int. J. Mol. Sci. 2018, 19, 2448. [Google Scholar] [CrossRef] [PubMed]
- Getts, D.R.; Martin, A.J.; McCarthy, D.P.; Terry, R.L.; Hunter, Z.N.; Yap, W.T.; Getts, M.T.; Pleiss, M.; Luo, X.; King, N.J.C.; et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat. Biotechnol. 2012, 30, 1217–1224. [Google Scholar] [CrossRef]


{kind=link}
| Molecule | Effects |
|---|---|
| BDNF | Promotion of remyelination |
| CTNF | Enhancement of OPC migration, protection from apoptosis and decrease myelin destruction |
| FGF2 | OPCs proliferation, disruption of OPC differentiation (in combination with PDGF) |
| PDGF | Proliferation and survival of oligodendrocytes and OPCs migration |
| LIF | Survival, differentiation and maturation of oligodendrocytes |
| IL-1 | Induction of apoptosis and obstruction of myelination |
| TNFα | Inhibition the differentiation and proliferation of OPCs |
| IFNγ | Reduction in OPCs proliferation, promotion of OPCs survival, and differentiation through induction of PDGF |
| IL-6 | Reduction in OPCs differentiation |
| CXCL1 | Inhibition of OPCs migration |
| Subpopulation Type | Markers |
|---|---|
| Pro-inflammatory | CD44, FOS, BLC6 |
| Pro-inflammatory | NRF2, MAFG, GM-CSF |
| Pro-inflammatory | C3 |
| Anti-inflammatory | LAMP1, TRAIL |
| Drug Class | Active Substance | Mechanism of Action |
|---|---|---|
| Interferons | IFNβ-1a, IFNβ-1b, pegylated IFNs | Increase in anti-inflammatory and trophic factors |
| Immunomodulators | Glatiramer acetate | Alteration of immune response towards the anti-inflammatory T helper 2 type |
| Teriflunomide | Inhibition of active T and B lymphocytes proliferation | |
| Dimethyl fumarate | Unknown mechanism, possible enhancement of oxidative stress protection | |
| Sphingosine 1-Phosphate (S1P) Receptor (S1PR) modulators | Fingolimod, Ozanimod, Siponimod | Prevention of T, B and NK cells lymph node mobilization |
| Monoclonal antibodies | Natalizumab | Blockage of CNS infiltration |
| Alemtuzumab | Depletion of T and B cells by targeting CD52 surface molecule | |
| Ocrelizumab | Depletion of B cells by targeting CD20 surface molecule | |
| Cell-depleting agents | Cladribine | Depletion of T, B cells and dendritic cells by targeting DNA metabolism |
| Mitoxantrone | Depletion of T, B cells and dendritic cells by targeting DNA metabolism and anti-inflammatory action |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barmpagiannos, K.; Theotokis, P.; Petratos, S.; Pagnin, M.; Einstein, O.; Kesidou, E.; Boziki, M.; Artemiadis, A.; Bakirtzis, C.; Grigoriadis, N. The Diversity of Astrocyte Activation during Multiple Sclerosis: Potential Cellular Targets for Novel Disease Modifying Therapeutics. Healthcare 2023, 11, 1585. https://doi.org/10.3390/healthcare11111585
Barmpagiannos K, Theotokis P, Petratos S, Pagnin M, Einstein O, Kesidou E, Boziki M, Artemiadis A, Bakirtzis C, Grigoriadis N. The Diversity of Astrocyte Activation during Multiple Sclerosis: Potential Cellular Targets for Novel Disease Modifying Therapeutics. Healthcare. 2023; 11(11):1585. https://doi.org/10.3390/healthcare11111585
Chicago/Turabian StyleBarmpagiannos, Konstantinos, Paschalis Theotokis, Steven Petratos, Maurice Pagnin, Ofira Einstein, Evangelia Kesidou, Marina Boziki, Artemios Artemiadis, Christos Bakirtzis, and Nikolaos Grigoriadis. 2023. "The Diversity of Astrocyte Activation during Multiple Sclerosis: Potential Cellular Targets for Novel Disease Modifying Therapeutics" Healthcare 11, no. 11: 1585. https://doi.org/10.3390/healthcare11111585