

Bayesian Inference Algorithm for Estimating Heterogeneity of Regulatory Mechanisms Based on Single-Cell Data

Abstract
:1. Introduction
2. Materials and Methods
2.1. Mathematical Model
2.2. Approximation Bayesian Computation
2.3. ABC-SMC with Adaptive Tolerance Threshold
| Algorithm 1 ABC-SMC algorithm. |
|
| Algorithm 2 ABC-PMC with adaptive tolerance threshold (ABC-CPMC). |
|
3. Results and Discussion
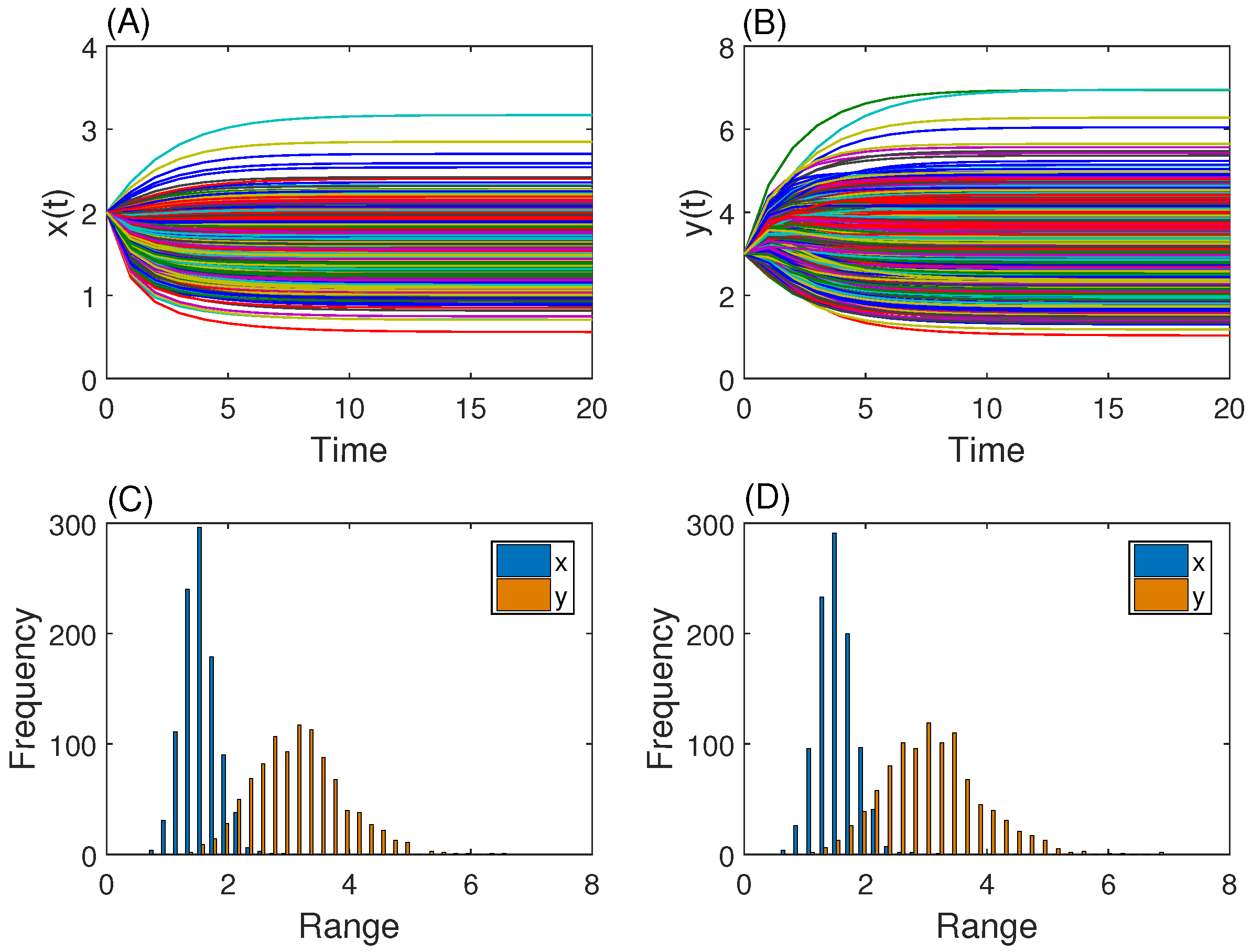
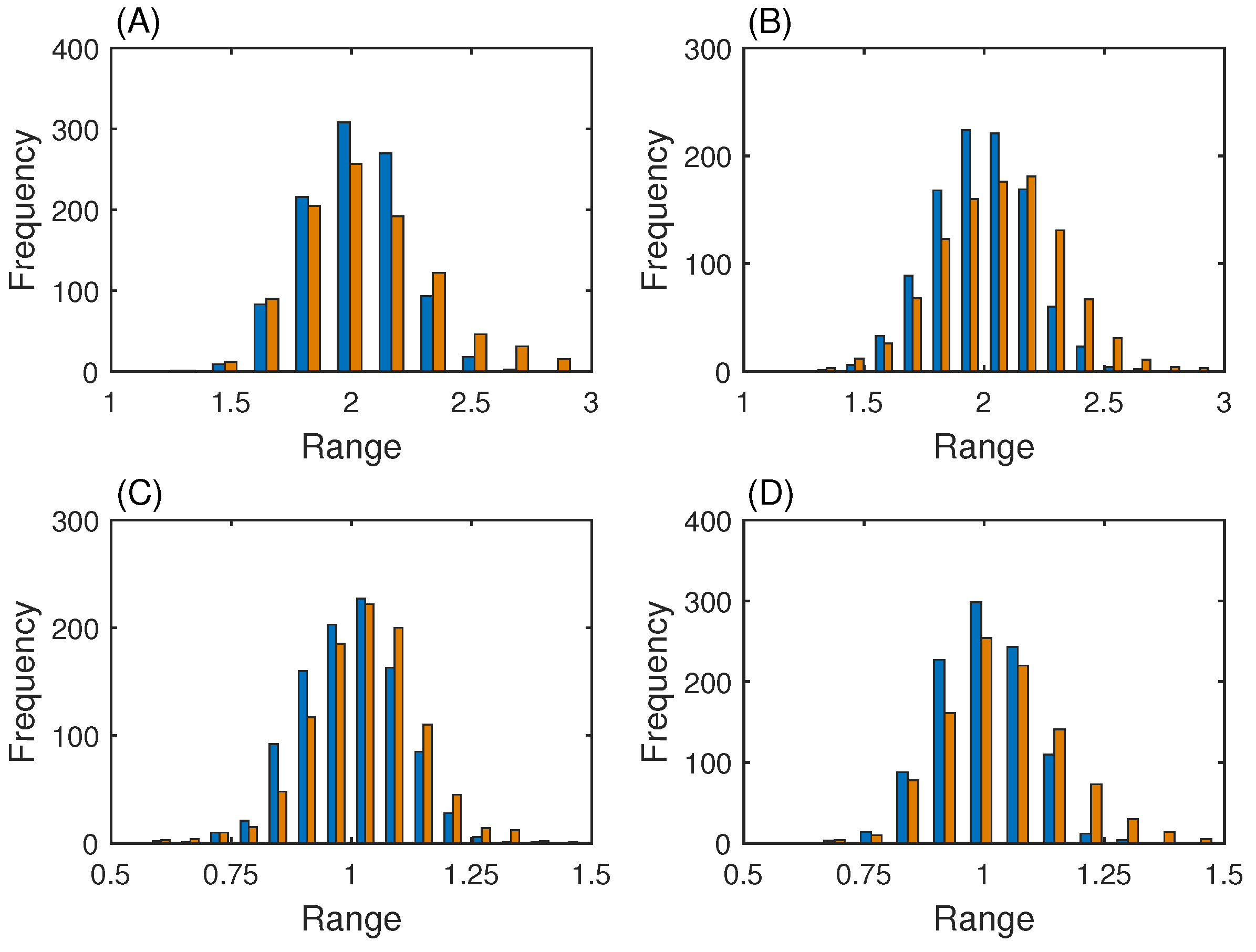
3.1. Gene Network with One Steady State
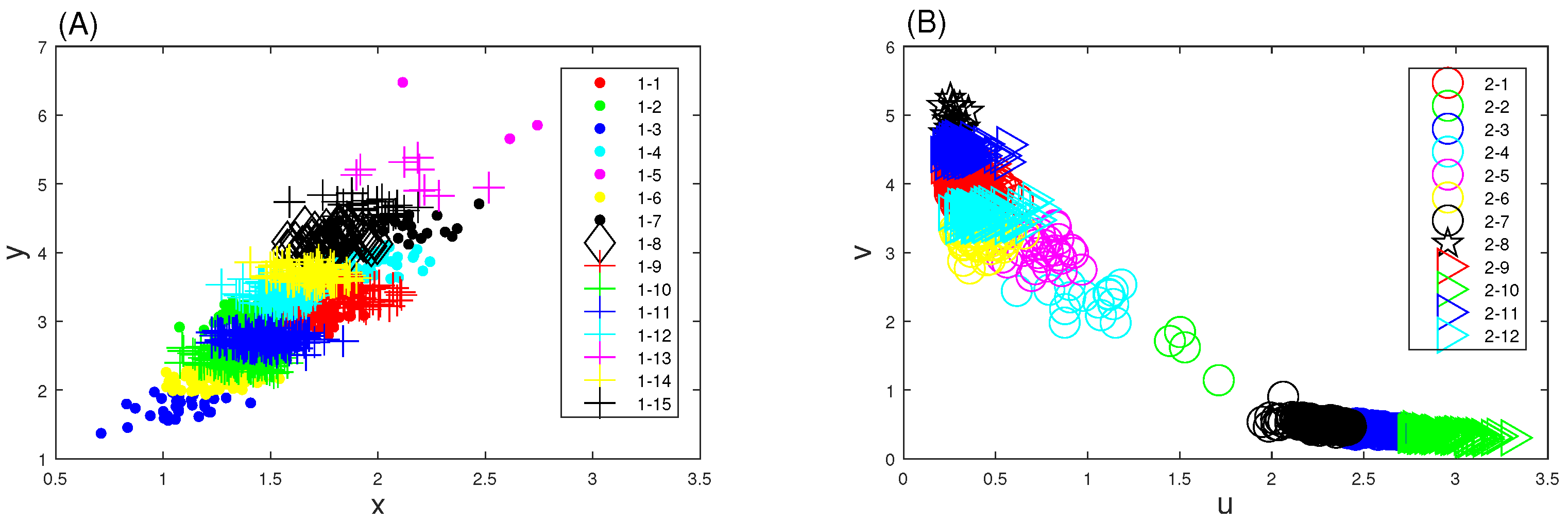
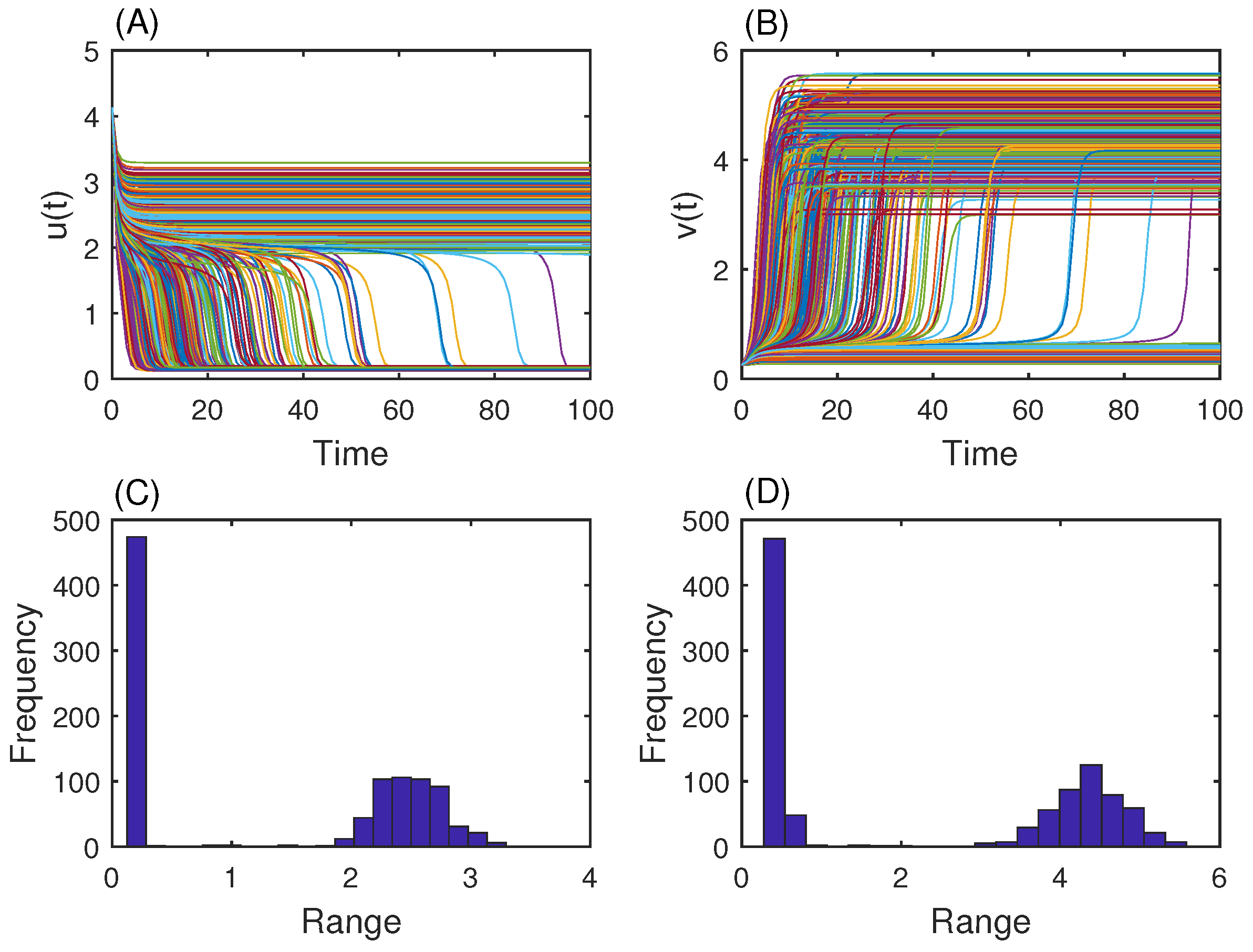
3.2. Genetic Toggle Switch Showing Bistability
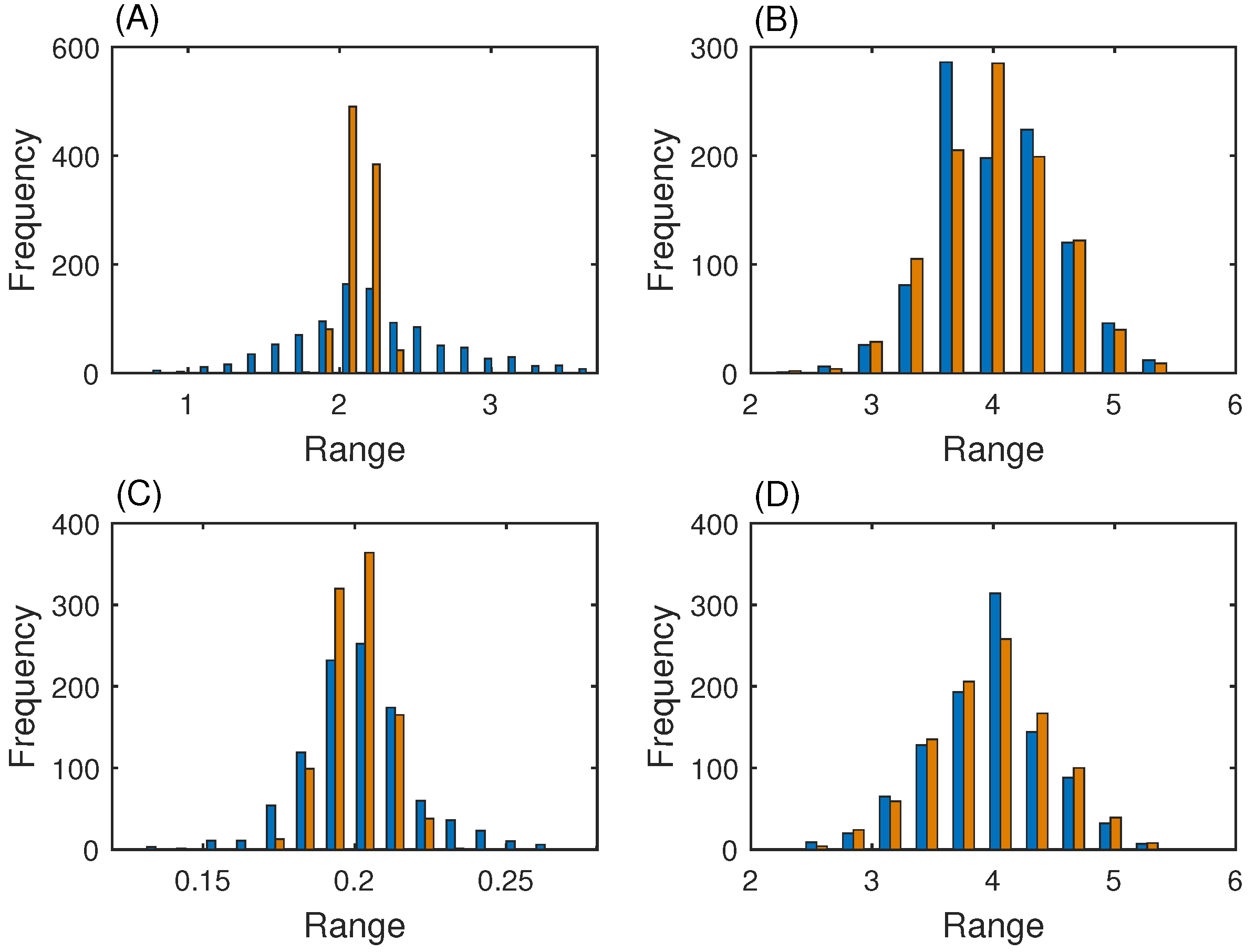
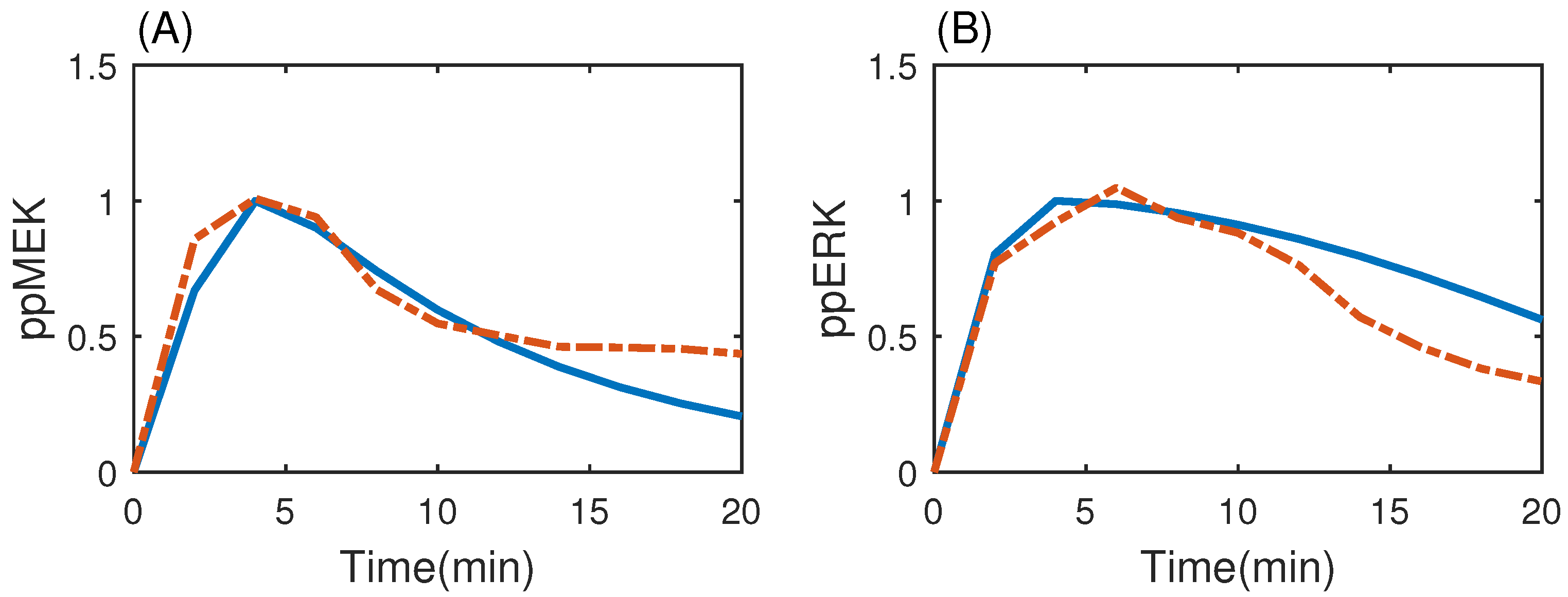
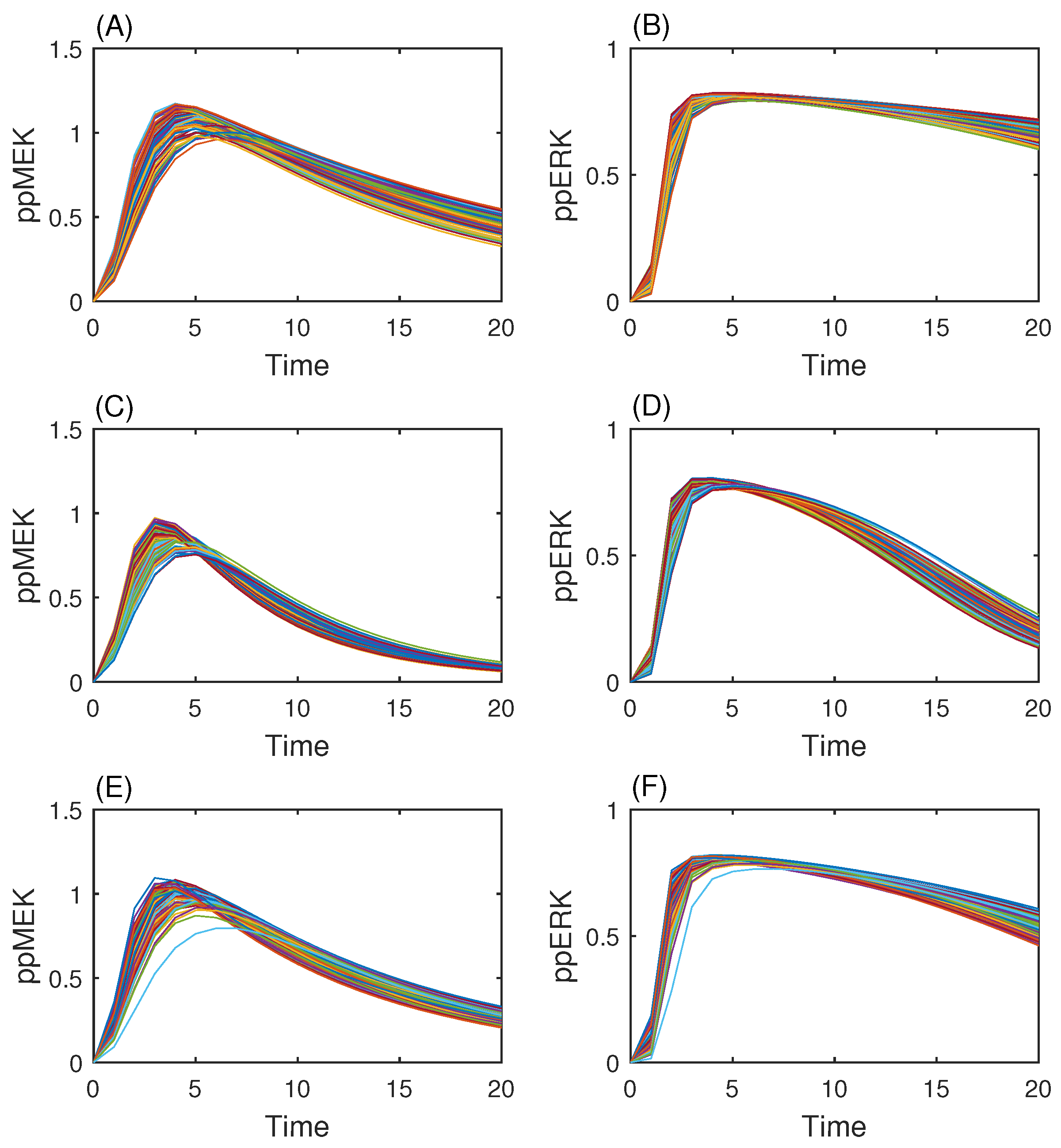
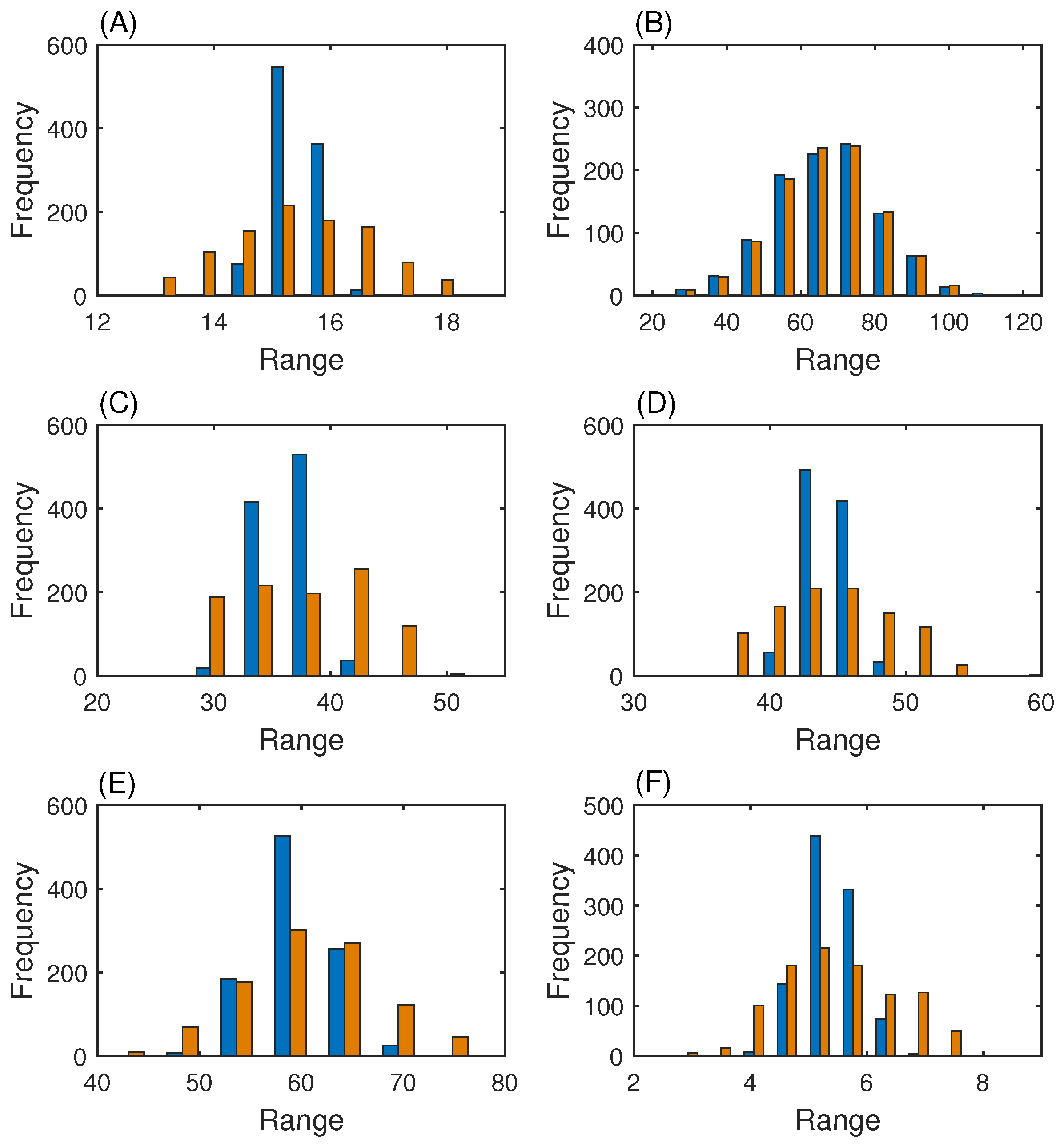
3.3. MAP Kinase Pathway for Efficiency Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taniguchi, Y.; Choi, P.J.; Li, G.W.; Chen, H.; Babu, M.; Hearn, J.; Emili, A.; Xie, X.S. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 2010, 329, 533–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junker, J.P.; van Oudenaarden, A. Every cell is special: Genome-wide studies add a new dimension to single-cell biology. Cell 2014, 157, 8–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, A.J.; Spelke, D.P.; Xu, Z.; Kang, C.C.; Schaffer, D.V.; Herr, A.E. Single-cell western blotting. Nat. Methods 2014, 11, 749–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Q.; Ramsköld, D.; Reinius, B.; Sandberg, R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 2014, 343, 193–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, T. Long-term single-cell imaging of mammalian stem cells. Nat. Methods 2011, 8, S30–S35. [Google Scholar] [CrossRef]
- Bodenmiller, B.; Zunder, E.R.; Finck, R.; Chen, T.J.; Savig, E.S.; Bruggner, R.V.; Simonds, E.F.; Bendall, S.C.; Sachs, K.; Krutzik, P.O.; et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat. Biotechnol. 2012, 30, 858–867. [Google Scholar] [CrossRef]
- Davey, H.M.; Kell, D.B. Flow cytometry and cell sorting of heterogeneous microbial populations: The importance of single-cell analyses. Microbiol. Res. 1996, 60, 641–696. [Google Scholar]
- Gaudet, S.; Miller-Jensen, K. Redefining Signaling Pathways with an Expanding Single-Cell Toolbox. Trends Biotechnol. 2016, 34, 458–469. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, M.; Janzén, D.L.; Durrieu, L.; Colman-Lerner, A.; Kjellsson, M.C.; Cedersund, G. Nonlinear mixed-effects modelling for single cell estimation: When, why, and how to use it. BMC Syst. Biol. 2015, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Dharmarajan, L.; Kaltenbach, H.M.; Rudolf, F.; Stelling, J. A Simple and Flexible Computational Framework for Inferring Sources of Heterogeneity from Single-Cell Dynamics. Cell Syst. 2019, 8, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Llamosi, A.; González-Vargas, A.M.; Versari, C.; Cinquemani, E.; Ferrari-Trecate, G.; Hersen, P.; Batt, G. What Population Reveals about Individual Cell Identity: Single-Cell Parameter Estimation of Models of Gene Expression in Yeast. PLoS Comput. Biol. 2016, 12, e1004706. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, F.; Reiser, A.; Fink, L.; Woschée, D.; Ligon, T.; Theis, F.J.; Rädler, J.O.; Hasenauer, J. Multi-experiment nonlinear mixed effect modeling of single-cell translation kinetics after transfection. NPJ Syst. Biol. Appl. 2018, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Seok, S.C.; Vieland, V.J.; Das, J. Cell responses only partially shape cell-to-cell variations in protein abundances in Escherichia coli chemotaxis. Proc. Natl. Acad. Sci. USA 2013, 110, 18531–18536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippi, S.; Barnes, C.P.; Kirk, P.D.; Kudo, T.; Kunida, K.; McMahon, S.S.; Tsuchiya, T.; Wada, T.; Kuroda, S.; Stumpf, M.P. Robustness of MEK-ERK Dynamics and Origins of Cell-to-Cell Variability in MAPK Signaling. Cell Rep. 2016, 15, 2524–2535. [Google Scholar] [CrossRef] [Green Version]
- Hasenauer, J.; Hasenauer, C.; Hucho, T.; Theis, F.J. ODE constrained mixture modelling: A method for unraveling subpopulation structures and dynamics. PLoS Comput. Biol. 2014, 10, e1003686. [Google Scholar] [CrossRef] [Green Version]
- Bijman, E.Y.; Kaltenbach, H.M.; Stelling, J. Experimental analysis and modeling of single-cell time-course data. Curr. Opin. Syst. Biol. 2021, 28, 100359. [Google Scholar] [CrossRef]
- Loos, C.; Hasenauer, J. Mathematical modeling of variability in intracellular signaling. Curr. Opin. Syst. Biol. 2019, 16, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Lillacci, G.; Khammash, M. Parameter Estimation and Model Selection in Computational Biology. PLoS Comput. Biol. 2010, 6, e1000696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moles, C.G.; Mendes, P.; Banga, J.R. Parameter estimation in biochemical pathways: A comparison of global optimization methods. Genome Res. 2003, 13, 2467–2474. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, D.J. Bayesian methods in bioinformatics and computational systems biology. Briefings Bioinform. 2007, 8, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, A.; Lu, L.; Raissi, M.; Karniadakis, G.E. Systems biology informed deep learning for inferring parameters and hidden dynamics. PLoS Comput. Biol. 2020, 16, e1007575. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.M.; Van Zandt, T. A tutorial on approximate Bayesian computation. J. Math. Psychol. 2012, 56, 69–85. [Google Scholar] [CrossRef]
- Sisson, S.A.; Fan, Y.; Tanaka, M.M. Sequential Monte Carlo without likelihoods. Proc. Natl. Acad. Sci. USA 2007, 104, 1760–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toni, T.; Welch, D.; Strelkowa, N.; Ipsen, A.; Stumpf, M.P. Approximate Bayesian computation scheme for parameter inference and model selection in dynamical systems. J. R. Soc. Interface 2009, 6, 187–202. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.Q.; Smith-Miles, K.; Tian, T. Approximate Bayesian computation schemes for parameter inference of discrete stochastic models using simulated likelihood density. BMC Bioinform. 2014, 15, S3. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Zhang, X.; Tian, T. Inference of model parameters using particle filter algorithm and Copula distributions. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 1231–1240. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J.; Tian, T. Bayesian inference of stochastic dynamic models using early-rejection methods based on sequential stochastic simulations. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 1484–1495. [Google Scholar] [CrossRef]
- Lenormand, M.; Jabot, F.; Deffuant, G. Adaptive approximate Bayesian computation for complex models. Comput. Stat. 2013, 28, 2777–2796. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Xia, P.; Zhang, X.; Tian, T. A Bayesian framework for inferring heterogeneity of cellular processes using single-cell data. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine, Houston, TX, USA, 9–12 December 2021; pp. 2142–2146. [Google Scholar]
- Toni, T.; Stumpf, M.P.H. Parameter inference and model selection in signaling pathway models. Methods Mol. Biol. 2010, 673, 283–295. [Google Scholar]
- Tian, T.; Burrage, K. Stochastic models for regulatory networks of the genetic toggle switch. Proc. Natl. Acad. Sci. USA 2006, 103, 8372–8377. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Kærn, M.; Araki, M.; Chung, K.; Gardner, T.S.; Cantor, C.R.; Collins, J.J. Programmable cells: Interfacing natural and engineered gene networks. Proc. Natl. Acad. Sci. USA 2004, 101, 8414–8419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Song, J. Mathematical modelling of the MAP kinase pathway using proteomic datasets. PLoS ONE 2012, 7, e42230. [Google Scholar] [CrossRef]
- Schilling, M.; Maiwald, T.; Hengl, S.; Winter, D.; Kreutz, C.; Kolch, W.; Lehmann, W.D.; Timmer, J.; Klingmüller, U. Theoretical and experimental analysis links isoform-specific ERK signaling to cell fate decisions. Mol. Syst. Biol. 2009, 5, 334. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Harding, A. How MAP kinase modules function as robust, yet adaptable, circuits. Cell Cycle 2014, 13, 2379–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujioka, A.; Terai, K.; Itoh, R.E.; Aoki, K.; Nakamura, T.; Kuroda, S.; Nishida, E.; Matsuda, M. Dynamics of the Ras/ERK MAPK Cascade as Monitored by Fluorescent Probes. J. Biol. Chem. 2006, 281, 8917–8926. [Google Scholar] [CrossRef] [Green Version]
- Schoeberl, B.; Eichler-Jonsson, C.; Gilles, E.D.; Müller, G. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat. Biotechnol. 2002, 20, 370–375. [Google Scholar] [CrossRef]
- Gao, H. Applied Multivariate Statistical Analysis; Peking University Press: Beijing, China, 2005. [Google Scholar]
- Molla, V.M.G. Sensitivity Analysis for ODEs and DAEs. MATLAB Central File Exchange. 2021. Available online: https://www.mathworks.com/matlabcentral/fileexchange/1480-sensitivity-analysis-for-odes-and-daes (accessed on 8 July 2021).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
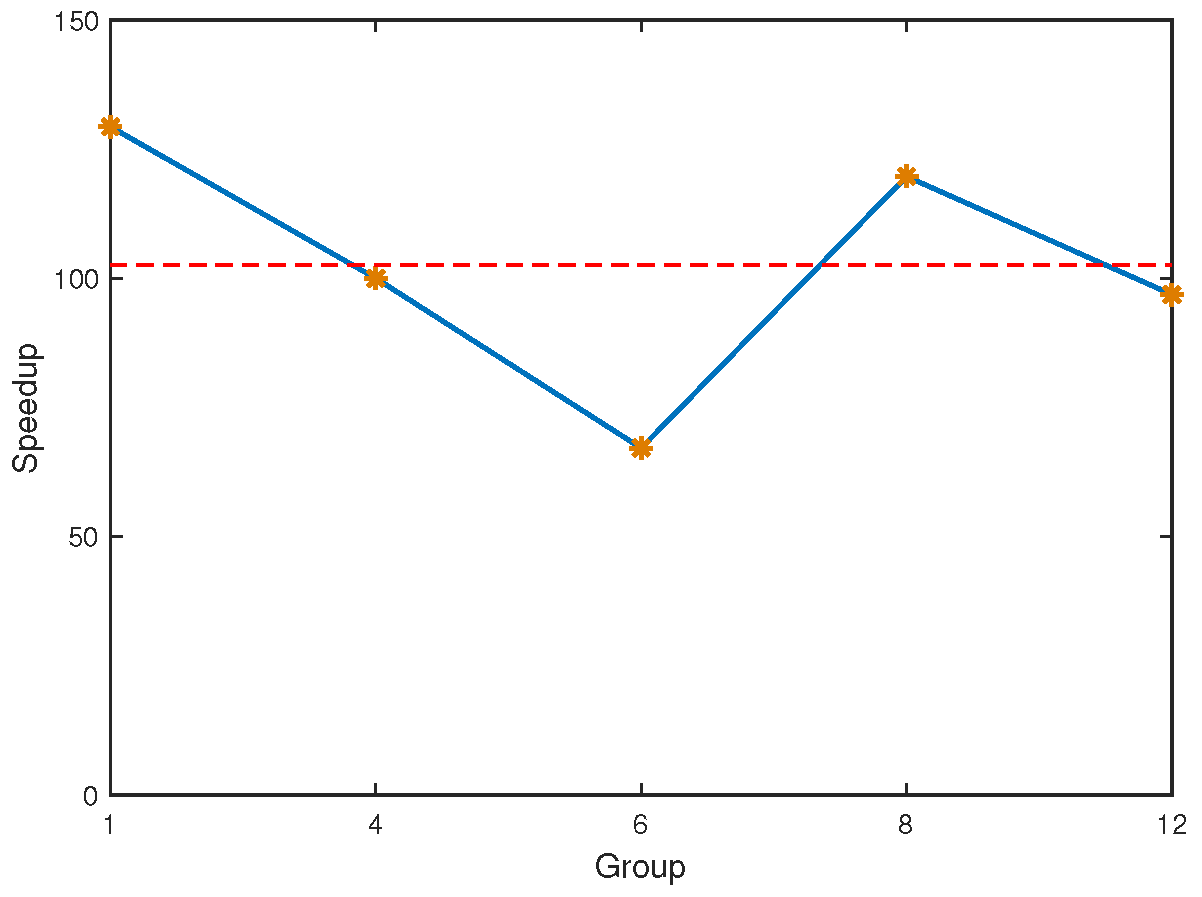{kind=link}
| Kinetic rates | |||||||
| Estimated value | 66.0452 | 0.9584 | 0.0121 | 15.3943 | 35.7607 | 5.7297 | 5.0556 |
| STD | 13.7871 | 0.6568 | 0.0031 | 0.3849 | 0.9216 | 0.1316 | 2.0026 |
| 20 | 0 | 0 | 10 | 25 | 1 | 0 | |
| 120 | 5 | 0.05 | 20 | 50 | 10 | 15 | |
| Kinetic rates | |||||||
| Estimated value | 0.1176 | 60.4114 | 35.3809 | 36.2956 | 16.3216 | 44.2443 | 2.4649 |
| STD | 0.08 | 3.2385 | 1.2341 | 2.0504 | 0.6656 | 1.5363 | 0.0958 |
| 0 | 30 | 25 | 25 | 10 | 30 | 1 | |
| 0.5 | 80 | 50 | 50 | 25 | 60 | 5 | |
| Kinetic rates | |||||||
| Estimated value | 25.248 | 12.1591 | 5.3689 | 59.3748 | 29.3347 | 28.6955 | 27.5407 |
| STD | 0.7066 | 0.4916 | 0.4452 | 3.551 | 1.6659 | 1.2889 | 0.553 |
| 20 | 5 | 1 | 40 | 20 | 20 | 20 | |
| 50 | 20 | 10 | 80 | 50 | 50 | 50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, W.; Xia, P.; Zhang, X.; Tian, T. Bayesian Inference Algorithm for Estimating Heterogeneity of Regulatory Mechanisms Based on Single-Cell Data. Mathematics 2022, 10, 4748. https://doi.org/10.3390/math10244748
He W, Xia P, Zhang X, Tian T. Bayesian Inference Algorithm for Estimating Heterogeneity of Regulatory Mechanisms Based on Single-Cell Data. Mathematics. 2022; 10(24):4748. https://doi.org/10.3390/math10244748
Chicago/Turabian StyleHe, Wenlong, Peng Xia, Xinan Zhang, and Tianhai Tian. 2022. "Bayesian Inference Algorithm for Estimating Heterogeneity of Regulatory Mechanisms Based on Single-Cell Data" Mathematics 10, no. 24: 4748. https://doi.org/10.3390/math10244748