
Microsatellite Content in 397 Nuclear Exons and Their Flanking Regions in the Fern Family Ophioglossaceae

Abstract
:1. Introduction
2. Results
2.1. SSR Number and Density-Exons
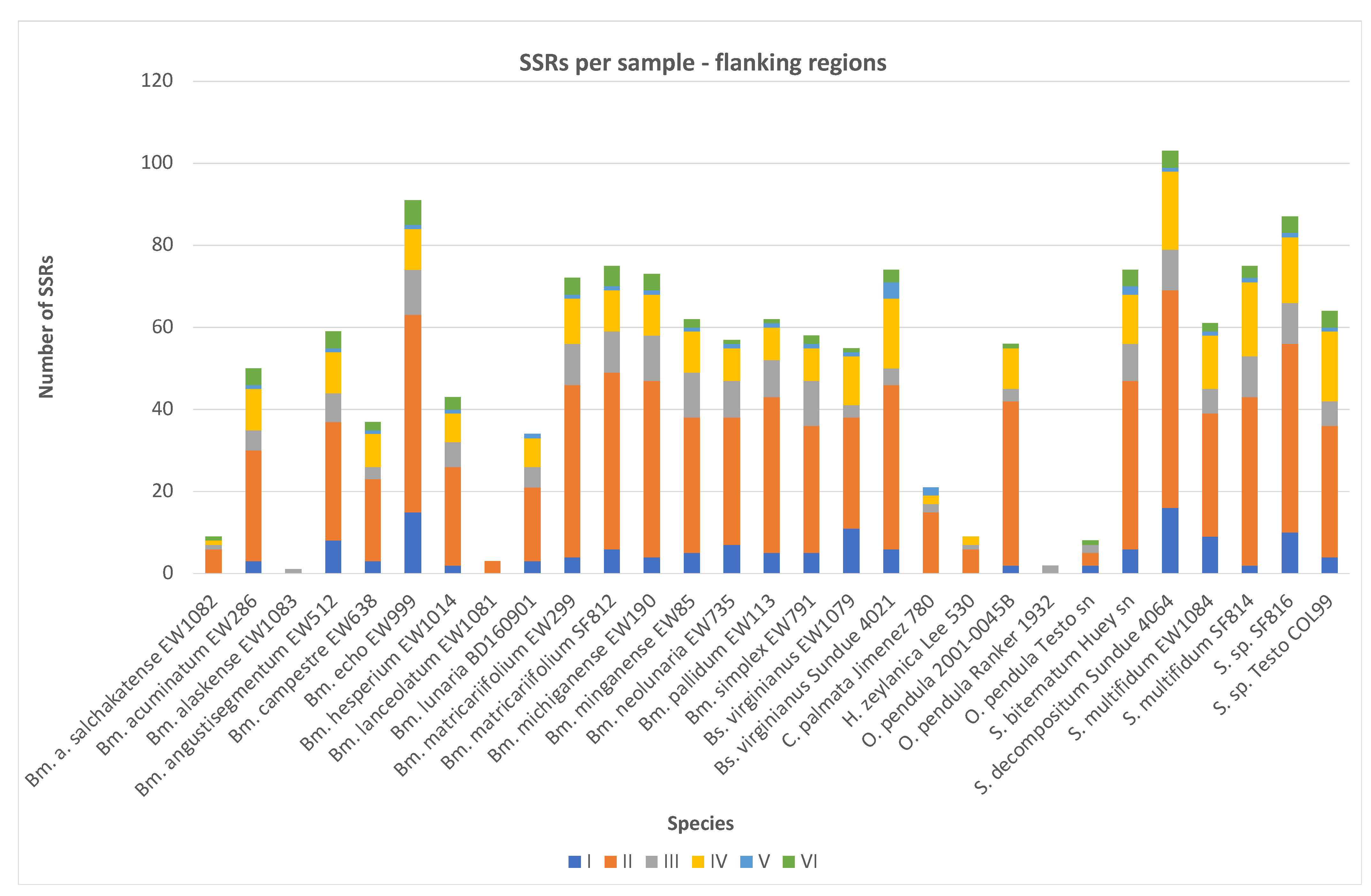
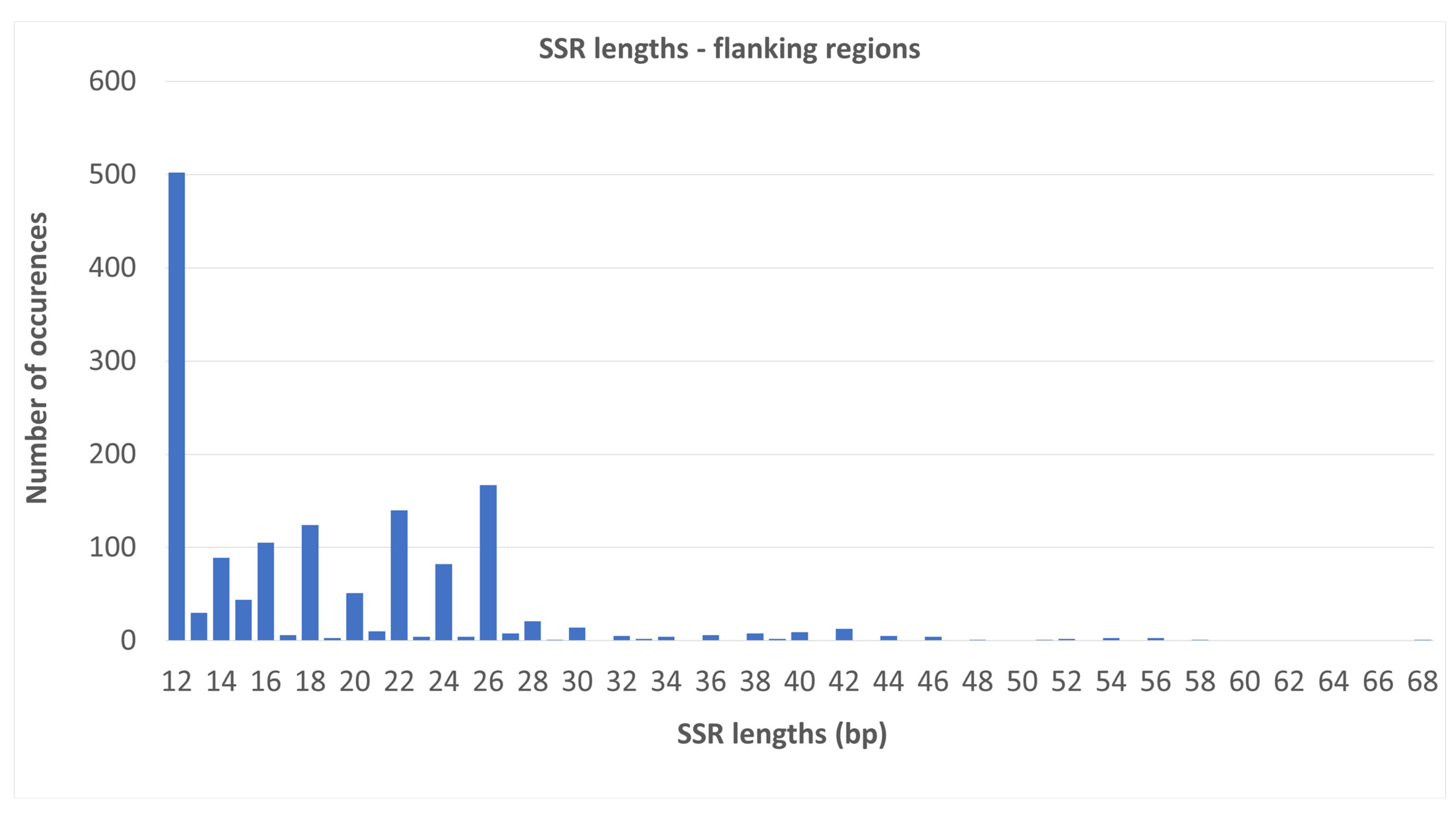
2.2. SSR Number and Density—Flanking Regions
2.3. Unique, Shared and Polymorphic SSRs
2.4. Compound Form
2.5. Primer Design
3. Discussion
4. Materials and Methods
4.1. Samples
4.2. SSR Mining and Primer Design
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jouffroy, O.; Saha, S.; Mueller, L.; Quesneville, H.; Maumus, F. Comprehensive Repeatome Annotation Reveals Strong Potential Impact of Repetitive Elements on Tomato Ripening. BMC Genom. 2016, 17, 624. [Google Scholar] [CrossRef]
- Vieira, M.L.C.; Santini, L.; Diniz, A.L.; de Munhoz, C.F. Microsatellite Markers: What They Mean and Why They Are so Useful. Genet. Mol. Biol. 2016, 39, 312–328. [Google Scholar] [CrossRef]
- Li, Y.-C.; Korol, A.B.; Fahima, T.; Nevo, E. Microsatellites within Genes: Structure, Function, and Evolution. Mol. Biol. Evol. 2004, 21, 991–1007. [Google Scholar] [CrossRef]
- Moran, C. Microsatellite Repeats in Pig (Sus domestica) and Chicken (Gallus domesticus) Genomes. J. Hered. 1993, 84, 274–280. [Google Scholar] [CrossRef]
- Song, X.; Yang, Q.; Bai, Y.; Gong, K.; Wu, T.; Yu, T.; Pei, Q.; Duan, W.; Huang, Z.; Wang, Z.; et al. Comprehensive Analysis of SSRs and Database Construction Using All Complete Gene-Coding Sequences in Major Horticultural and Representative Plants. Hortic. Res. 2021, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Wells, R.D. Non-B DNA Conformations, Genomic Rearrangements, and Human Disease. J. Biol. Chem. 2004, 279, 47411–47414. [Google Scholar] [CrossRef] [PubMed]
- Rohilla, K.J.; Gagnon, K.T. RNA Biology of Disease-Associated Microsatellite Repeat Expansions. Acta Neuropathol. Commun. 2017, 5, 63. [Google Scholar] [CrossRef]
- Gao, C.; Ren, X.; Mason, A.S.; Li, J.; Wang, W.; Xiao, M.; Fu, D.; Gao, C.; Ren, X.; Mason, A.S.; et al. Revisiting an Important Component of Plant Genomes: Microsatellites. Funct. Plant Biol. 2013, 40, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic Distribution, Putative Functions and Mutational Mechanisms: A Review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Germain-Aubrey, C.C.; Nelson, C.; Soltis, D.E.; Soltis, P.S.; Gitzendanner, M.A. Are Microsatellite Fragment Lengths Useful for Population-Level Studies? The Case of Polygala lewtonii (Polygalaceae). Appl. Plant Sci. 2016, 4, 1500115. [Google Scholar] [CrossRef]
- Putman, A.I.; Carbone, I. Challenges in Analysis and Interpretation of Microsatellite Data for Population Genetic Studies. Ecol. Evol. 2014, 4, 4399–4428. [Google Scholar] [CrossRef] [PubMed]
- Aiello, D.; Ferradini, N.; Torelli, L.; Volpi, C.; Lambalk, J.; Russi, L.; Albertini, E. Evaluation of Cross-Species Transferability of SSR Markers in Foeniculum vulgare. Plants 2020, 9, 175. [Google Scholar] [CrossRef]
- Ngangkham, U.; Dash, S.; Parida, M.; Samantaray, S.; Nongthombam, D.; Yadav, M.K.; Kumar, A.; Chidambaranathan, P.; Katara, J.L.; Patra, B.C.; et al. The Potentiality of Rice Microsatellite Markers in Assessment of Cross-Species Transferability and Genetic Diversity of Rice and Its Wild Relatives. 3 Biotech 2019, 9, 217. [Google Scholar] [CrossRef]
- Maurya, S.; Darshetkar, A.M.; Yi, D.-K.; Kim, J.; Lee, C.; Ali, M.A.; Choi, S.; Choudhary, R.K.; Kim, S.-Y. Plastome Comparison and Evolution within the Tribes of Plantaginaceae: Insights from an Asian Gypsyweed. Saudi J. Biol. Sci. 2020, 27, 3489–3498. [Google Scholar] [CrossRef]
- Yue, J.; Lu, Q.; Ni, Y.; Chen, P.; Liu, C. Comparative Analysis of the Plastid and Mitochondrial Genomes of Artemisia giraldii Pamp. Sci. Rep. 2022, 12, 13931. [Google Scholar] [CrossRef]
- PPG, I. A Community-Derived Classification for Extant Lycophytes and Ferns. J. Syst. Evol. 2016, 54, 563–603. [Google Scholar] [CrossRef]
- Kuo, L.-Y.; Tang, S.K.; Kao, T.-T.; Ebihara, A.; Fawcett, S.; Hsiao, M.-C.; Shinohara, W.; Dauphin, B. A Dormant Resource for Genome Size Estimation in Ferns: C-Value Inference of the Ophioglossaceae Using Herbarium Specimen Spores. Appl. Plant Sci. 2021, 9, e11452. [Google Scholar] [CrossRef]
- Khandelwal, S. Chromosome Evolution in the Genus Ophioglossum L. Bot. J. Linn. Soc. 1990, 102, 205–217. [Google Scholar] [CrossRef]
- Shinohara, W.; Nakato, N.; Yatabe-Kakugawa, Y.; Oka, T.; Kim, J.K.; Murakami, N.; Noda, H.; Sahashi, N. The Use of MatK in Ophioglossaceae Phylogeny and the Determination of Mankyua Chromosome Number Shed Light on Chromosome Number Evolution in Ophioglossaceae. SBOT 2013, 38, 564–570. [Google Scholar] [CrossRef]
- Fan, R.; Ma, W.; Liu, S.; Huang, Q. Integrated Analysis of Three Newly Sequenced Fern Chloroplast Genomes: Genome Structure and Comparative Analysis. Ecol. Evol. 2021, 11, 4550–4563. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xing, Y.; Wang, B.; Liu, C.; Wang, W.; Kang, T. Plastid Genome and Composition Analysis of Two Medical Ferns: Dryopteris crassirhizoma Nakai and Osmunda japonica Thunb. Chin. Med. 2019, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Z.; Su, Y.; Wang, T. Comparative Genomic Analysis of Polypodiaceae Chloroplasts Reveals Fine Structural Features and Dynamic Insertion Sequences. BMC Plant Biol. 2021, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Koubínová, D.; Kuo, L.-Y.; Ebihara, A.; Grant, J.R. Simple Sequence Repeat (SSR) Mining in the Chloroplast Genomes of the Ancient Fern Family Ophioglossaceae. 2024; in preparation. [Google Scholar]
- Liu, Y.; Xiao, X.; Li, G.; Zhu, C.; Yang, K.; Feng, X.; Lou, Y.; Gao, Z. Comprehensive Analyses of Simple Sequence Repeat (SSR) in Bamboo Genomes and Development of SSR Markers with Peroxidase Genes. Genes 2022, 13, 1518. [Google Scholar] [CrossRef]
- Scotti, I.; Magni, F.; Fink, R.; Powell, W.; Binelli, G.; Hedley, P.E. Microsatellite Repeats Are Not Randomly Distributed within Norway Spruce (Picea abies K.) Expressed Sequences. Genome 2000, 43, 41–46. [Google Scholar] [CrossRef]
- Dang, Z.; Huang, L.; Jia, Y.; Lockhart, P.J.; Fong, Y.; Tian, Y. Identification of Genic SSRs Provide a Perspective for Studying Environmental Adaptation in the Endemic Shrub Tetraena mongolica. Genes 2020, 11, 322. [Google Scholar] [CrossRef]
- Wang, F.-G.; Wang, A.-H.; Bai, C.-K.; Jin, D.-M.; Nie, L.-Y.; Harris, A.; Che, L.; Wang, J.-J.; Li, S.-Y.; Xu, L.; et al. Genome Size Evolution of the Extant Lycophytes and Ferns. Plant Divers. 2022, 44, 141–152. [Google Scholar] [CrossRef]
- Breinholt, J.W.; Carey, S.B.; Tiley, G.P.; Davis, E.C.; Endara, L.; McDaniel, S.F.; Neves, L.G.; Sessa, E.B.; von Konrat, M.; Chantanaorrapint, S.; et al. A Target Enrichment Probe Set for Resolving the Flagellate Land Plant Tree of Life. Appl. Plant Sci. 2021, 9, e11406. [Google Scholar] [CrossRef]
- Carpenter, E.J.; Matasci, N.; Ayyampalayam, S.; Wu, S.; Sun, J.; Yu, J.; Jimenez Vieira, F.R.; Bowler, C.; Dorrell, R.G.; Gitzendanner, M.A.; et al. Access to RNA-Sequencing Data from 1173 Plant Species: The 1000 Plant Transcriptomes Initiative (1KP). GigaScience 2019, 8, giz126. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-Web: A Web Server for Microsatellite Prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Schneider, A.; Dessimoz, C.; Gonnet, G.H. OMA Browser—Exploring Orthologous Relations across 352 Complete Genomes. Bioinformatics 2007, 23, 2180–2182. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New Capabilities and Interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Species | Voucher | Sequencing ID | Repeat Category-Exons | Total Number | No. of Nucleotides in Exons (bp) | SSR Density | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | ||||||
| Botrychium acuminatum | EW286 | P015 WG11 | 0 | 1 | 1 | 1 | 0 | 0 | 3 | 66,866 | 0.0449 |
| Botrychium alaskense | EW1083 | P03 WD06 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5522 | 0 |
| Botrychium alaskense salchakatense | EW1082 | P03 WC06 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 21,221 | 0 |
| Botrychium angustisegmentum | EW512 | P015 WA12 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 68,226 | 0.0147 |
| Botrychium campestre | EW638 | P015 WB12 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 66,839 | 0.015 |
| Botrychium echo | EW999 | P015 WE12 | 0 | 1 | 1 | 0 | 0 | 0 | 2 | 71,166 | 0.0281 |
| Botrychium hesperium | EW1014 | P015 WF12 | 0 | 1 | 1 | 1 | 0 | 0 | 3 | 67,467 | 0.0445 |
| Botrychium lanceolatum | EW1081 | P03 WB06 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6905 | 0 |
| Botrychium lunaria | BD160901 | P015 WG12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 60,524 | 0 |
| Botrychium matricariifolium | EW299 | P015 WH11 | 0 | 1 | 1 | 1 | 0 | 0 | 3 | 69,113 | 0.0434 |
| Botrychium matricariifolium | SF812 | P028 WD08 | 0 | 1 | 1 | 1 | 0 | 0 | 3 | 70,879 | 0.0423 |
| Botrychium michiganense | EW190 | P015 WF11 | 0 | 1 | 1 | 1 | 0 | 0 | 3 | 70,189 | 0.0427 |
| Botrychium minganense | EW85 | P015 WD11 | 0 | 1 | 1 | 0 | 0 | 0 | 2 | 70,243 | 0.0285 |
| Botrychium neolunaria | EW735 | P015 WC12 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 70,226 | 0.0142 |
| Botrychium pallidum | EW113 | P015 WE11 | 0 | 1 | 1 | 0 | 0 | 0 | 2 | 70,056 | 0.0285 |
| Botrychium simplex | EW791 | P015 WD12 | 0 | 0 | 1 | 1 | 0 | 0 | 2 | 69,654 | 0.0287 |
| Botrypus virginianus | Sundue 4021 | P055 WB10 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 72,233 | 0.0138 |
| Botrypus virginianus | EW1079 | P03 WA06 | 0 | 1 | 1 | 0 | 0 | 0 | 2 | 72,435 | 0.0276 |
| Cheiroglossa palmata | Jimenez 780 | P071 WF12 | 0 | 0 | 1 | 1 | 0 | 0 | 2 | 36,029 | 0.0555 |
| Helminthostachys zeylanica | Lee 530 | P069 WF05 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 52,149 | 0.0384 |
| Ophioderma pendula | 2001-0045B | P016 WH10 | 0 | 0 | 1 | 4 | 0 | 0 | 5 | 70,580 | 0.0708 |
| Ophioderma pendula | Ranker 1932 | P067 WA10 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 17,091 | 0.0585 |
| Ophioderma pendula | Testo sn | P069 WH03 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 25,543 | 0 |
| Sceptridium biternatum | Huey sn | P072 WC08 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 72,420 | 0.0138 |
| Sceptridium decompositum | Sundue 4064 | P063 WA11 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 71,323 | 0.014 |
| Sceptridium multifidum | SF814 | P028 WE08 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 70,552 | 0.0142 |
| Sceptridium multifidum | EW1084 | P03 WE06 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 71,229 | 0.014 |
| Sceptridium sp. | SF816 | P028 WG08 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 71,359 | 0.014 |
| Sceptridium sp. | Testo COL99 | P058 WH09 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 69,912 | 0.0143 |
| Total number of SSRs per category | 0 | 16 | 18 | 11 | 0 | 0 | 45 | ||||
| Species | Voucher | Sequencing ID | Repeat Category-Flanking Region | Total Number | Compound | No. of Nucleotides | SSR Density | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | |||||||
| Botrychium acuminatum | EW286 | P015 WG11 | 3 | 27 | 5 | 10 | 1 | 4 | 50 | 1 | 124,140 | 0.4028 |
| Botrychium alaskense | EW1083 | P03 WD06 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 2608 | 0.3834 |
| Botrychium alaskense salchakatense | EW1082 | P03 WC06 | 0 | 6 | 1 | 1 | 0 | 1 | 9 | 0 | 12,969 | 0.694 |
| Botrychium angustisegmentum | EW512 | P015 WA12 | 8 | 29 | 7 | 10 | 1 | 4 | 59 | 0 | 148,071 | 0.3985 |
| Botrychium campestre | EW638 | P015 WB12 | 3 | 20 | 3 | 8 | 1 | 2 | 37 | 0 | 106,754 | 0.3466 |
| Botrychium echo | EW999 | P015 WE12 | 15 | 48 | 11 | 10 | 1 | 6 | 91 | 6 | 198,825 | 0.4577 |
| Botrychium hesperium | EW1014 | P015 WF12 | 2 | 24 | 6 | 7 | 1 | 3 | 43 | 3 | 134,642 | 0.3194 |
| Botrychium lanceolatum | EW1081 | P03 WB06 | 0 | 3 | 0 | 0 | 0 | 0 | 3 | 0 | 3714 | 0.8078 |
| Botrychium lunaria | BD160901 | P015 WG12 | 3 | 18 | 5 | 7 | 1 | 0 | 34 | 1 | 106,522 | 0.3192 |
| Botrychium matricariifolium | EW299 | P015 WH11 | 4 | 42 | 10 | 11 | 1 | 4 | 72 | 3 | 164,355 | 0.4381 |
| Botrychium matricariifolium | SF812 | P028 WD08 | 6 | 43 | 10 | 10 | 1 | 5 | 75 | 3 | 202,143 | 0.371 |
| Botrychium michiganense | EW190 | P015 WF11 | 4 | 43 | 11 | 10 | 1 | 4 | 73 | 4 | 172,060 | 0.4243 |
| Botrychium minganense | EW85 | P015 WD11 | 5 | 33 | 11 | 10 | 1 | 2 | 62 | 0 | 166,287 | 0.3728 |
| Botrychium neolunaria | EW735 | P015 WC12 | 7 | 31 | 9 | 8 | 1 | 1 | 57 | 2 | 154,150 | 0.3698 |
| Botrychium pallidum | EW113 | P015 WE11 | 5 | 38 | 9 | 8 | 1 | 1 | 62 | 0 | 177,548 | 0.3492 |
| Botrychium simplex | EW791 | P015 WD12 | 5 | 31 | 11 | 8 | 1 | 2 | 58 | 1 | 135,075 | 0.4294 |
| Botrypus virginianus | EW1079 | P03 WA06 | 11 | 27 | 3 | 12 | 1 | 1 | 55 | 1 | 152,100 | 0.3616 |
| Botrypus virginianus | Sundue 4021 | P055 WB10 | 6 | 40 | 4 | 17 | 4 | 3 | 74 | 7 | 198,398 | 0.373 |
| Cheiroglossa palmata | Jimenez 780 | P071 WF12 | 0 | 15 | 2 | 2 | 2 | 0 | 21 | 0 | 32,181 | 0.6526 |
| Helminthostachys zeylanica | Lee 530 | P069 WF05 | 0 | 6 | 1 | 2 | 0 | 0 | 9 | 1 | 46,126 | 0.1951 |
| Ophioderma pendula | 2001-0045B | P016 WH10 | 2 | 40 | 3 | 10 | 0 | 1 | 56 | 2 | 112,807 | 0.4964 |
| Ophioderma pendula | Ranker 1932 | P067 WA10 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 6704 | 0.2983 |
| Ophioderma pendula | Testo sn | P069 WH03 | 2 | 3 | 2 | 0 | 0 | 1 | 8 | 1 | 20,763 | 0.3853 |
| Sceptridium biternatum | Huey sn | P072 WC08 | 6 | 41 | 9 | 12 | 2 | 4 | 74 | 2 | 193,043 | 0.3833 |
| Sceptridium decompositum | Sundue 4064 | P063 WA11 | 16 | 53 | 10 | 19 | 1 | 4 | 103 | 7 | 237,178 | 0.4343 |
| Sceptridium multifidum | EW1084 | P03 WE06 | 9 | 30 | 6 | 13 | 1 | 2 | 61 | 4 | 146,786 | 0.4156 |
| Sceptridium multifidum | SF814 | P028 WE08 | 2 | 41 | 10 | 18 | 1 | 3 | 75 | 5 | 205,941 | 0.3642 |
| Sceptridium sp. | SF816 | P028 WG08 | 10 | 46 | 10 | 16 | 1 | 4 | 87 | 5 | 213,200 | 0.4081 |
| Sceptridium sp. | Testo COL99 | P058 WH09 | 4 | 32 | 6 | 17 | 1 | 4 | 64 | 2 | 151,207 | 0.4233 |
| Total number of SSRs per category | 138 | 810 | 178 | 256 | 27 | 66 | 1475 | 61 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koubínová, D.; GoFlag Consortium; Grant, J.R. Microsatellite Content in 397 Nuclear Exons and Their Flanking Regions in the Fern Family Ophioglossaceae. Plants 2024, 13, 713. https://doi.org/10.3390/plants13050713
Koubínová D, GoFlag Consortium, Grant JR. Microsatellite Content in 397 Nuclear Exons and Their Flanking Regions in the Fern Family Ophioglossaceae. Plants. 2024; 13(5):713. https://doi.org/10.3390/plants13050713
Chicago/Turabian StyleKoubínová, Darina, GoFlag Consortium, and Jason R. Grant. 2024. "Microsatellite Content in 397 Nuclear Exons and Their Flanking Regions in the Fern Family Ophioglossaceae" Plants 13, no. 5: 713. https://doi.org/10.3390/plants13050713