
Multi-Omics of Corynebacterium Pseudotuberculosis 12CS0282 and an In Silico Reverse Vaccinology Approach Reveal Novel Vaccine and Drug Targets

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacteria and Growth Conditions
2.2. Genome Sequencing and Analysis
2.3. Sample Preparation of Proteomic Analyses
2.4. Mass Spectrometry
2.5. Characterization and Visualization of Proteome Data
2.6. Reverse Vaccinology
2.7. Interaction of C. Pseudotuberculosis Strain 12CS0282 with Human Macrophages
2.8. Statistical Considerations
3. Results
3.1. Genome Analysis
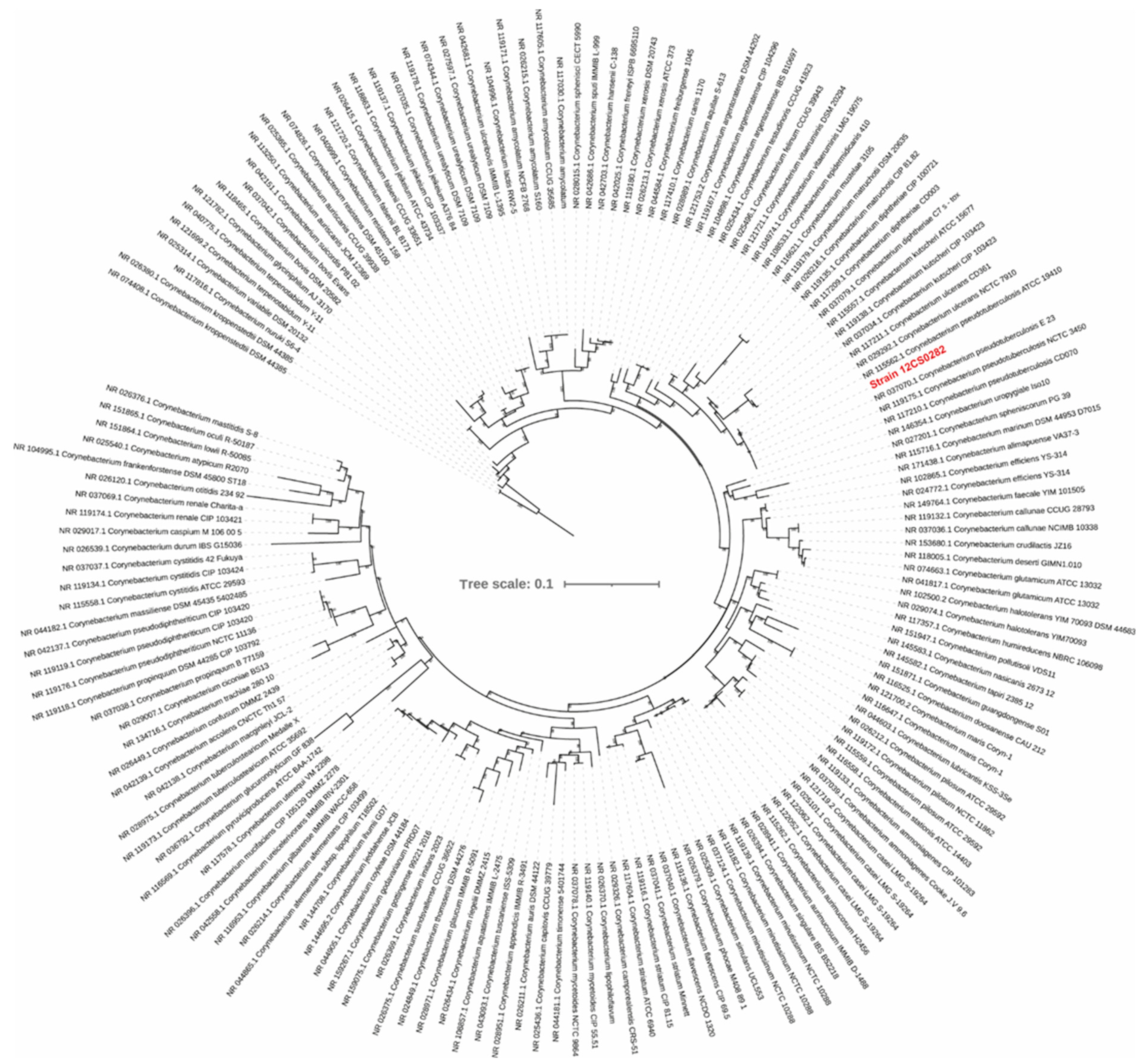
3.1.1. Phylogenomic Characteristics of Strain 12CS0282
3.1.2. Virulence Genes in Strain 12CS0282
3.2. Proteome Analyses
3.2.1. C. Pseudotuberculosis 12CS0282 Whole Cell Proteome, Surface Fraction and Secreted Proteome Fraction
3.2.2. Metabolic Pathway Analysis
3.2.3. Identification of Virulence Proteins
3.2.4. Reverse Vaccinology
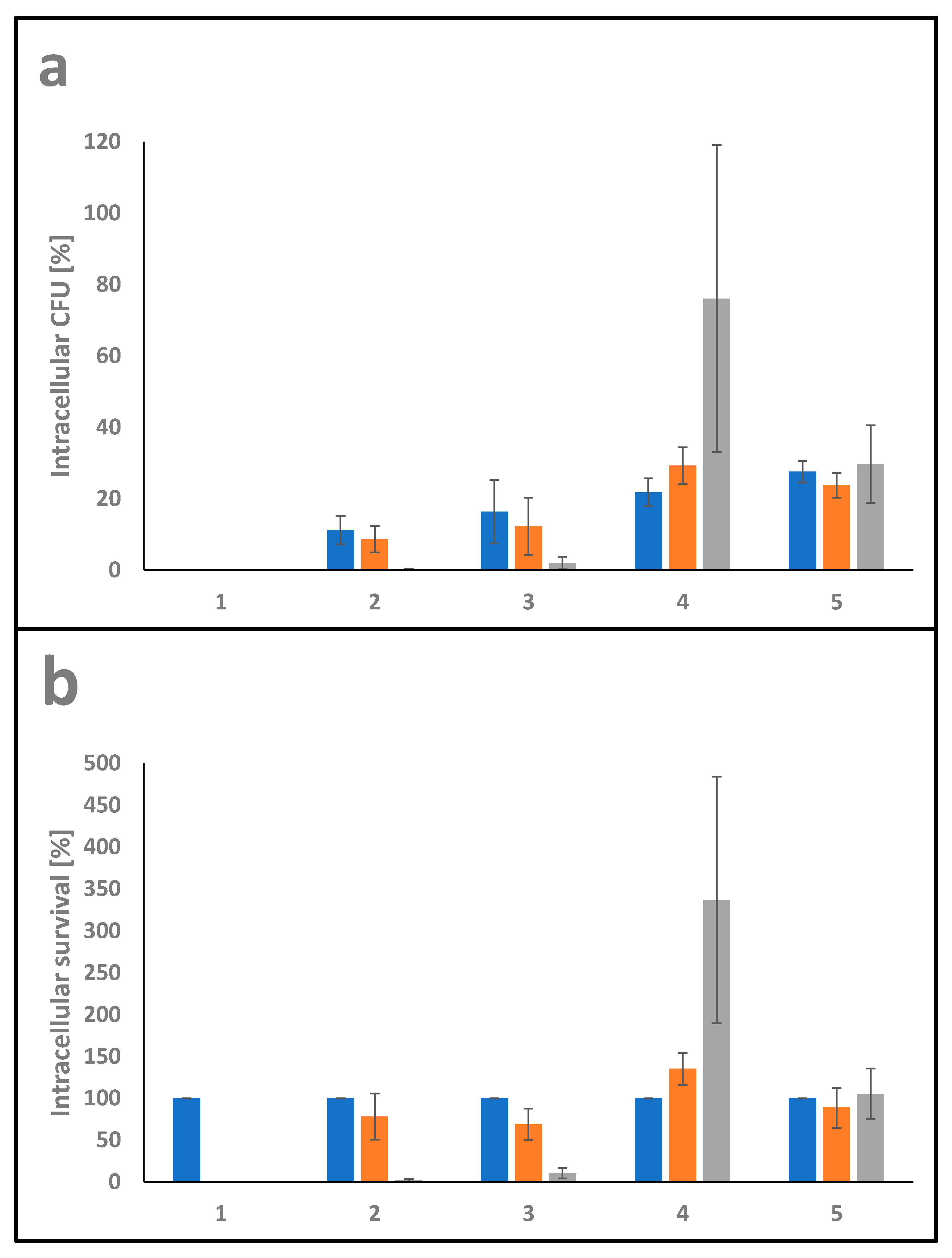
3.2.5. Interaction with Macrophages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sangal, V.; Hoskisson, P.A. Corynephages: Infections of the infectors. In Diphtheria and its Etiological Agents; Burkovski, A., Ed.; Springer: Dordrecht, The Netherlands, 2014; pp. 67–82. [Google Scholar]
- Riegel, P.; Ruimy, R.; Brie, D.; De Prkost, G.; Jehl, F.; Christen, R. Taxonomy of Corynebacterium diphtheriae and related taxa with recognition of Corynebacterium ulcerans sp. nov. nom. rev. FEMS Microbiol. Lett. 1995, 126, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Dorella, F.A.; Pacheco, L.G.C.; Oliveira, S.C.; Miyoshi, A.; Azevedo, V. Corynebacterium pseudotuberculosis: Microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Vet. Res. 2006, 37, 201–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, G.J.; Fontaine, M.C. Corynebacterium pseudotuberculosis and its role in ovine caseous lymphadenitis. J. Comp. Pathol. 2007, 137, 179–201. [Google Scholar] [PubMed]
- Silva, A.; Schneider, M.P.; Cerdeira, L.; Barbosa, M.S.; Ramos, R.T.; Carneiro, A.R.; Santos, R.; Lima, M.; D’Afonseca, V.; Almeida, S.S.; et al. Complete genome sequence of Corynebacterium pseudotuberculosis I19, a strain isolated from a cow in Israel with bovine mastitis. J. Bacteriol. 2011, 193, 323–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windsor, P.A.; Bush, R.D. Caseous lymphadenitis: Present and near forgotten from persistent. Small Ruminant Res. 2016, 142, 6–10. [Google Scholar] [CrossRef]
- Selim, S.A. Oedematous skin disease of buffalo in Egypt. J. Vet. Med. B Infect. Dis. Vet. Public Health. 2001, 48, 241–258. [Google Scholar] [CrossRef]
- Peel, M.M.; Palmer, G.G.; Stacpoole, A.M.; Kerr, T.G. Human lymphadenitis due to Corynebacterium pseudotuberculosis: Report of ten cases from Australia and review. Clin. Infect. Dis. 1997, 24, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Trost, E.; Ott, L.; Schneider, J.; Schröder, J.; Jaenicke, S.; Goesmann, A.; Husemann, P.; Stoye, J.; Dorella, F.A.; Rocha, F.S.; et al. The complete genome sequence of Corynebacterium pseudotuberculosis FRC41 isolated from a 12-year-old girl with necrotizing lymphadenitis reveals insights into gene- regulatory networks contributing to virulence. BMC Genom. 2010, 11, 728. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, L.G.; Slade, S.E.; Seyffert, N.; Santos, A.R.; Castro, T.L.; Silva, W.M.; Santos, A.V.; Santos, S.G.; Farias, L.M.; Carvalho, M.A.; et al. A combined approach for comparative exoproteome analysis of Corynebacterium pseudotuberculosis. BMC Microbiol. 2011, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Silva, W.M.; Seyffert, N.; Ciprandi, A.; Santos, A.V.; Castro, T.L.; Pacheco, L.G.; Barh, D.; Le Loir, Y.; Pimenta, A.M.; Miyoshi, A.; et al. Differential exoproteome analysis of two Corynebacterium pseudotuberculosis biovar ovis strains isolated from goat (1002) and sheep (C231). Curr. Microbiol. 2013, 67, 460–465. [Google Scholar] [CrossRef]
- Soares, S.C.; Silva, A.; Trost, E.; Blom, J.; Ramos, R.; Carneiro, A.; Ali, A.; Santos, A.R.; Pinto, A.C.; Diniz, C.; et al. The Pan-Genome of the animal pathogen Corynebacterium pseudotuberculosis reveals differences in genome plasticity between the biovar ovis and equi strains. PLoS ONE 2013, 8, e53818. [Google Scholar]
- Silva, W.M.; Carvalho, R.D.; Soares, S.C.; Bastos, I.F.; Folador, E.L.; Souza, G.H.; Le Loir, Y.; Miyoshi, A.; Silva, A.; Azevedo, V. Label-free proteomic analysis to confirm the predicted proteome of Corynebacterium pseudotuberculosis under nitrosative stress mediated by nitric oxide. BMC Genom. 2014, 15, 1065. [Google Scholar]
- Silva, J.W.; Droppa-Almeida, D.; Borsuk, S.; Azevedo, V.; Portela, R.W.; Miyoshi, A.; Rocha, F.S.; Dorella, F.A.; Vivas, W.L.; Padilha, F.F.; et al. Corynebacterium pseudotuberculosis Cp09 mutant and Cp40 recombinant protein partially protect mice against caseous lymphadenitis. BMC Vet. Res. 2014, 10, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droppa-Almeida, D.; da Silva, G.A.; Gaspar, L.M.D.A.C.; Pereyra, B.B.S.; Nascimento, R.J.M.; Borsuk, S.; Franceschi, E.; Padilha, F.F. Peptide vaccines designed with the aid of immunoinformatic against Caseous Lymphadenitis promotes humoral and cellular response induction in mice. PLoS ONE 2021, 16, e0256864. [Google Scholar]
- Parise, D.; Teixeira Dornelles Parise, M.; Pinto Gomide, A.C.; Figueira Aburjaile, F.; Bentes Kato, R.; Salgado-Albarrán, M.; Tauch, A.; Ariston de Carvalho Azevedo, V.; Baumbach, J. The Transcriptional Regulatory Network of Corynebacterium pseudotuberculosis. Microorganisms 2021, 9, 415. [Google Scholar] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.F.; Lio, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A multi-draft based scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Goker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Goker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tange, O. GNU Parallel—The command-line power tool. USENIX Mag. 2011, 36, 42–47. [Google Scholar]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Möller, J.; Schorlemmer, S.; Hofmann, J.; Burkovski, A. Cellular and extracellular proteome of the animal pathogen Corynebacterium silvaticum, a close relative of zoonotic Corynebacterium ulcerans and Corynebacterium pseudotuberculosis. Proteomes 2020, 8, 19. [Google Scholar] [CrossRef]
- Kraner, M.E.; Müller, C.; Sonnewald, U. Comparative proteomic profiling of the choline transporter-like1 (CHER1) mutant provides insights into plasmodesmata composition of fully developed Arabidopsis thaliana leaves. Plant J. 2017, 92, 696–709. [Google Scholar]
- Bittel, M.; Gastiger, S.; Amin, B.; Hofmann, J.; Burkovski, A. Surface and extracellular proteome of the emerging pathogen Corynebacterium ulcerans. Proteomes 2018, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Meth. 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Möller, J.; Kraner, M.; Sonnewald, U.; Sangal, V.; Tittlbach, H.; Winkler, J.; Winkler, T.H.; Melnikov, V.; Lang, R.; Sing, A.; et al. Proteomics of diphtheria toxoid vaccines reveals multiple proteins that are immunogenic and may contribute to protection of humans against Corynebacterium diphtheriae. Vaccine 2019, 37, 3061–3070. [Google Scholar] [PubMed]
- Tjalsma, H.; Lambooy, L.; Hermans, P.W.; Swinkels, D.W. Shedding & shaving: Disclosure of proteomic expressions on a bacterial face. Proteomics 2008, 8, 1415–1428. [Google Scholar]
- Schäfer, W.; Eckart, R.A.; Schmid, B.; Cagköylü, H.; Hof, K.; Muller, Y.A.; Amin, B.; Lührmann, A. Nuclear trafficking of the anti-apoptotic Coxiella burnetii effector protein AnkG requires binding to p32 and Importin-α 1. Cell. Microbiol. 2017, 19, e12634. [Google Scholar] [CrossRef] [PubMed]
- Laird, M.R.; Melli, G.; Sahinalp, S.C.; Yu, N.Y.; Lo, R.; Dao, P.; Brinkman, F.S.L.; Wagner, J.R.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar]
- Juncker, A.S.; Willenbrock, H.; von Heijne, G.; Nielsen, H.; Brunak, S.; Krogh, A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003, 12, 1652–1662. [Google Scholar] [CrossRef] [Green Version]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Bernhardt, J.; Funke, S.; Hecker, M.; Siebourg, J. Visualizing Gene Expression Data via Voronoi Treemaps. In Proceedings of the Sixth International Symposium on Voronoi Diagrams, Copenhagen, Denmark, 23–26 June 2009; pp. 233–241. [Google Scholar] [CrossRef]
- Otto, A.; Bernhardt, J.; Meyer, H.; Schaffer, M.; Herbst, F.-A.; Siebourg, J.; Mäder, U.; Lalk, M.; Hecker, M.; Becher, D. Systems-wide temporal proteomic profiling in glucose-starved Bacillus subtilis. Nature Commun. 2010, 1, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual account of protein investment in cellular functions. Proc. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo, C.L.; Alves, J.; Nogueira, W.; Pereira, L.C.; Gomide, A.C.; Ramos, R.; Azevedo, V.; Silva, A.; Folador, A. Prediction of new vaccine targets in the core genome of Corynebacterium pseudotuberculosis through omics approaches and reverse vaccinology. Gene 2019, 702, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Chukwu-Osazuwa, J.; Cao, T.; Vasquez, I.; Gnanagobal, H.; Hossain, A.; Machimbirike, V.I.; Santander, J. Comparative Reverse Vaccinology of Piscirickettsia salmonis, Aeromonas salmonicida, Yersinia ruckeri, Vibrio anguillarum and Moritella viscosa, Frequent Pathogens of Atlantic Salmon and Lumpfish Aquaculture. Vaccines 2022, 10, 473. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Lin, Y.; Gao, F.; Zhang, C.T.; Zhang, R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014, 42, D574–D580. [Google Scholar] [CrossRef] [Green Version]
- Barh, D.; Jain, N.; Tiwari, S.; Parida, B.P.; D’Afonseca, V.; Li, L.; Ali, A.; Santos, A.R.; Guimarães, L.C.; de Castro Soares, S.; et al. A novel comparative genomics analysis for common drug and vaccine targets in Corynebacterium pseudotuberculosis and other CMN group of human pathogens. Chem. Biol. Drug Des. 2011, 78, 73–84. [Google Scholar] [CrossRef]
- Ong, E.; Cooke, M.F.; Huffman, A.; Xiang, Z.; Wong, M.U.; Wang, H.; Seetharaman, M.; Valdez, N.; He, Y. Vaxign2: The second generation of the first Web-based vaccine design program using reverse vaccinology and machine learning. Nucleic Acids Res. 2021, 49, W671–W678. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. Predicting class I major histocompatibility complex (MHC) binders using multivariate statistics: Comparison of discriminant analysis and multiple linear regression. J. Chem. Inf. Model. 2007, 47, 234–238. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Goker, M.; Sproer, C.; Klenk, H.P. When should a DDH experiment be mandatory in microbial taxonomy? Arch. Microbiol. 2013, 195, 413–418. [Google Scholar] [PubMed]
- Be, N.A.; Bishai, W.R.; Jain, S.K. Role of Mycobacterium tuberculosis PknD in the pathogenesis of central nervous system tuberculosis. BMC Microbiol. 2012, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thais de Oliveira Silva, M.; Barros de Pinho, R.; da Rocha Fonseca, B.; Silvestre Brilhante Bezerra, F.; Severo Sabedra Sousa, F.; Kommling Seixas, F.; Collares, T.; Meyer Nascimento, J.R.; Wagner Portela, R.; Ariston Carvalho Azevedo, V.; et al. NanH and PknG putative virulence factors as a recombinant subunit immunogen against Corynebacterium pseudotuberculosis infection in mice. Vaccine 2020, 38, 8099–8106. [Google Scholar] [PubMed]
- Trost, E.; Al-Dilaimi, A.; Papavasiliou, P.; Schneider, J.; Viehoever, P.; Burkovski, A.; de Castro Soares, S.; Silva Almeida, S.; Alves Dorella, F.; Miyoshi, A.; et al. Comparative analysis of two complete Corynebacterium ulcerans genomes and detection of candidate virulence factors. BMC Genom. 2011, 12, 383. [Google Scholar]
- Hacker, E.; Ott, L.; Schulze-Luehrmann, J.; Lührmann, A.; Wiesmann, V.; Wittenberg, T.; Burkovski, A. The killing of macrophages by Corynebacterium ulcerans. Virulence 2016, 7, 45–55. [Google Scholar]
- Möller, J.; Musella, L.; Melnikov, V.; Geißdörfer, W.; Burkovski, A.; Sangal, V. Phylogenomic characterisation of a novel corynebacterial species pathogenic to animals. Antonie Van Leeuwenhoek 2020, 113, 1225–1239. [Google Scholar]
- Ott, L.; Hacker, E.; Kunert, T.; Karrington, I.; Etschel, P.; Lang, R.; Wiesmann, V.; Wittenberg, T.; Singh, A.; Varela, C.; et al. Analysis of Corynebacterium diphtheriae macrophage interaction: Dispensability of corynomycolic acids for inhibition of phagolysosome maturation and identification of a new gene involved in synthesis of the corynomycolic acid layer. PLoS ONE 2017, 12, e0180105. [Google Scholar]
- Weerasekera, D.; Hahn, J.; Herrmann, M.; Burkovski, A. Induction of necrosis in human macrophage cell lines by Corynebacterium diphtheriae and Corynebacterium ulcerans strains isolated from fatal cases of systemic infections. Int. J. Mol. Sci. 2019, 20, 4109. [Google Scholar] [CrossRef] [Green Version]
- McKean, S.C.; Davies, J.K.; Moore, R.J. Expression of phospholipase D, the major virulence factor of Corynebacterium pseudotuberculosis, is regulated by multiple environmental factors and plays a role in macrophage death. Microbiology 2007, 153, 2203–2211. [Google Scholar]
- Tauch, A.; Burkovski, A. Molecular armory or niche factors: Virulence determinants of Corynebacterium species. FEMS Microbiol. Lett. 2015, 362, fnv185. [Google Scholar] [CrossRef] [Green Version]
- Slayden, R.A.; Belisle, J.T. Morphological features and signature gene response elicited by inactivation of FtsI in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2009, 63, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.K.; Zulauf, K.E.; Braunstein, B. The Sec Pathways and Exportomes of Mycobacterium tuberculosis. Microbiol. Spec. 2017, 2, 24. [Google Scholar]
- Burg-Golani, T.; Pozniak, Y.; Rabinovich, L.; Sigal, N.; Nir Paz, R.; Herskovits, A.A. Membrane chaperone SecDF plays a role in the secretion of Listeria monocytogenes major virulence factors. J. Bacteriol. 2013, 195, 5262–5272. [Google Scholar] [PubMed] [Green Version]
- Quiblier, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Berger-Bächi, B.; Senn, M.M. Contribution of SecDF to Staphylococcus aureus resistance and expression of virulence factors. BMC Microbiol. 2011, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Petriman, N.A.; Jauß, B.; Hufnagel, A.; Franz, L.; Sachelaru, I.; Drepper, F.; Warscheid, B.; Koch, H.G. The interaction network of the YidC insertase with the SecYEG translocon, SRP and the SRP receptor FtsY. Sci. Rep. 2018, 8, 578. [Google Scholar]
- Thakur, P.; Choudhary, E.; Pareek, M.; AgarWal, N. Regulation and overexpression studies of YidC in Mycobacterium tuberculosis. Sci. Rep. 2018, 8, 17114. [Google Scholar]
- Mayer, R.L.; Impens, F. Immunopeptidomics for next-generation bacterial vaccine development. Trends Microbiol. 2021, 29, 1034–1045. [Google Scholar]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, 442–450. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Genome | DDH | Distance | G + C Difference |
|---|---|---|---|
| C. belfanti DSM 105776 T | 20.6 | 0.2129 | 1.44 |
| C. diphtheriae DSM44123 T | 20.8 | 0.2116 | 1.35 |
| C. pseudotuberculosis ATCC 19410 T | 99.9 | 0.0003 | 0 |
| C. pseudotuberculosis DSM 20689 T | 99.9 | 0.0002 | 0 |
| C. silvaticum KL0182 T | 28.5 | 0.1509 | 2.26 |
| C. silvaticum W25 | 28.5 | 0.1509 | 2.25 |
| C. ulcerans FRC11 | 27.6 | 0.1563 | 1.17 |
| C. ulcerans NCTC 7910 T | 27.5 | 0.1568 | 1.13 |
| Virulence Gene | Function | Identifier in Strain 12CS0282 |
|---|---|---|
| cpfrc_00029 (pld) | phospholipase D (sphingomyelin-degrading enzyme) | cp12CS0282_00124 |
| cpfrc_00128 (nor) | nitric oxide reductase | cp12CS0282_00230 |
| cpfrc_00386 (nanH) | neuraminidase H (sialidase) | cp12CS0282_00502 |
| cpfrc_00397 | secreted subtilisin-like serine protease | cp12CS0282_00513 |
| cpfrc_00491 (dtsR2) | acyl-CoA carboxylase b-subunit involved in mycolic acid synthesis | cp12CS0282_00606 |
| cpfrc_00492 (dtsR1) | acetyl-CoA carboxylase b-subunit involved in fatty acid synthesis | cp12CS0282_00607 |
| cpfrc_00536 | secreted SGNH-hydrolase | cp12CS0282_00649 |
| cpfrc_00562 | secreted trypsin-like serine protease | cp12CS0282_00678 |
| cpfrc_00565 (nrpS1) | nonribosomal peptide synthetase 1 | cp12CS0282_00682 |
| cpfrc_00594 (rpfA) | resuscitation-promoting factor A (muralytic enzyme) | cp12CS0282_02168 |
| cpfrc_00679 (rpfB) | resuscitation-promoting factor B (muralytic enzyme) | cp12CS0282_02083 |
| cpfrc_01079 (rpfI) | resuscitation-promoting factor interacting protein (D,L-endopeptidase) | cp12CS0282_01163 |
| cpfrc_01634 | secreted subtilisin-like serine protease | cp12CS0282_01728 |
| cpfrc_01801 | nonribosomal peptide synthetase 2 | cp12CS0282_00926 |
| cpfrc_01895 (cpp) | corynebacterial protease CP40 (serine protease) | cp12CS0282_00833 |
| cpfrc_01953 (accD3) | acyl-CoA carboxylase b-subunit involved in mycolic acid synthesis | cp12CS0282_00773 |
| Pathway | Theoretical Proteome | Identified Proteins | n = 3 |
|---|---|---|---|
| Cellular processes and signaling | 235 [11.1%] | 141 [9.8%] | 97 [9.5%] |
| Environmental information processing | 67 [3.2%] | 37 [2.6%] | 25 [2.4%] |
| Genetic information processing | 52 [2.5%] | 38 [2.6%] | 24 [2.3%] |
| Information storage and processing | 224 [10.6%] | 142 [9.8%] | 100 [9.8%] |
| Metabolism | 563 [26.6%] | 382 [26.5%] | 267 [26.1%] |
| Pathogenicity | 53 [3.1%] | 45 [3.1%] | 40 [3.9%] |
| Poorly characterized | 253 [11.9%] | 187 [13.0%] | 142 [13.9%] |
| Uncharacterized | 671 [31.7%] | 472 [32.7%] | 328 [32.1%] |
| Total | 2118 | 1444 | 1023 |
| Designation | Function | Localization and Relative Abundance |
|---|---|---|
| cp12CS0282_00124 (pld) | phospholipase D | E (0.5%), W (0.4%) |
| cp12CS0282_00230 (nor) | nitric oxide reductase | - |
| cp12CS0282_00502 (nanH) | neuraminidase H | S (0.6%) |
| cp12CS0282_00513 | secreted subtilisin-like serine protease | - |
| cp12CS0282_00606 (dtsR2) | acyl-CoA carboxylase b-subunit involved in mycolic acid synthesis | W (0.5%) |
| cp12CS0282_00607 (dtsR1) | acetyl-CoA carboxylase b-subunit involved in fatty acid synthesis | W (0.4%) |
| cp12CS0282_00649 | secreted SGNH-hydrolase | S (6.3%), W (0.2%) |
| cp12CS0282_00678 | secreted trypsin-like serine protease | E, S, W |
| cp12CS0282_00682 (nrpS1) | nonribosomal peptide synthetase 1 | - |
| cp12CS0282_02168 (rpfA) | resuscitation-promoting factor A (muralytic enzyme) | E (4.2%) |
| cp12CS0282_02083 (rpfB) | resuscitation-promoting factor B (muralytic enzyme) | S (0.3%) |
| cp12CS0282_01163 (rpfI) | resuscitation-promoting factor interacting protein (D,L-endopeptidase) | - |
| cp12CS0282_01728 | secreted subtilisin-like serine protease | E (0.04%), S (0.3%), W (0.2%) |
| cp12CS0282_0092 | nonribosomal peptide synthetase 2 | - |
| cp12CS0282_00833 (cpp) | corynebacterial protease CP40 | S (0.2%) |
| cp12CS0282_00773 (accD3) | acyl-CoA carboxylase b-subunit involved in mycolic acid synthesis | W (0.6%) |
| Protein ID | Protein Name | MW (Da) | Stability |
|---|---|---|---|
| cp12CS0282_00093 | membrane protein insertase | 36,402.13 | − |
| cp12CS0282_00370 | signal-transduction histidine kinase | 44,858.97 | + |
| cp12CS0282_00394 | cytochrome C biogenesis protein | 60,045.54 | + |
| cp12CS0282_00666 | hypothetical protein | 109,088.9 | + |
| cp12CS0282_00740 | putative cell wall biosynthesis protein | 46,819.89 | − |
| cp12CS0282_00766 | diacylglycerol acyltransferase/mycolyltransferase | 36,582.47 | + |
| cp12CS0282_00770 | hypothetical protein | 32,832.33 | + |
| cp12CS0282_00932 | hypothetical protein | 41,309.86 | + |
| cp12CS0282_00991 | adaptive-response sensory-kinase | 54,403.91 | − |
| cp12CS0282_01097 | NADH dehydrogenase-like protein | 49,062.65 | + |
| cp12CS0282_01233 | endolytic murein transglycosylase | 41,096.36 | + |
| cp12CS0282_01244 | FMN reductase | 19,296.33 | − |
| cp12CS0282_01259 | protein translocase subunit | 65,933.18 | + |
| cp12CS0282_01491 | penicillin-binding protein | 73,053.21 | + |
| cp12CS0282_01513 | cytochrome Bc1 complex cytochrome B subunit | 110,964 | + |
| cp12CS0282_01515 | cytochrome Bc1 complex cytochrome C subunit | 31,394.71 | + |
| cp12CS0282_01518 | cytochrome C oxidase subunit 2 | 40,192.72 | + |
| cp12CS0282_01584 | bifunctional protein | 41,939.33 | + |
| cp12CS0282_01590 | hypothetical protein | 34,917.49 | − |
| cp12CS0282_01839 | hypothetical protein | 34,482.87 | − |
| cp12CS0282_02102 | signal transduction histidine-protein kinase/phosphatase | 56,585.5 | − |
| cp12CS0282_02120 | hypothetical protein | 24,721.76 | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Möller, J.; Bodenschatz, M.; Sangal, V.; Hofmann, J.; Burkovski, A. Multi-Omics of Corynebacterium Pseudotuberculosis 12CS0282 and an In Silico Reverse Vaccinology Approach Reveal Novel Vaccine and Drug Targets. Proteomes 2022, 10, 39. https://doi.org/10.3390/proteomes10040039
Möller J, Bodenschatz M, Sangal V, Hofmann J, Burkovski A. Multi-Omics of Corynebacterium Pseudotuberculosis 12CS0282 and an In Silico Reverse Vaccinology Approach Reveal Novel Vaccine and Drug Targets. Proteomes. 2022; 10(4):39. https://doi.org/10.3390/proteomes10040039
Chicago/Turabian StyleMöller, Jens, Mona Bodenschatz, Vartul Sangal, Jörg Hofmann, and Andreas Burkovski. 2022. "Multi-Omics of Corynebacterium Pseudotuberculosis 12CS0282 and an In Silico Reverse Vaccinology Approach Reveal Novel Vaccine and Drug Targets" Proteomes 10, no. 4: 39. https://doi.org/10.3390/proteomes10040039