
WAVECNV: A New Approach for Detecting Copy Number Variation by Wavelet Clustering




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
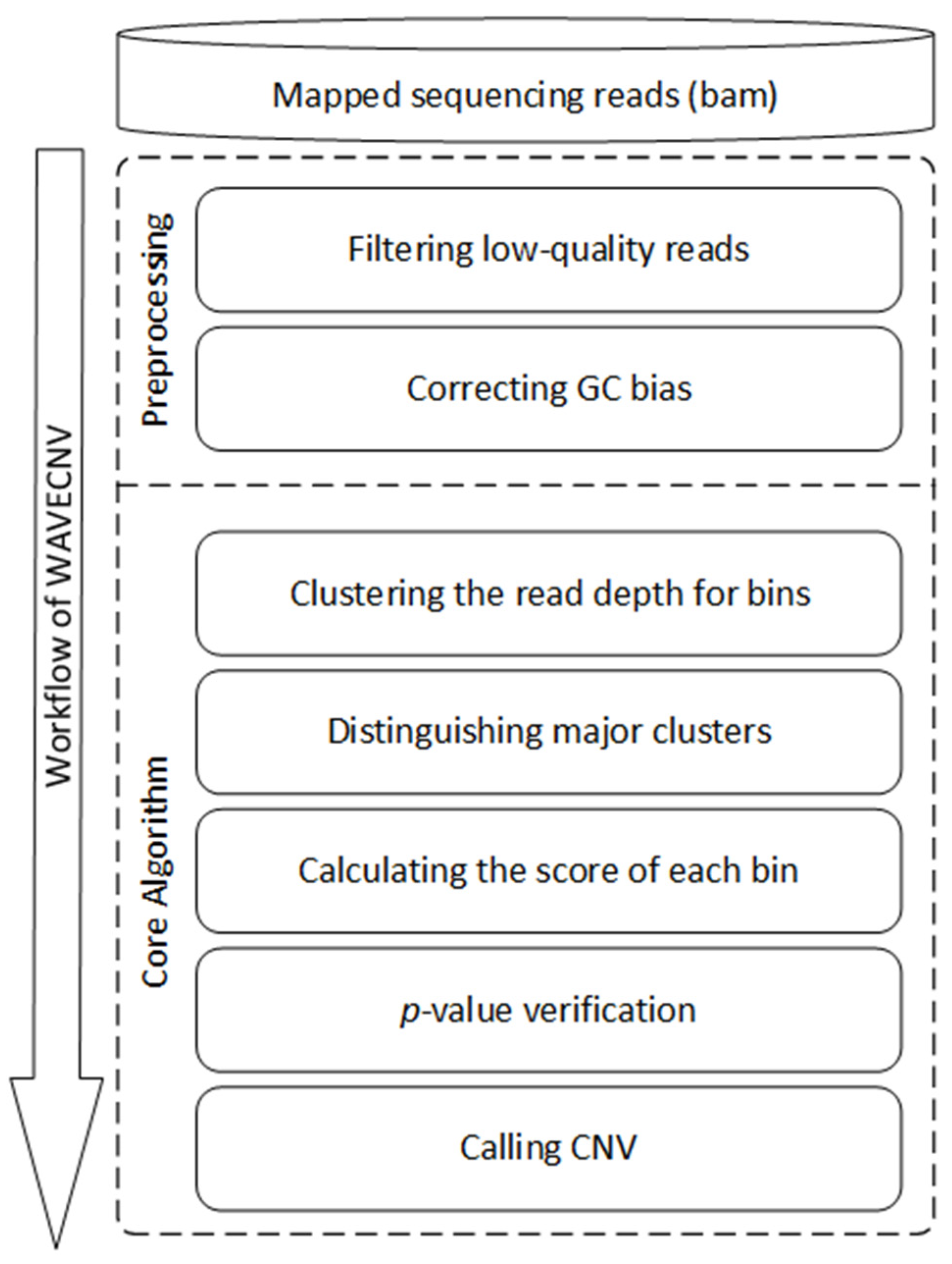
2.1. Workflow of WAVECNV
2.2. Preprocessing
2.3. Clustering the RDs of Bins by Wavelet Clustering
2.4. Distinguishing Normal and Abnormal Clusters
2.5. Calculating the Scores of Each Bin
2.6. Calling CNVs by p-Value Verification
3. Results
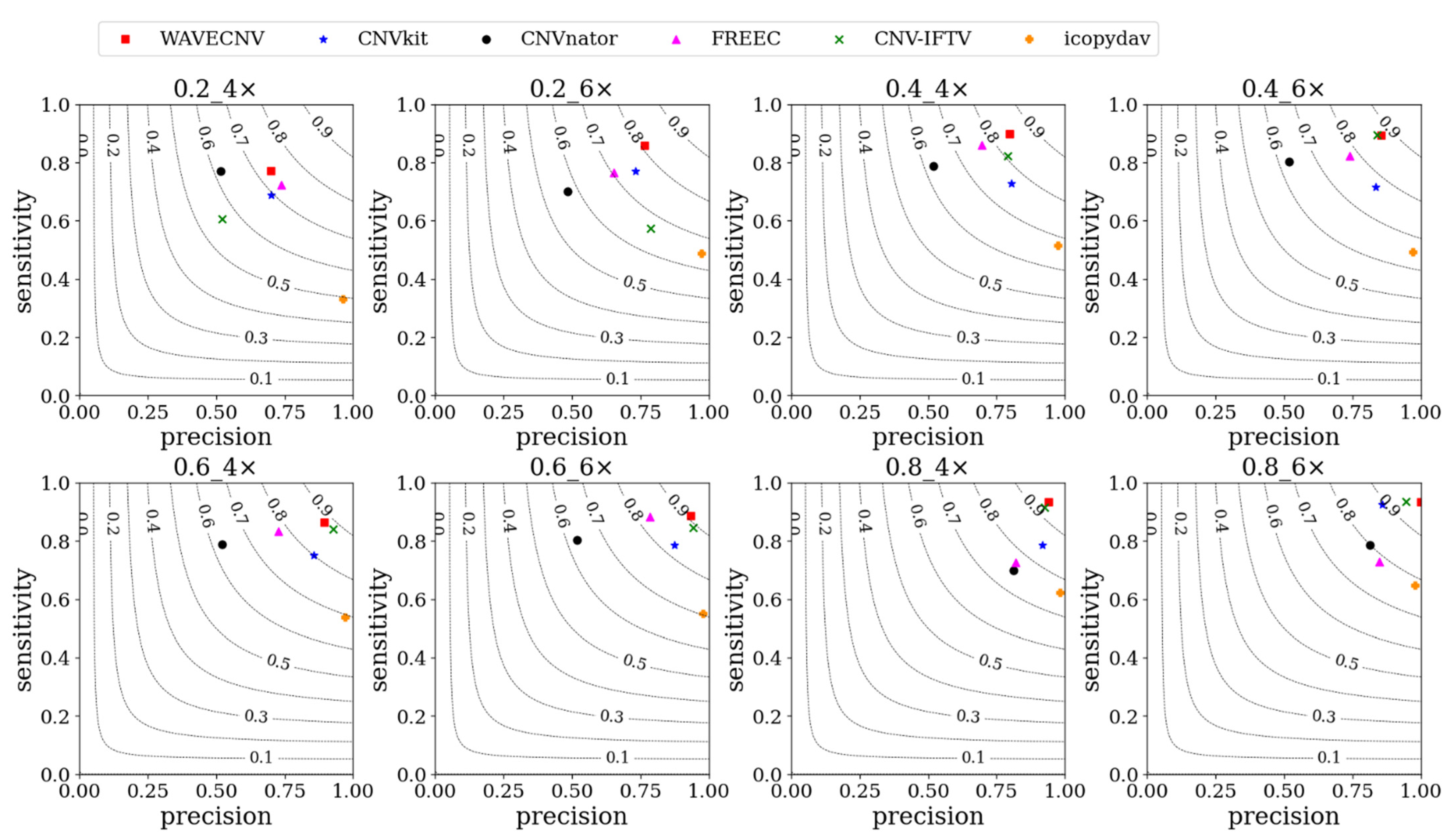
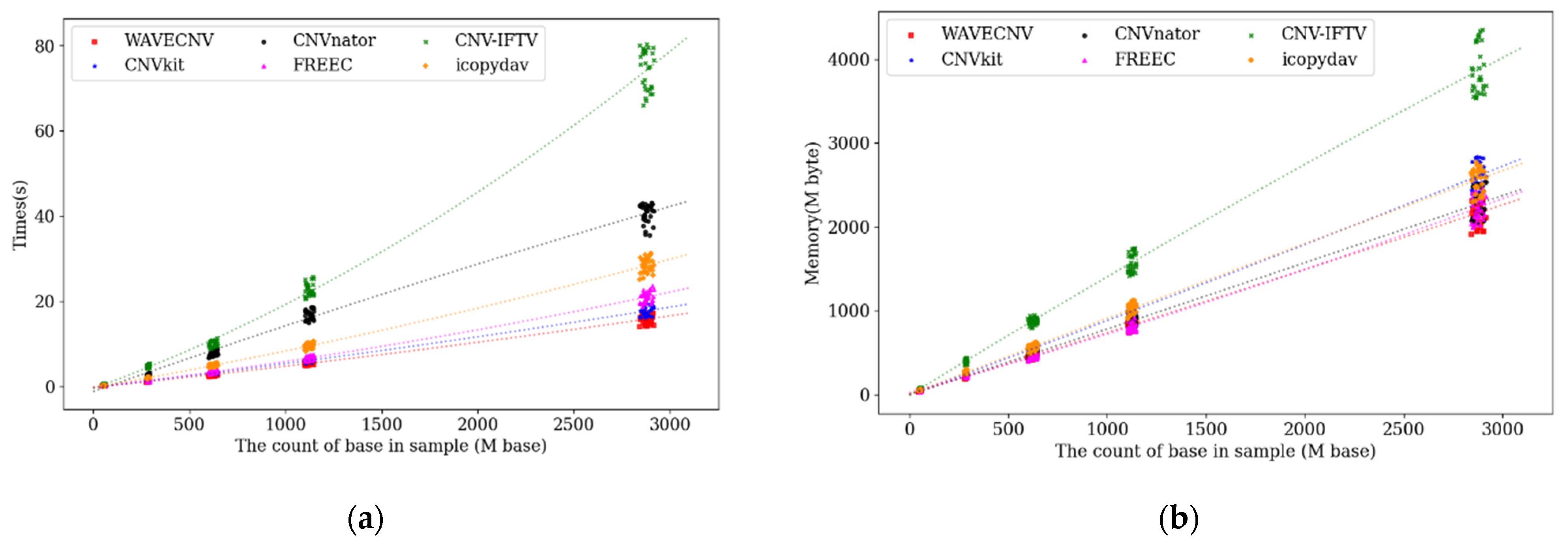
3.1. Simulation Studies
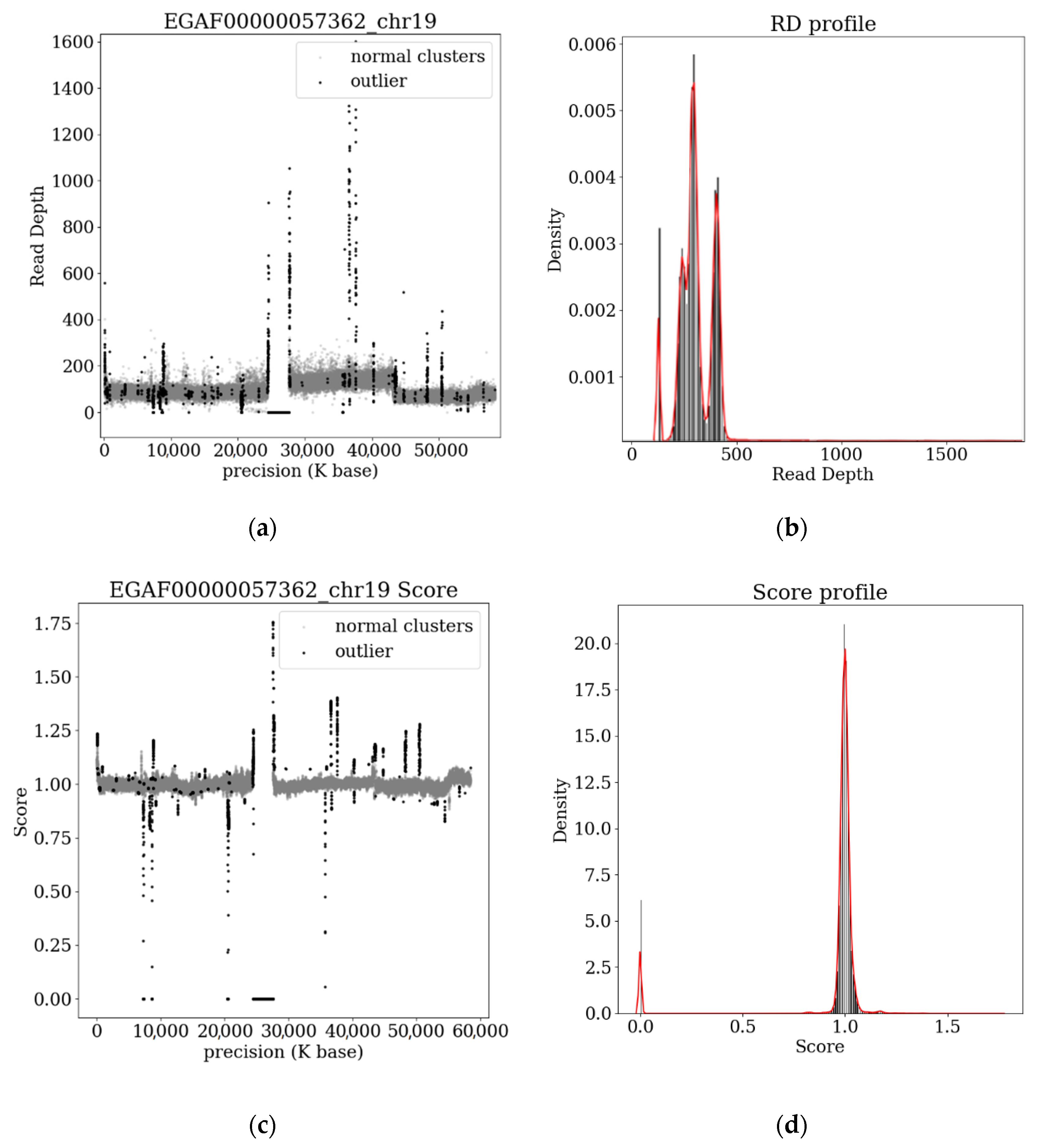
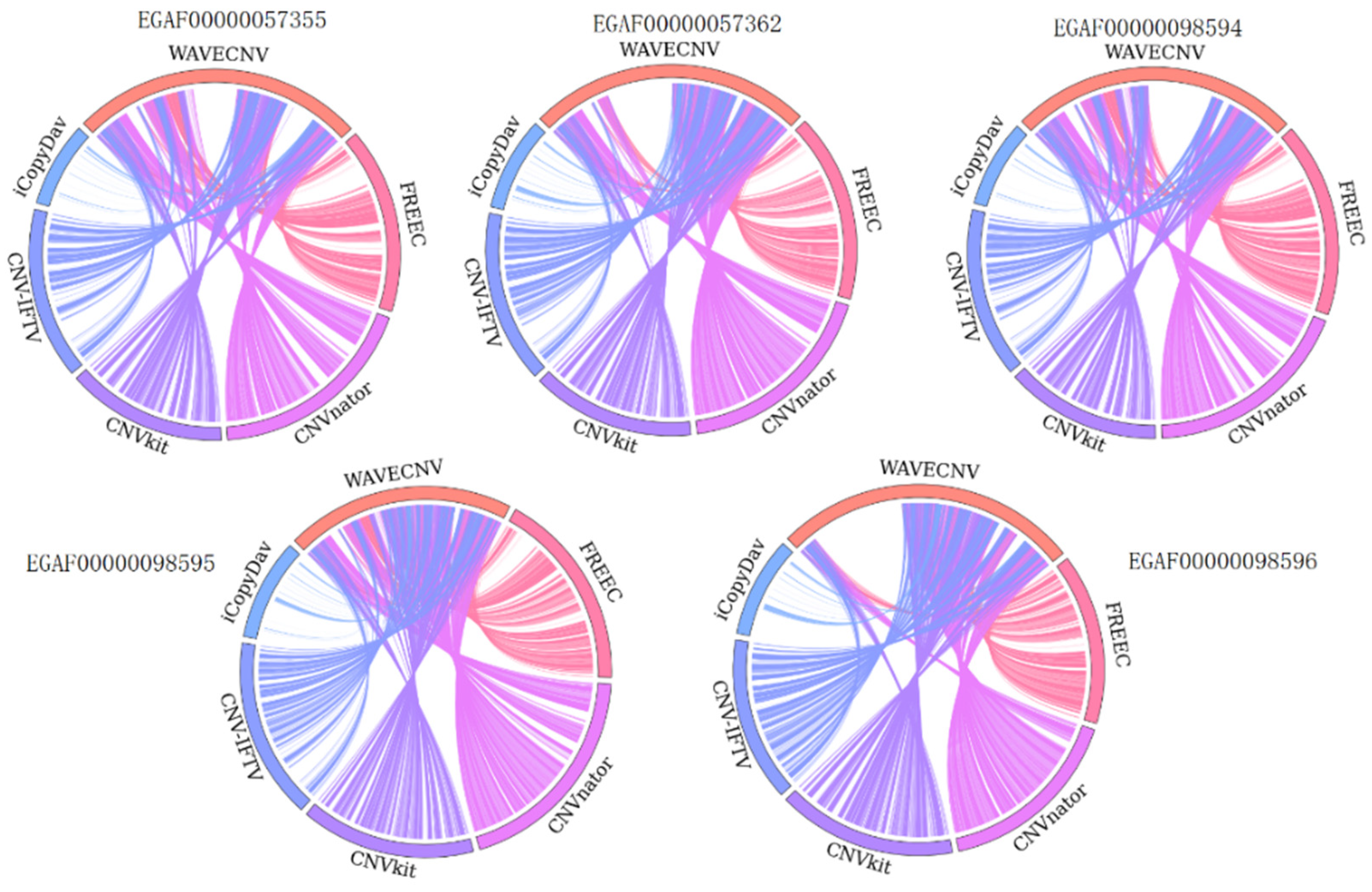
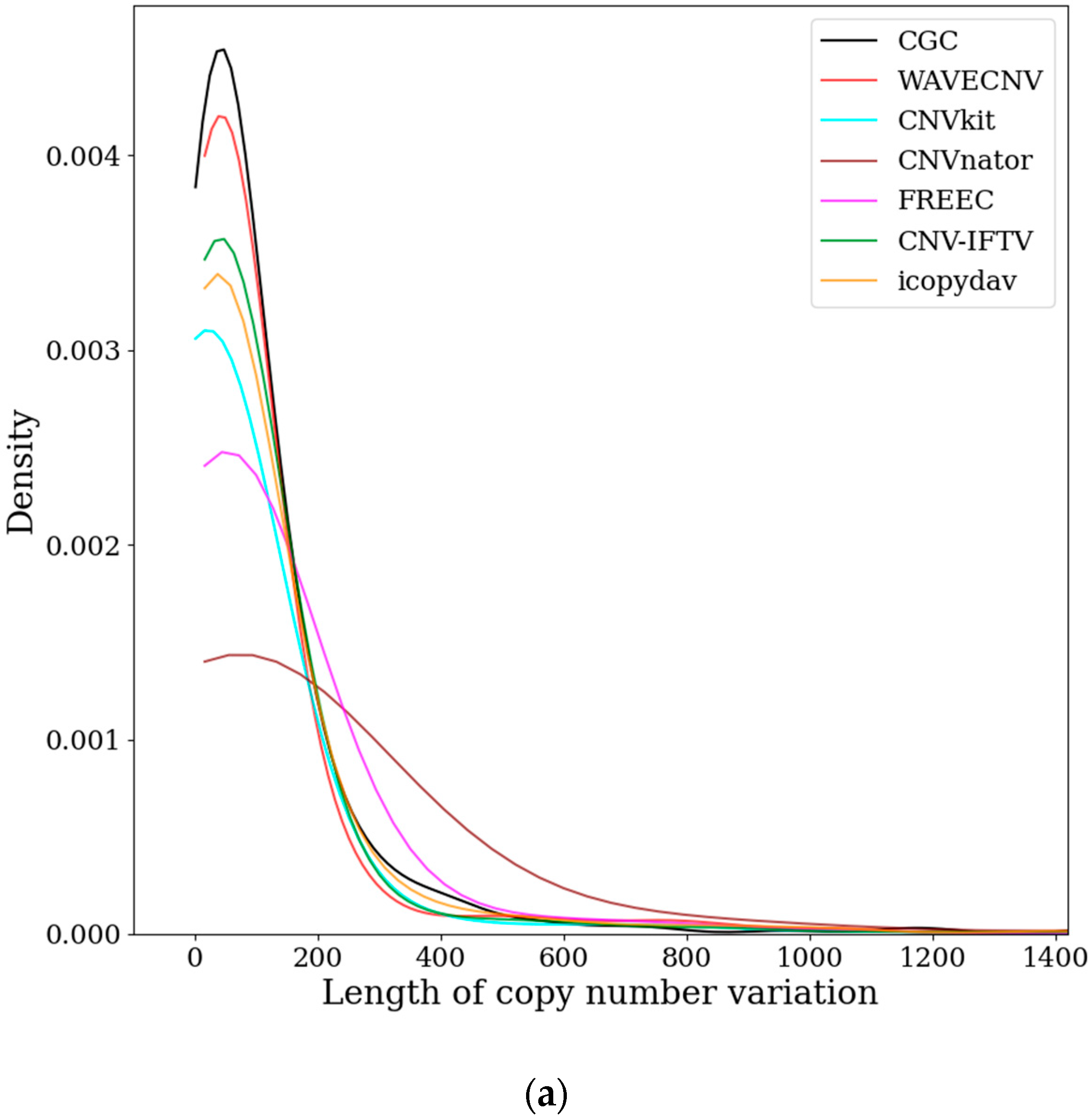
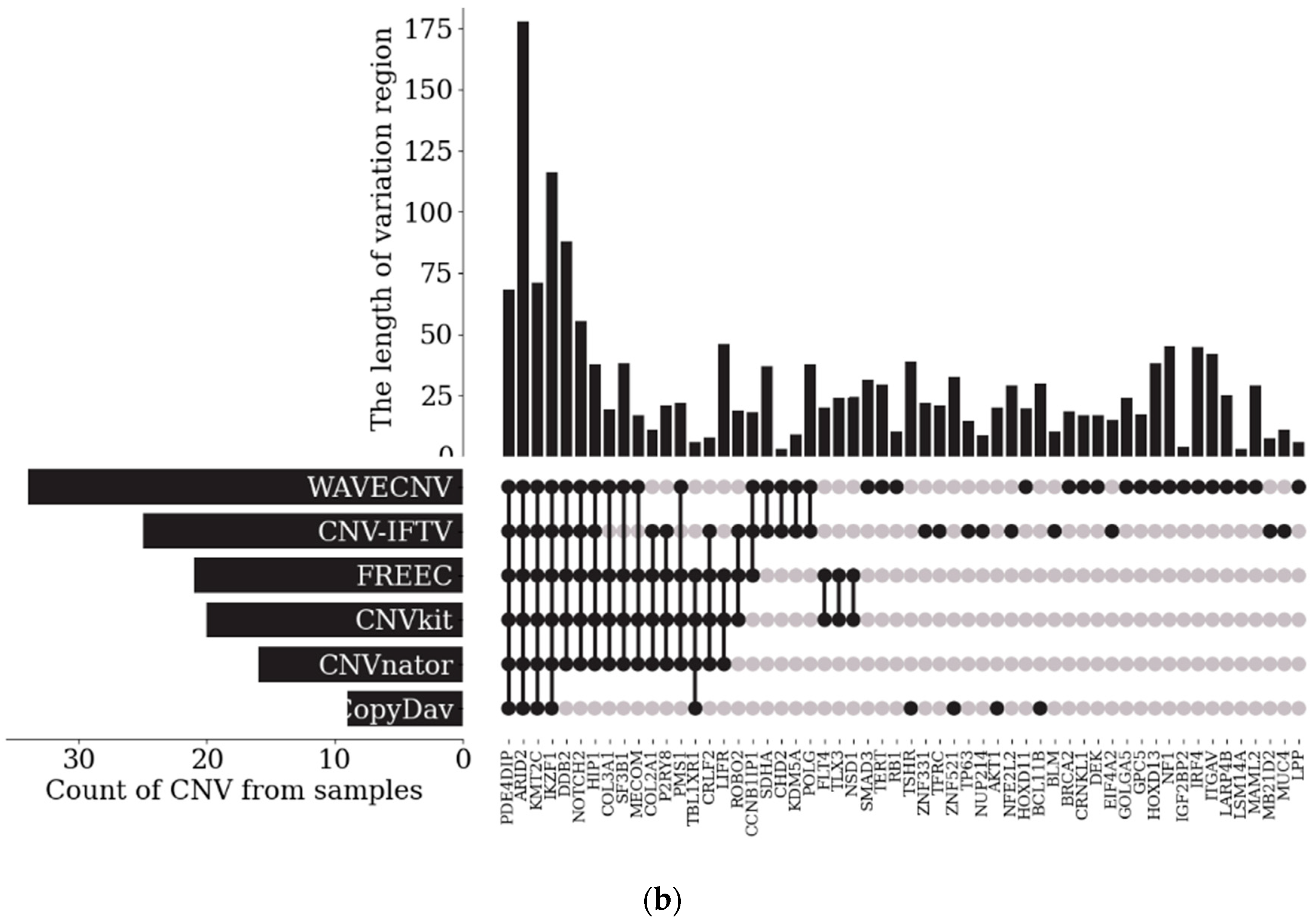
3.2. Analysis of Samples from the EGA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Handsaker, R.E.; Doren, V.V.; Berman, J.R.; Genovese, G.; Kashin, S.; Boettger, L.M.; McCarroll, S.A. Large multiallelic copy number variations in humans. Nat. Genet. 2015, 47, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Redon, R.; Ishikawa, S.; Fitch, K.; Feuk, L.; Perry, G.; Andrews, T.; Fiegler, H.; Shapero, M.; Carson, A.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.-F.; Yuan, X.-G.; Cun, Y.-P. A novel machine learning approach (svmSomatic) to distinguish somatic and germline mutations using next-generation sequencing data. J. Zool. Res. 2021, 42, 246–249. [Google Scholar] [CrossRef]
- Tarabichi, M.; Salcedo, A.; Deshwar, A.G.; Ni Leathlobhair, M.; Wintersinger, J.; Wedge, D.C.; Van Loo, P.; Morris, Q.D.; Boutros, P.C. A practical guide to cancer subclonal reconstruction from DNA sequencing. Nat. Methods 2021, 18, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Prashanthi, D.; Sriharsha, V.; Nita, P.; Ulrich, M.J.P.O. iCopyDAV: Integrated platform for copy number variations—Detection, annotation and visualization. PLoS ONE 2018, 13, e0195334. [Google Scholar]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Zaccaria, S.; Raphael, B.J. Accurate quantification of copy-number aberrations and whole-genome duplications in multi-sample tumor sequencing data. Nat. Commun. 2020, 11, 4301. [Google Scholar] [CrossRef]
- Yuan, X.; Yu, J.; Xi, J.; Yang, L.; Shang, J.; Li, Z.; Duan, J. CNV_IFTV: An isolation forest and total variation-based detection of CNVs from short-read sequencing data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 18, 539–549. [Google Scholar] [CrossRef]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M.J.G.R. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comaniciu, D.; Meer, P. Mean shift: A robust approach toward feature space analysis. IEEE Trans. Pattern Anal. Mach. Intell. 2002, 24, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Li, J.; Bai, J.; Xi, J. A Local outlier factor-based detection of copy number variations from NGS data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 18, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.R.; Johnson, M.D.; Kucherlapati, R.; Park, P.J. Comparative analysis of algorithms for identifying amplifications and deletions in array CGH data. Bioinformatics 2005, 21, 3763–3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikholeslami, G.; Chatterjee, S.; Zhang, A. WaveCluster: A wavelet-based clustering approach for spatial data in very large databases. VLDB J. 2000, 8, 289–304. [Google Scholar] [CrossRef]
- Liu, F.T.; Ting, K.; Zhou, Z.-H. Isolation Forest. In Proceedings of the 2008 Eighth IEEE International Conference on Data Mining, Pisa, Italy, 15–19 December 2008; pp. 413–422. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E.J.B. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2011, 28, 423–425. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Speed, T.P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012, 40, e72. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.A.; Hampton, O.; Coarfa, C.; Milosavljevic, A. ReadDepth: A parallel r package for detecting copy number alterations from short sequencing reads. PLoS ONE 2011, 6, e16327. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Li, A.; Wang, M. CloneCNA: Detecting subclonal somatic copy number alterations in heterogeneous tumor samples from whole-exome sequencing data. BMC Bioinform. 2016, 17, 310. [Google Scholar] [CrossRef] [Green Version]
- Poell, J.B.; Mendeville, M.; Sie, D.; Brink, A.; Brakenhoff, R.H.; Ylstra, B.J.B. ACE: Absolute copy number estimation from low-coverage whole-genome sequencing data. Bioinformatics 2019, 35, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Freeberg, M.A.; Fromont, L.A.; D’Altri, T.; Romero, A.F.; Ciges, J.I.; Jene, A.; Kerry, G.; Moldes, M.; Ariosa, R.; Bahena, S.; et al. The European Genome-phenome Archive in 2021. Nucleic Acids Res. 2021, 50, D980–D987. [Google Scholar] [CrossRef] [PubMed]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, J.; Yang, L. IntSIM: An Integrated Simulator of Next-Generation Sequencing Data. IEEE Trans. Biomed. Eng. 2016, 64, 441–451. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, L.; Wang, Y.; Cao, M.; Gelowani, V.; Xu, M.; Agrawal, S.A.; Li, Y.; Daiger, S.P.; Gibbs, R.; et al. SeqCNV: A novel method for identification of copy number variations in targeted next-generation sequencing data. BMC Bioinform. 2017, 18, 147. [Google Scholar] [CrossRef] [Green Version]
- Cmero, M.; Yuan, K.; Ong, C.S.; Schröder, J.; Adams, D.J.; Anur, P.; Beroukhim, R.; Boutros, P.C.; Bowtell, D.D.L.; Campbell, P.J.; et al. Inferring structural variant cancer cell fraction. Nat. Commun. 2020, 11, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshwar, A.G.; Vembu, S.; Yung, C.K.; Jang, G.H.; Stein, L.; Morris, Q. PhyloWGS: Reconstructing subclonal composition and evolution from whole-genome sequencing of tumors. Genome Biol. 2015, 16, 35. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Wang, S.; Haque, A.K.A.; Yuan, X. WAVECNV: A New Approach for Detecting Copy Number Variation by Wavelet Clustering. Mathematics 2022, 10, 2151. https://doi.org/10.3390/math10122151
Guo Y, Wang S, Haque AKA, Yuan X. WAVECNV: A New Approach for Detecting Copy Number Variation by Wavelet Clustering. Mathematics. 2022; 10(12):2151. https://doi.org/10.3390/math10122151
Chicago/Turabian StyleGuo, Yang, Shuzhen Wang, A. K. Alvi Haque, and Xiguo Yuan. 2022. "WAVECNV: A New Approach for Detecting Copy Number Variation by Wavelet Clustering" Mathematics 10, no. 12: 2151. https://doi.org/10.3390/math10122151