
The Need for Biomarkers in the ALS–FTD Spectrum: A Clinical Point of View on the Role of Proteomics
 ,
,  ,
,
Abstract
:1. Introduction
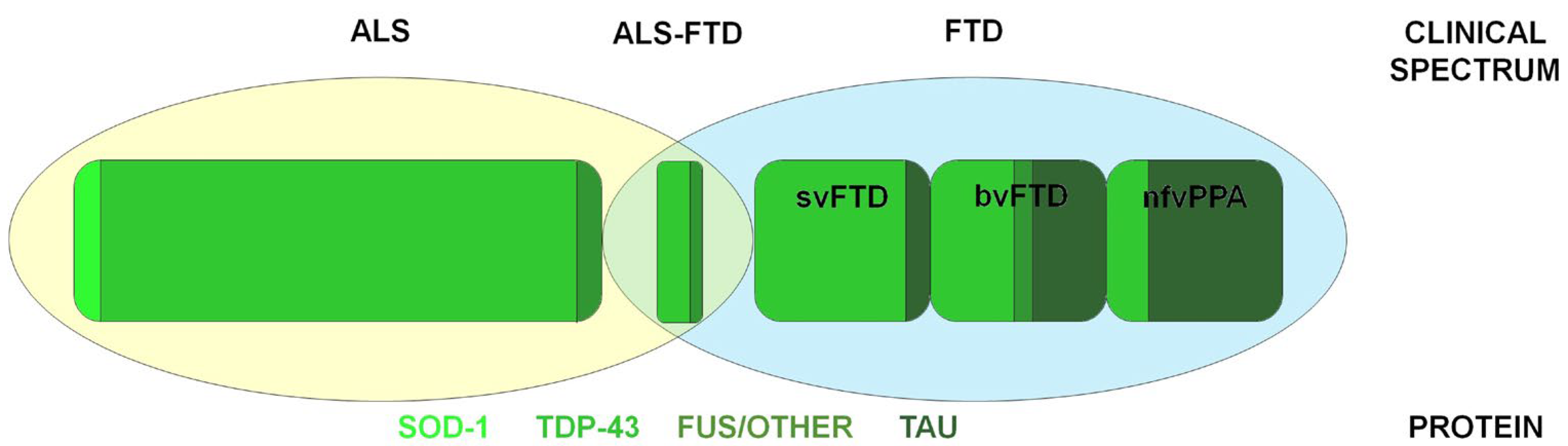
2. ALS–FTD Spectrum Disorders
2.1. Frontotemporal Dementia: Clinical and Pathogenesis
2.2. Amyotrophic Lateral Sclerosis: Clinical and Pathogenesis
3. Biomarkers in ALS–FTD Spectrum Disorders
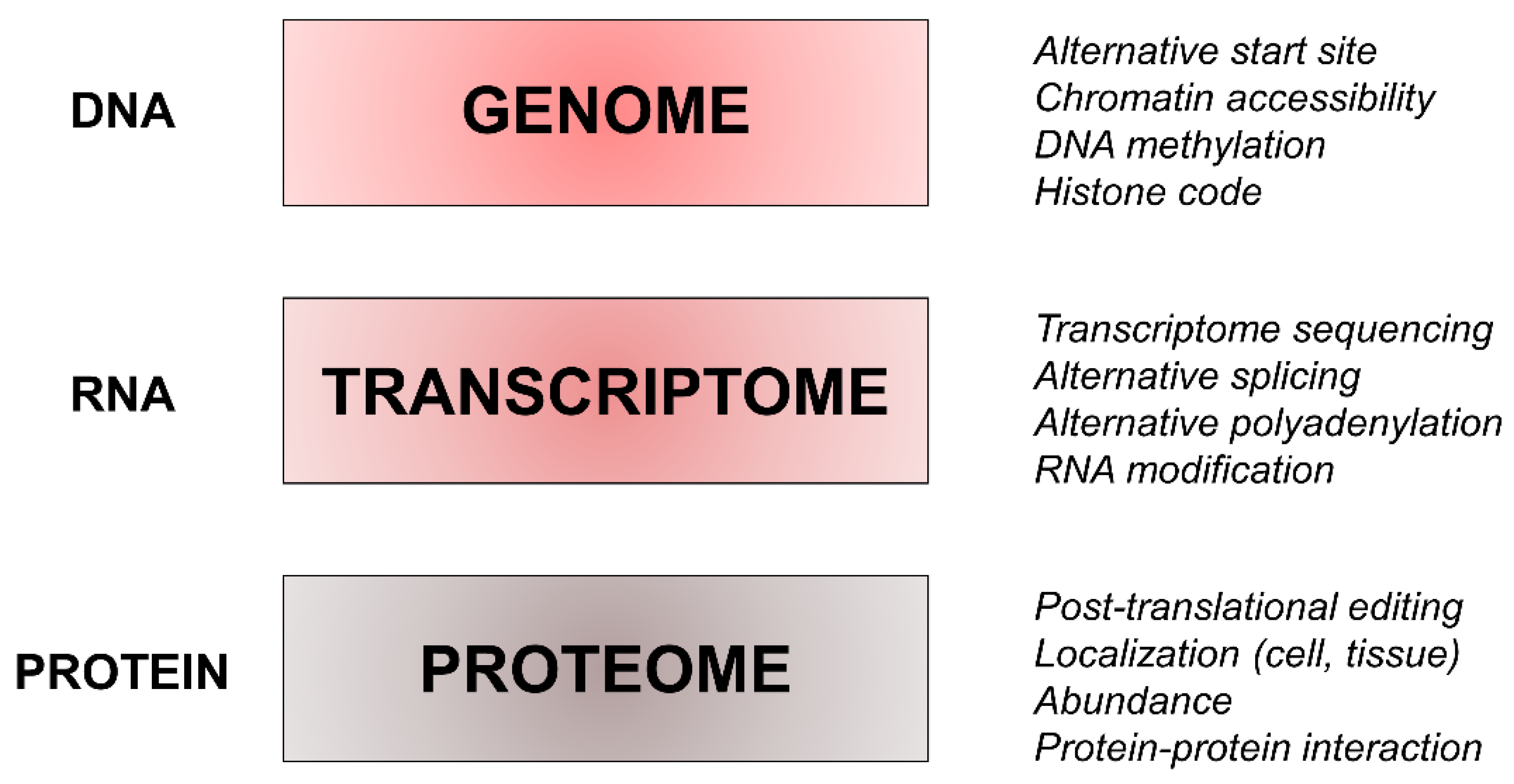
4. Proteomics and its Complexity
5. Proteomics in the ALS–FTD Spectrum Disorders
5.1. Proteomics in Cellular and Animal Models
5.2. Proteomics in Human Samples
5.2.1. Cerebrospinal Fluid
5.2.2. Blood

{kind=link}
{kind=link}
| Disease | Year | Method | Main Findings |
|---|---|---|---|
| Cerebrospinal Fluid | |||
| ALS vs. HC | 2005 | surface-enhanced laser desorption ionization time-of-flight mass spectrometry |
|
| ALS vs. HC | 2012 | two-dimensional difference in gel electrophoresis with matrix-assisted laser desorption ionization time-of-flight mass spectrometry |
|
| ALS (fast vs. slow) | 2010 | two-dimensional difference in gel electrophoresis with matrix-assisted laser desorption ionization time-of-flight mass spectrometry |
|
| ALS vs. HC | 2022 | ultra-sensitive proximity extension assay |
|
| ALS vs. HC | 2013 | liquid chromatography-tandem mass spectrometry |
|
| ALS vs. HC | 2012 | paramagnetic bead chromatography with matrix-assisted laser desorption ionization time-of-flight mass spectrometry |
|
| ALS vs. HC and other neurodegenerative diseases | 2015 | label-free liquid chromatography-tandem mass spectrometry |
|
| ALS vs. other neurological diseases | 2020 | liquid chromatography-tandem mass spectrometry |
|
| ALS vs. HC and Parkinson’s disease | 2019 | targeted multiple reaction monitoring (MRM) mass spectrometry |
|
| ALS vs. HC and other neurodegenerative diseases | 2016 | two-dimensional liquid chromatography mass spectrometry |
|
| ALS vs. HC and neuropathies | 2008 | two-dimensional gel electrophoresis |
|
| ALS vs. other neurological diseases | 2009 | Bio-Plex human 27-plex panel of cytokines and growth factors with atomic absorption spectroscopy |
|
| ALS | 2020 | shotgun proteomics and data-independent acquisition mass spectrometry |
|
| FTD vs. HC | 2004 | prefractionation method with two-dimensional electrophoresis |
|
| FTD vs. HC and AD | 2002 | Two-dimensional gel electrophoresis with mass spectrometry |
|
| FTD (GRN carriers vs. non-carriers) | 2019 | parallel reaction monitoring mass spectrometry |
|
| Blood | |||
| ALS vs. HC | 2022 | cytometric bead array and proteome profiling |
|
| ALS vs. HC | 2017 | bi-dimensional electrophoresis and mass spectrometry |
|
| ALS vs. HC | 2018 | nano-liquid chromatography and time-of-flight mass spectrometry |
|
| ALS vs. FTD vs. HC | 2020 | nano-capillary liquid chromatography–tandem mass spectrometry |
|
5.2.3. Other Tissues
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bang, J.; Spina, S.; Miller, B.L. Non-Alzheimer’s dementia 1: Frontotemporal dementia. Lancet 2015, 386, 1672. [Google Scholar] [CrossRef] [Green Version]
- Mol, M.O.; Miedema, S.S.M.; van Swieten, J.C.; van Rooij, J.G.J.; Dopper, E.G.P. Molecular Pathways Involved in Frontotemporal Lobar Degeneration with TDP-43 Proteinopathy: What Can We Learn from Proteomics? Int. J. Mol. Sci. 2021, 22, 10298. [Google Scholar] [CrossRef] [PubMed]
- Lashley, T.; Rohrer, J.D.; Mead, S.; Revesz, T. An update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol. Appl. Neurobiol. 2015, 41, 858–881. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, F.; Carrarini, C.; De Martino, A.; Diamanti, L.; Fasano, A.; Lupica, A.; Russo, M.; Salemme, S.; Spinelli, E.G.; Bombaci, A. Cognitive dysfunction in amyotrophic lateral sclerosis: Can we predict it? Neurol. Sci. 2021, 42, 2211–2222. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Elahi, F.M.; Miller, B.L. A clinicopathological approach to the diagnosis of dementia. Nat. Rev. Neurol. 2017, 13, 457–476. [Google Scholar] [CrossRef] [Green Version]
- Perani, D.; Iaccarino, L.; Lammertsma, A.A.; Windhorst, A.D.; Edison, P.; Boellaard, R.; Hansson, O.; Nordberg, A.; Jacobs, A.H.; IMBI Project. A new perspective for advanced positron emission tomography–based molecular imaging in neurodegenerative proteinopathies. Alzheimer’s Dement. 2019, 15, 1081–1103. [Google Scholar] [CrossRef]
- Hedl, T.J.; San Gil, R.; Cheng, F.; Rayner, S.L.; Davidson, J.M.; De Luca, A.; Hansson, O.; Nordberg, A.; Jacobs, A.H.; IMBI Project. Proteomics approaches for biomarker and drug target discovery in ALS and FTD. Front. Neurosci. 2019, 13, 548. [Google Scholar] [CrossRef]
- Davidsson, P.; Sjögren, M. The use of proteomics in biomarker discovery in neurodegenerative diseases. Dis. Markers. 2005, 21, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroksveen, A.C.; Opsahl, J.A.; Aye, T.T.; Ulvik, R.J.; Berven, F.S. Proteomics of human cerebrospinal fluid: Discovery and verification of biomarker candidates in neurodegenerative diseases using quantitative proteomics. J. Proteom. 2011, 74, 371–388. [Google Scholar] [CrossRef]
- Olney, N.T.; Spina, S.; Miller, B.L. Frontotemporal dementia. Neurol. Clin. 2017, 35, 339–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeve, B.F.; Boylan, K.B.; Graff-Radford, N.R.; DeJesus-Hernandez, M.; Knopman, D.S.; Pedraza, O.; Vemuri, P.; Jones, D.; Lowe, V.; Murray, M.E.; et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012, 135, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Tondo, G.; De Marchi, F.; Terazzi, E.; Sacchetti, M.; Cantello, R. Frontotemporal Dementia Presenting as Gambling Disorder: When a Psychiatric Condition Is the Clue to a Neurodegenerative Disease. Cogn. Behav. Neurol. 2017, 30, 62–67. [Google Scholar] [CrossRef]
- Liljegren, M.; Naasan, G.; Temlett, J.; Perry, D.C.; Rankin, K.P.; Merrilees, J.; Grinberg, L.T.; Seeley, W.W.; Englund, E.; Miller, B.L. Criminal behavior in frontotemporal dementia and Alzheimer disease. JAMA Neurol. 2015, 72, 295–300. [Google Scholar] [CrossRef]
- Mendez, M.F. Pathological stealing in dementia: Poor response to SSRI medications. J. Clin. Psychiatry 2011, 72, 13927. [Google Scholar] [CrossRef] [Green Version]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef]
- Chow, T.W.; Binns, M.A.; Cummings, J.L.; Lam, I.; Black, S.E.; Miller, B.L.; Freedman, M.; Stuss, D.T.; van Reekum, R. Apathy symptom profile and behavioral associations in frontotemporal dementia vs. dementia of Alzheimer type. Arch. Neurol. 2009, 66, 888–893. [Google Scholar] [CrossRef] [Green Version]
- Cerami, C.; Cappa, S.F. The behavioral variant of frontotemporal dementia: Linking neuropathology to social cognition. Neurol. Sci. 2013, 34, 1267–1274. [Google Scholar] [CrossRef]
- Tippett, D.C. Classification of primary progressive aphasia: Challenges and complexities. F1000Research 2020, 9, 32047619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorno-Tempini, M.L.; Hillis, A.E.; Weintraub, S.; Kertesz, A.; Mendez, M.; Cappa, S.F.; Ogar, J.M.; Rohrer, J.D.; Black, S.; Boeve, B.F.; et al. Classification of primary progressive aphasia and its variants. Neurology 2011, 76, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snowden, J.S.; Harris, J.M.; Thompson, J.C.; Kobylecki, C.; Jones, M.; Richardson, A.M.; Neary, D. Semantic dementia and the left and right temporal lobes. Cortex 2018, 107, 188–203. [Google Scholar] [CrossRef] [PubMed]
- Boccalini, C.; Carli, G.; Tondo, G.; Polito, C.; Catricalà, E.; Berti, V.; Bessi, V.; Sorbi, S.; Iannaccone, S.; Esposito, V.; et al. Brain metabolic connectivity reconfiguration in the semantic variant of primary progressive aphasia. Cortex 2022, 154, 1–14. [Google Scholar] [CrossRef]
- Rademakers, R.; Cruts, M.; Van Broeckhoven, C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat. 2004, 24, 277–295. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Neumann, M. Molecular neuropathology of frontotemporal dementia: Insights into disease mechanisms from postmortem studies. J. Neurochem. 2016, 138, 54–70. [Google Scholar] [CrossRef]
- Chare, L.; Hodges, J.R.; Leyton, C.E.; McGinley, C.; Tan, R.H.; Kril, J.J.; Halliday, G.M. New criteria for frontotemporal dementia syndromes: Clinical and pathological diagnostic implications. J. Neurol. Neurosurg. Psychiatry 2014, 85, 865–870. [Google Scholar] [CrossRef]
- Benussi, A.; Padovani, A.; Borroni, B. Phenotypic heterogeneity of monogenic frontotemporal dementia. Front. Aging Neurosci. 2015, 7, 171. [Google Scholar] [CrossRef]
- Olszewska, D.A.; Lonergan, R.; Fallon, E.M.; Lynch, T. Genetics of frontotemporal dementia. Curr. Neurol. Neurosci. Rep. 2016, 16, 1–15. [Google Scholar] [CrossRef]
- Sieben, A.; Van Langenhove, T.; Engelborghs, S.; Martin, J.-J.; Boon, P.; Cras, P.; de Deyn, P.P.; Santens, P.; Van Broeckhoven, C.; Cruts, M. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012, 124, 353–372. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.A.; Neumann, M.; Baborie, A.; Sampathu, D.M.; Du Plessis, D.; Jaros, E.; Perry, R.H.; Trojanowski, J.Q.; Mann, D.M.; Lee, V.M. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011, 122, 111–113. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Clark, C.M.; Miller, B.L.; Lee, V.M.-Y.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.A.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, J.W.; Seeley, W.W.; Huang, E.J. RNA binding proteins and the pathogenesis of frontotemporal lobar degeneration. Annu. Rev. Pathol. 2019, 14, 469. [Google Scholar] [CrossRef] [PubMed]
- Nolan, M.; Talbot, K.; Ansorge, O. Pathogenesis of FUS-associated ALS and FTD: Insights from rodent models. Acta Neuropathol. Commun. 2016, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tiryaki, E.; Horak, H.A. ALS and other motor neuron diseases. Contin. Lifelong Learn. Neurol. 2014, 20, 1185–1207. [Google Scholar] [CrossRef]
- Huynh, W.; Ahmed, R.; Mahoney, C.J.; Nguyen, C.; Tu, S.; Caga, J.; Loh, P.; Lin, C.S.; Kiernan, M.C. The impact of cognitive and behavioral impairment in amyotrophic lateral sclerosis. Expert Rev. Neurother. 2020, 20, 281–293. [Google Scholar] [CrossRef]
- Murphy, J.; Factor-Litvak, P.; Goetz, R.; Lomen-Hoerth, C.; Nagy, P.L.; Hupf, J.; Singleton, J.; Woolley, S.; Andrews, H.; Heitzman, D.; et al. Cognitive-behavioral screening reveals prevalent impairment in a large multicenter ALS cohort. Neurology 2016, 86, 813–820. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotter, E.L.; Chen, H.-J.; Shaw, C.E. TDP-43 proteinopathy and ALS: Insights into disease mechanisms and therapeutic targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umoh, M.E.; Dammer, E.B.; Dai, J.; Duong, D.M.; Lah, J.J.; Levey, A.I.; Glass, J.D.; Seyfried, N.T. A proteomic network approach across the ALS-FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol. Med. 2018, 10, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 2005, 6, 891–898. [Google Scholar] [CrossRef]
- Biomarkers Definitions Working Group; Atkinson, A.J.J.; Colburn, W.A.; DeGruttola, V.G.; DeMets, D.L.; Downing, G.J.; Zeger, S.L. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar]
- Bibl, M.; Gallus, M.; Welge, V.; Esselmann, H.; Wolf, S.; Rüther, E.; Wiltfang, J. Cerebrospinal fluid amyloid-β 2-42 is decreased in Alzheimer’s, but not in frontotemporal dementia. J. Neural Transm. 2012, 119, 805–813. [Google Scholar] [CrossRef] [Green Version]
- Tondo, G.; Mazzini, L.; Caminiti, S.P.; Sarnelli, M.F.; Corrado, L.; Matheoud, R.; D’Alfonso, S.; Cantello, R.; Sacchetti, G.M.; Perani, D.; et al. Clinical relevance of single-subject brain metabolism patterns in amyotrophic lateral sclerosis mutation carriers. NeuroImage Clin. 2022, 36, 103222. [Google Scholar] [CrossRef]
- Tondo, G.; De Marchi, F. From Biomarkers to Precision Medicine in Neurodegenerative Diseases: Where Are We? J. Clin. Med. 2022, 11, 4515. [Google Scholar] [CrossRef]
- Aronson, J.K.; Ferner, R.E. Biomarkers—A general review. Curr. Protoc. Pharmacol. 2017, 76, 9–23. [Google Scholar] [CrossRef]
- Schünemann, H.; Hill, S.; Guyatt, G.; Akl, E.A.; Ahmed, F. The GRADE approach and Bradford Hill’s criteria for causation. J. Epidemiol. Community Health 2011, 65, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Howick, J.; Glasziou, P.; Aronson, J.K. The evolution of evidence hierarchies: What can Bradford Hill’s ‘guidelines for causation’contribute? J. R. Soc. Med. 2009, 102, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O. Biomarkers for neurodegenerative diseases. Nat. Med. 2021, 27, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; Fang, T.; De Marchi, F.; Neel, D.; Van Weehaeghe, D.; Berry, J.D.; Paganoni, S. Pharmacotherapy for Amyotrophic Lateral Sclerosis: A Review of Approved and Upcoming Agents. Drugs 2022, 82, 1367–1388. [Google Scholar] [CrossRef] [PubMed]
- Trostchansky, A. Overview of lipid biomarkers in amyotrophic lateral sclerosis (ALS). Role Bioact. Lipids Cancer Inflamm. Relat. Dis. 2019, 1161, 233–241. [Google Scholar]
- Ryberg, H.; Bowser, R. Protein biomarkers for amyotrophic lateral sclerosis. Expert Rev. Proteom. 2008, 5, 249–262. [Google Scholar] [CrossRef]
- Turner, M.R.; Kiernan, M.C.; Leigh, P.N.; Talbot, K. Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol. 2009, 8, 94–109. [Google Scholar] [CrossRef]
- Lehnert, S.; Costa, J.; De Carvalho, M.; Kirby, J.; Kuzma-Kozakiewicz, M.; Morelli, C.; Robberecht, W.; Shaw, P.; Silani, V.; Steinacker, P.; et al. Multicentre quality control evaluation of different biomarker candidates for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 344–350. [Google Scholar] [CrossRef]
- Robelin, L.; Gonzalez De Aguilar, J.L. Blood biomarkers for amyotrophic lateral sclerosis: Myth or reality? Biomed. Res. Int. 2014, 2014, 525097. [Google Scholar] [CrossRef] [Green Version]
- Ganesalingam, J.; An, J.; Bowser, R.; Andersen, P.M.; Shaw, C.E. pNfH is a promising biomarker for ALS. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 146–149. [Google Scholar] [CrossRef]
- Kuhle, J.; Lindberg, R.L.P.; Regeniter, A.; Mehling, M.; Steck, A.J.; Kappos, L.; Czaplinski, A. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur. J. Neurol. 2009, 16, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.M.; Simmons, Z.; Beard, J.L.; Stephens, H.E.; Connor, J.R. Plasma biomarkers associated with ALS and their relationship to iron homeostasis. Muscle Nerve 2010, 42, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Zhang, M.; Quiroz Zárate, A.; Jaffa, M.; Yu, H.; Cudkowicz, M.E.; Wills, A.M. Uric acid levels predict survival in men with amyotrophic lateral sclerosis. J. Neurol. 2012, 259, 1923–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-W.; Lin, T.-S.; Lai, M.-L. The correlation between uric acid levels and amyotrophic lateral sclerosis. Am. J. Clin. Med. Res. 2011, 1, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Siddiqui, S.; Gabriely, G.; Lanser, A.J.; Dake, B.; Murugaiyan, G.; Doykan, C.E.; Wu, P.M.; Gali, R.R.; Iyer, L.K.; et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J. Clin. Investig. 2012, 122, 3063–3087. [Google Scholar] [CrossRef] [Green Version]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Huang, A.; Wen, S.; Liao, B.; Appel, S.H. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 2011, 134, 1293–1314. [Google Scholar] [CrossRef]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The need for multi-omics biomarker signatures in precision medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef] [Green Version]
- Domon, B.; Aebersold, R. Mass spectrometry and protein analysis. Science 2006, 312, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef]
- Raghunathan, R.; Turajane, K.; Wong, L.C. Biomarkers in Neurodegenerative Diseases: Proteomics Spotlight on ALS and Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 9299. [Google Scholar] [CrossRef]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and their applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungbauer, A.; Hahn, R. Ion-exchange chromatography. Methods Enzymol. 2009, 463, 349–371. [Google Scholar] [PubMed]
- Hage, D.S.; Anguizola, J.A.; Bi, C.; Li, R.; Matsuda, R.; Papastavros, E.; Pfaunmiller, E.; Vargas, J.; Zheng, X. Pharmaceutical and biomedical applications of affinity chromatography: Recent trends and developments. J. Pharm. Biomed. Anal. 2012, 69, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lequin, R.M. Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). Clin. Chem. 2005, 51, 2415–2418. [Google Scholar] [CrossRef] [Green Version]
- Kurien, B.T.; Scofield, R.H. Western blotting. Methods 2006, 38, 283–293. [Google Scholar] [CrossRef]
- Yates, J.R., III. A century of mass spectrometry: From atoms to proteomes. Nat. Methods 2011, 8, 633–637. [Google Scholar] [CrossRef]
- Smith, J.B. Peptide Sequencing by Edman Degradation; Macmillan Publishers Ltd, Nature Publishing Group: London, UK, 2001; pp. 1–3. [Google Scholar] [CrossRef]
- Shiio, Y.; Aebersold, R. Quantitative proteome analysis using isotope-coded affinity tags and mass spectrometry. Nat. Protoc. 2006, 1, 139–145. [Google Scholar] [CrossRef]
- Wiese, S.; Reidegeld, K.A.; Meyer, H.E.; Warscheid, B. Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research. Proteomics 2007, 7, 340–350. [Google Scholar] [CrossRef]
- Schmidt, A.; Forne, I.; Imhof, A. Bioinformatic analysis of proteomics data. BMC Syst. Biol. 2014, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Monti, C.; Zilocchi, M.; Colugnat, I.; Alberio, T. Proteomics turns functional. J. Proteom. 2019, 198, 36–44. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Kurtishi, A.; Rosen, B.; Patil, K.S.; Alves, G.W.; Møller, S.G. Cellular proteostasis in neurodegeneration. Mol. Neurobiol. 2019, 56, 3676–3689. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Teichmann, S.A. Structure, dynamics, assembly, and evolution of protein complexes. Annu. Rev. Biochem. 2015, 84, 551–575. [Google Scholar] [CrossRef]
- Cheng, F.; De Luca, A.; Hogan, A.L.; Rayner, S.L.; Davidson, J.M.; Watchon, M.; Stevens, C.H.; Muñoz, S.S.; Ooi, L.; Yerbury, J.J.; et al. Unbiased label-free quantitative proteomics of cells expressing amyotrophic lateral sclerosis (ALS) mutations in CCNF reveals activation of the apoptosis pathway: A workflow to screen pathogenic gene mutations. Front. Mol. Neurosci. 2021, 14, 627740. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, H.; Hornburg, D.; Czuppa, M.; Bader, J.; Michaelsen, M.; Farny, D.; Arzberger, T.; Mann, M.; Meissner, F.; Edbauer, D. Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci. Alliance 2018, 1, e201800070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeynaems, S.; Bogaert, E.; Kovacs, D.; Konijnenberg, A.; Timmerman, E.; Volkov, A.; Guharoy, M.; De Decker, M.; Jaspers, T.; Ryan, V.H.; et al. Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol. Cell 2017, 65, 1044–1055. [Google Scholar] [CrossRef] [Green Version]
- Lualdi, M.; Shafique, A.; Pedrini, E.; Pieroni, L.; Greco, V.; Castagnola, M.; Cucina, G.; Corrado, L.; Di Pierro, A.; De Marchi, F.; et al. C9ORF72 repeat expansion affects the proteome of primary skin fibroblasts in ALS. Int. J. Mol. Sci. 2021, 22, 10385. [Google Scholar] [CrossRef]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef]
- McAlary, L.; Aquilina, J.A.; Yerbury, J.J. Susceptibility of mutant SOD1 to form a destabilized monomer predicts cellular aggregation and toxicity but not in vitro aggregation propensity. Front. Neurosci. 2016, 10, 499. [Google Scholar] [CrossRef] [Green Version]
- Štalekar, M.; Yin, X.; Rebolj, K.; Darovic, S.; Troakes, C.; Mayr, M.; Shaw, C.E.; Rogelj, B. Proteomic analyses reveal that loss of TDP-43 affects RNA processing and intracellular transport. Neuroscience 2015, 293, 157–170. [Google Scholar] [CrossRef]
- Stella, R.; Bonadio, R.S.; Cagnin, S.; Massimino, M.L.; Bertoli, A.; Peggion, C. Perturbations of the Proteome and of Secreted Metabolites in Primary Astrocytes from the hSOD1 (G93A) ALS Mouse Model. Int. J. Mol. Sci. 2021, 22, 7028. [Google Scholar] [CrossRef]
- Ruegsegger, C.; Maharjan, N.; Goswami, A.; Filézac de L’Etang, A.; Weis, J.; Troost, D.; Heller, M.; Gut, H.; Saxena, S. Aberrant association of misfolded SOD1 with Na+/K+ ATPase-α3 impairs its activity and contributes to motor neuron vulnerability in ALS. Acta Neuropathol. 2016, 131, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Hogan, A.L.; Don, E.K.; Rayner, S.L.; Lee, A.; Laird, A.S.; Watchon, M.; Winnick, C.; Tarr, I.S.; Morsch, M.; Fifita, J.A.; et al. Expression of ALS/FTD-linked mutant CCNF in zebrafish leads to increased cell death in the spinal cord and an aberrant motor phenotype. Hum. Mol. Genet. 2017, 26, 2616–2626. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Mogel, H.; Lehmensiek, V.; Ahlert, T.; Süssmuth, S.; Ludolph, A.C.; Tumani, H. Proteome analysis of cerebrospinal fluid in amyotrophic lateral sclerosis (ALS). Neurochem. Res. 2008, 33, 2358–2363. [Google Scholar] [CrossRef]
- Ranganathan, S.; Williams, E.; Ganchev, P.; Gopalakrishnan, V.; Lacomis, D.; Urbinelli, L.; Newhall, K.; Cudkowicz, M.E.; Brown, R.H., Jr.; Bowser, R. Proteomic profiling of cerebrospinal fluid identifies biomarkers for amyotrophic lateral sclerosis. J. Neurochem. 2005, 95, 1461–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendonça, D.M.F.; Pizzati, L.; Mostacada, K.; de S. Martins, S.C.; Higashi, R.; Ayres Sá, L.; Moura Neto, V.; Chimelli, L.; Martinez, A.M. Neuroproteomics: An insight into ALS. Neurol. Res. 2012, 34, 937–943. [Google Scholar] [CrossRef]
- Brettschneider, J.; Lehmensiek, V.; Mogel, H.; Pfeifle, M.; Dorst, J.; Hendrich, C.; Ludolph, A.C.; Tumani, H. Proteome analysis reveals candidate markers of disease progression in amyotrophic lateral sclerosis (ALS). Neurosci. Lett. 2010, 468, 23–27. [Google Scholar] [CrossRef]
- Sjoqvist, S.; Otake, K. A pilot study using proximity extension assay of cerebrospinal fluid and its extracellular vesicles identifies novel amyotrophic lateral sclerosis biomarker candidates. Biochem. Biophys. Res. Commun. 2022, 613, 166–173. [Google Scholar] [CrossRef]
- Varghese, A.M.; Sharma, A.; Mishra, P.; Vijayalakshmi, K.; Harsha, H.C.; Sathyaprabha, T.N.; Bharath, S.M.; Nalini, A.; Alladi, P.A.; Raju, T.R. Chitotriosidase-a putative biomarker for sporadic amyotrophic lateral sclerosis. Clin. Proteom. 2013, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Von Neuhoff, N.; Oumeraci, T.; Wolf, T.; Kollewe, K.; Bewerunge, P.; Neumann, B.; Brors, B.; Bufler, J.; Wurster, U.; Schlegelberger, B.; et al. Monitoring CSF proteome alterations in amyotrophic lateral sclerosis: Obstacles and perspectives in translating a novel marker panel to the clinic. PLoS ONE 2012, 7, e44401. [Google Scholar] [CrossRef]
- Collins, M.A.; An, J.; Hood, B.L.; Conrads, T.P.; Bowser, R.P. Label-Free LC–MS/MS proteomic analysis of cerebrospinal fluid identifies Protein/Pathway alterations and candidate biomarkers for amyotrophic lateral sclerosis. J. Proteome Res. 2015, 14, 4486–4501. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Benito, P.; Povedano, M.; Domínguez, R.; Marco, C.; Colomina, M.J.; López-Pérez, Ó.; Santana, I.; Baldeiras, I.; Martínez-Yelámos, S.; Zerr, I.; et al. Increased CXC motif chemokine ligand 12 levels in cerebrospinal fluid as a candidate biomarker in sporadic amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2020, 21, 8680. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wuolikainen, A.; Wu, J.; Öhman, A.; Wingsle, G.; Moritz, T.; Andersen, P.M.; Forsgren, L.; Trupp, M. Targeted multiple reaction monitoring analysis of CSF identifies UCHL1 and GPNMB as candidate biomarkers for ALS. J. Mol. Neurosci. 2019, 69, 643–657. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, X.; Wu, J.; Ren, H.; Wang, J.; Ding, Z.T.; Jiang, Y.P. Proteomic analysis of cerebrospinal fluid in amyotrophic lateral sclerosis. Exp. Ther. Med. 2016, 11, 2095–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, A.; Iannaccone, S.; Sferrazza, B.; De Monte, L.; Cappa, S.; Franciotta, D.; Olivieri, S.; Alessio, M. Differential expression of ceruloplasmin isoforms in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. PROTEOMICS–Clin. Appl. 2008, 2, 1628–1637. [Google Scholar] [CrossRef]
- Mitchell, R.M.; Freeman, W.M.; Randazzo, W.T.; Stephens, H.E.; Beard, J.L.; Simmons, Z.; Connor, J.R. A CSF biomarker panel for identification of patients with amyotrophic lateral sclerosis. Neurology 2009, 72, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Hansson, S.F.; Puchades, M.; Blennow, K.; Sjögren, M.; Davidsson, P. Validation of a prefractionation method followed by two-dimensional electrophoresis–Applied to cerebrospinal fluid proteins from frontotemporal dementia patients. Proteome Sci. 2004, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Davidsson, P.; Sjögren, M.; Andreasen, N.; Lindbjer, M.; Nilsson, C.L.; Westman-Brinkmalm, A.; Blennow, K. Studies of the pathophysiological mechanisms in frontotemporal dementia by proteome analysis of CSF proteins. Mol. Brain Res. 2002, 109, 128–133. [Google Scholar] [CrossRef]
- Van der Ende, E.L.; Meeter, L.H.; Stingl, C.; van Rooij, J.G.J.; Stoop, M.P.; Nijholt, D.A.T.; Sanchez-Valle, R.; Graff, C.; Öijerstedt, L.; Grossman, M.; et al. Novel CSF biomarkers in genetic frontotemporal dementia identified by proteomics. Ann. Clin. Transl. Neurol. 2019, 6, 698–707. [Google Scholar] [CrossRef]
- Mellinger, A.L.; Griffith, E.H.; Bereman, M.S. Peptide variability and signatures associated with disease progression in CSF collected longitudinally from ALS patients. Anal. Bioanal. Chem. 2020, 412, 5465–5475. [Google Scholar] [CrossRef]
- Comi, C.; Tondo, G. Insights into the protective role of immunity in neurodegenerative disease. Neural Regen. Res. 2017, 12, 64. [Google Scholar] [CrossRef]
- Cao, M.C.; Cawston, E.E.; Chen, G.; Brooks, C.; Douwes, J.; McLean, D.; Graham, E.S.; Dragunow, M.; Scotter, E.L. Serum biomarkers of neuroinflammation and blood-brain barrier leakage in amyotrophic lateral sclerosis. BMC Neurol. 2022, 22, 216. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, S.; Gianazza, E.; Banfi, C.; Marocchi, A.; Lunetta, C.; Penco, S.; Bonomi, F.; Iametti, S. Serum proteome in a sporadic amyotrophic lateral sclerosis geographical cluster. PROTEOMICS–Clin. Appl. 2017, 11, 1700043. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Lee, A.; Nouwens, A.; Henderson, R.D.; McCombe, P.A. Mass spectrometry analysis of plasma from amyotrophic lateral sclerosis and control subjects. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Katzeff, J.S.; Bright, F.; Lo, K.; Kril, J.J.; Connolly, A.; Crossett, B.; Ittner, L.M.; Kassiou, M.; Loy, C.T.; Hodges, J.R.; et al. Altered serum protein levels in frontotemporal dementia and amyotrophic lateral sclerosis indicate calcium and immunity dysregulation. Sci. Rep. 2020, 10, 13741. [Google Scholar] [CrossRef]
- Gozal, Y.M.; Dammer, E.B.; Duong, D.M.; Cheng, D.; Gearing, M.; Rees, H.D.; Peng, J.; Lah, J.J.; Levey, A.I. Proteomic analysis of hippocampal dentate granule cells in frontotemporal lobar degeneration: Application of laser capture technology. Front. Neurol. 2011, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Gozal, Y.M.; Seyfried, N.T.; Gearing, M.; Glass, J.D.; Heilman, C.J.; Wuu, J.; Duong, D.M.; Cheng, D.; Xia, Q.; Rees, H.D.; et al. Aberrant septin 11 is associated with sporadic frontotemporal lobar degeneration. Mol. Neurodegener. 2011, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Viodé, A.; Fournier, C.; Camuzat, A.; Fenaille, F.; Bank, N.B.; Latouche, M.; Elahi, F.; Le Ber, I.; Junot, C.; Lamari, F.; et al. New antibody-free mass spectrometry-based quantification reveals that C9ORF72 long protein isoform is reduced in the frontal cortex of hexanucleotide-repeat expansion carriers. Front. Neurosci. 2018, 12, 589. [Google Scholar] [CrossRef]
- Engelen-Lee, J.; Blokhuis, A.M.; Spliet, W.G.M.; Pasterkamp, R.J.; Aronica, E.; Demmers, J.A.A.; Broekhuizen, R.; Nardo, G.; Bovenschen, N.; Van Den Berg, L.H.; et al. Proteomic profiling of the spinal cord in ALS: Decreased ATP5D levels suggest synaptic dysfunction in ALS pathogenesis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Liu, C.; Li, J.; Azadzoi, K.; Yang, Y.; Fei, Z.; Dou, K.; Kowall, N.W.; Choi, H.P.; Vieira, F.; et al. Proteomic analysis reveals differentially regulated protein acetylation in human amyotrophic lateral sclerosis spinal cord. PLoS ONE 2013, 8, e80779. [Google Scholar] [CrossRef]
- Iridoy, M.O.; Zubiri, I.; Zelaya, M.V.; Martinez, L.; Ausín, K.; Lachen-Montes, M.; Santamaría, E.; Fernandez-Irigoyen, J.; Jericó, I. Neuroanatomical quantitative proteomics reveals common pathogenic biological routes between amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Int. J. Mol. Sci. 2018, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Boyarko, B.; Hook, V. Human Tau Isoforms and Proteolysis for Production of Toxic Tau Fragments in Neurodegeneration. Front. Neurosci. 2021, 15, 702788. [Google Scholar] [CrossRef] [PubMed]
- Virgilio, E.; De Marchi, F.; Contaldi, E.; Dianzani, U.; Cantello, R.; Mazzini, L.; Comi, C. The Role of Tau beyond Alzheimer’s Disease: A Narrative Review. Biomedicines 2022, 10, 760. [Google Scholar] [CrossRef] [PubMed]


| Technique | Primary Function |
|---|---|
| Proteomics | characterization of protein constituents in biological samples
|
| Genomics | set of DNA sequences provided by genome-wide association studies and, more recently, next-generation whole exome and whole genome sequencing data |
| Transcriptomics | representing gene expression patterns |
| Metabolomics | characterization of metabolic profiles |
| Lipidomics | characterization of the complete collection of lipids |
| Epigenomics | profile of the modifications to DNA that control gene expression |
| Exposomics | the sum of exposure an individual incurs over a period of time |
| Microbiomics | characterization of the microbes that reside in or on an individual |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vignaroli, F.; Mele, A.; Tondo, G.; De Giorgis, V.; Manfredi, M.; Comi, C.; Mazzini, L.; De Marchi, F. The Need for Biomarkers in the ALS–FTD Spectrum: A Clinical Point of View on the Role of Proteomics. Proteomes 2023, 11, 1. https://doi.org/10.3390/proteomes11010001
Vignaroli F, Mele A, Tondo G, De Giorgis V, Manfredi M, Comi C, Mazzini L, De Marchi F. The Need for Biomarkers in the ALS–FTD Spectrum: A Clinical Point of View on the Role of Proteomics. Proteomes. 2023; 11(1):1. https://doi.org/10.3390/proteomes11010001
Chicago/Turabian StyleVignaroli, Francesca, Angelica Mele, Giacomo Tondo, Veronica De Giorgis, Marcello Manfredi, Cristoforo Comi, Letizia Mazzini, and Fabiola De Marchi. 2023. "The Need for Biomarkers in the ALS–FTD Spectrum: A Clinical Point of View on the Role of Proteomics" Proteomes 11, no. 1: 1. https://doi.org/10.3390/proteomes11010001